
Structural Contour Map of the Iota Carbonic Anhydrase from the Diatom Thalassiosira pseudonana Using a Multiprong Approach

 , , and
, , and
Abstract
:1. Introduction
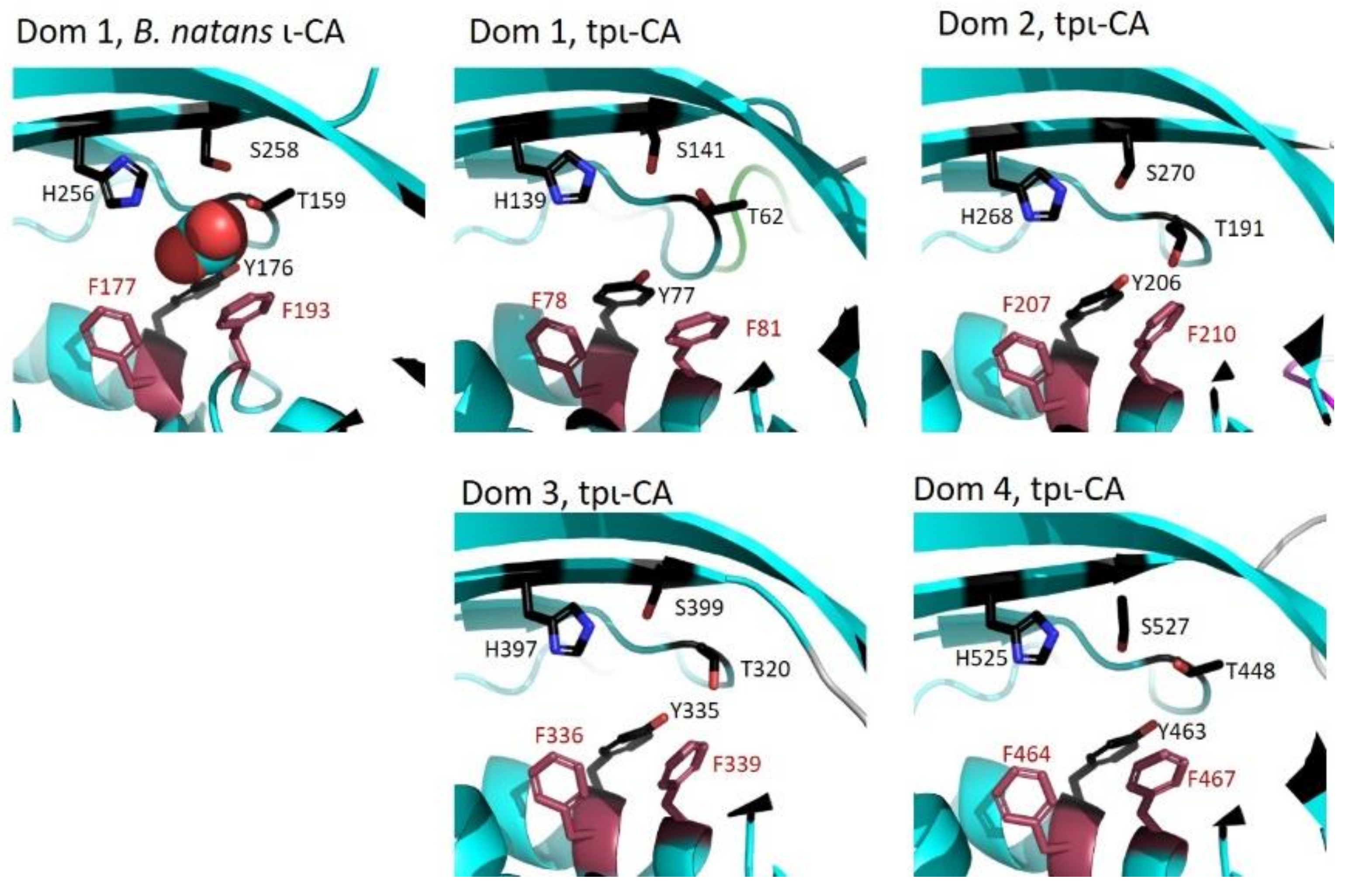
2. Results
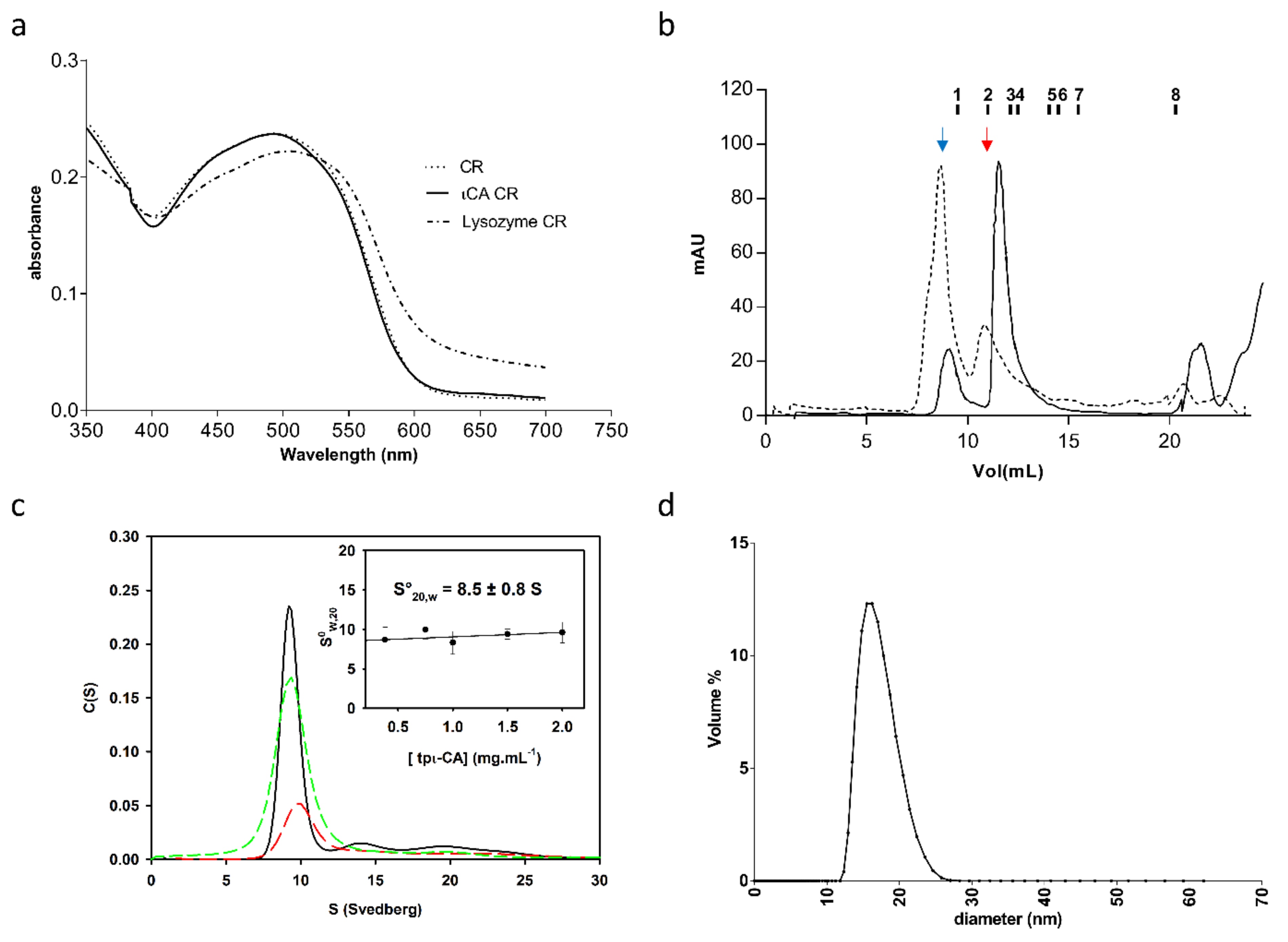
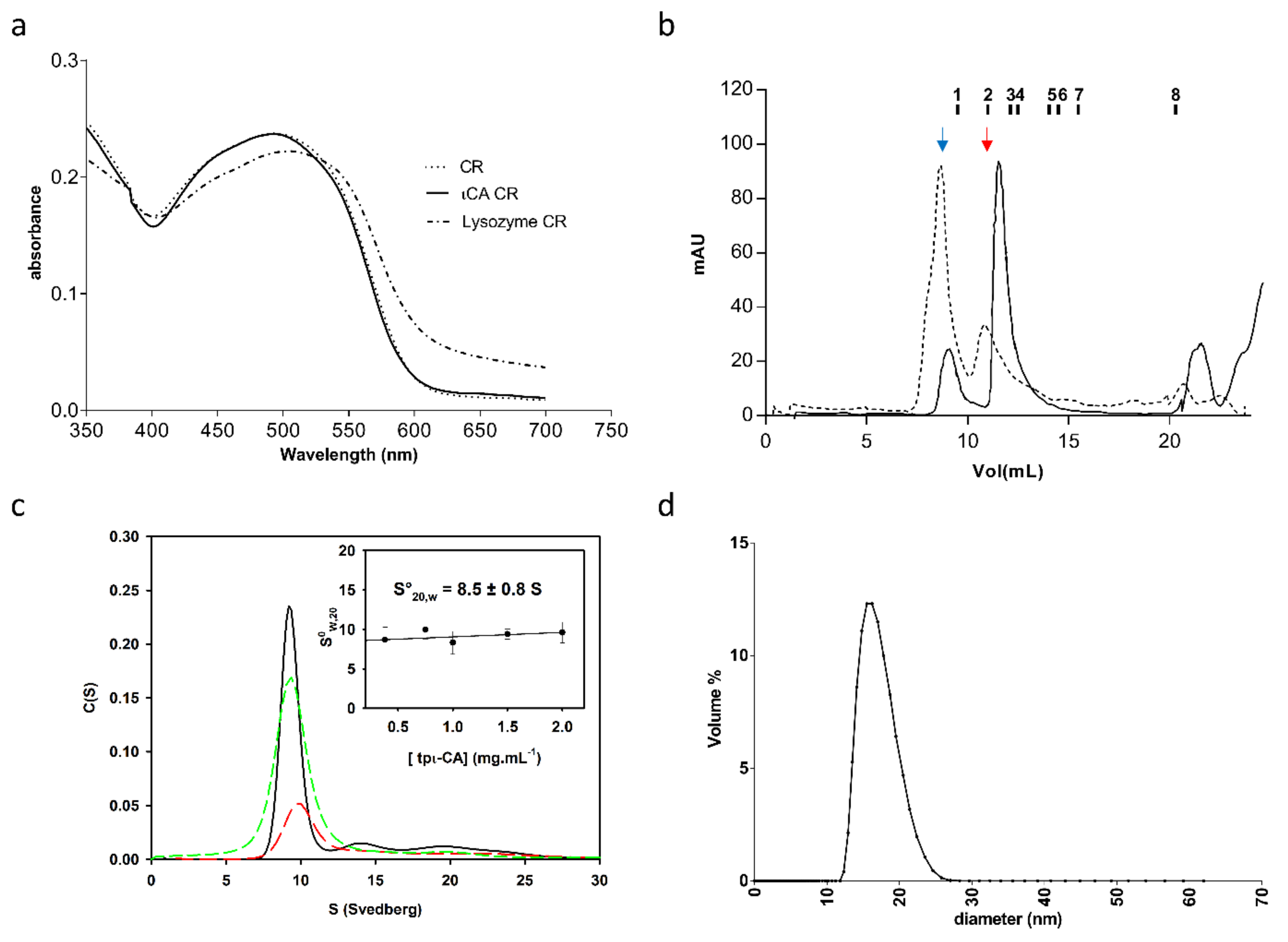
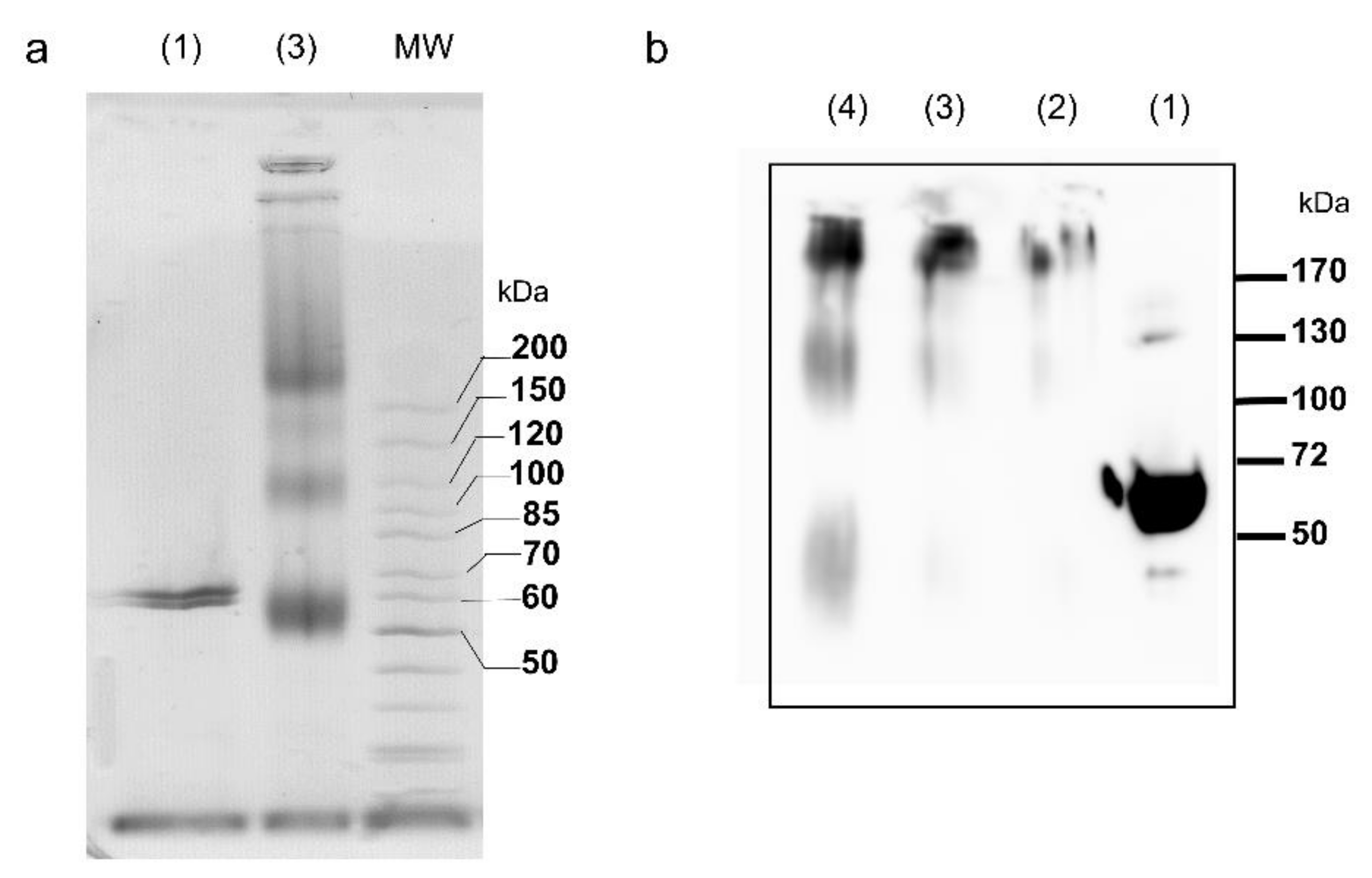
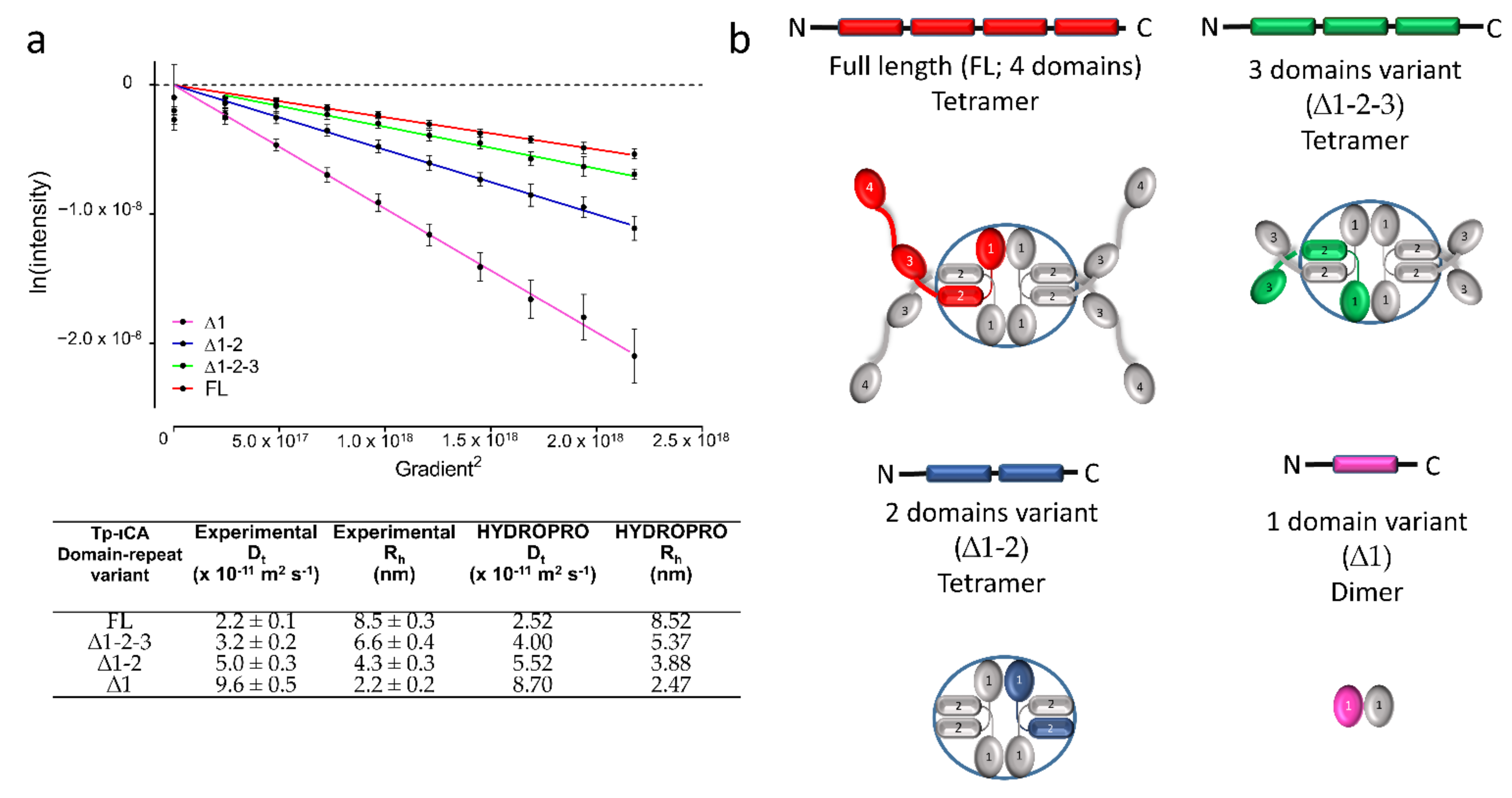
2.1. Oligomerization State of the tpι-CA
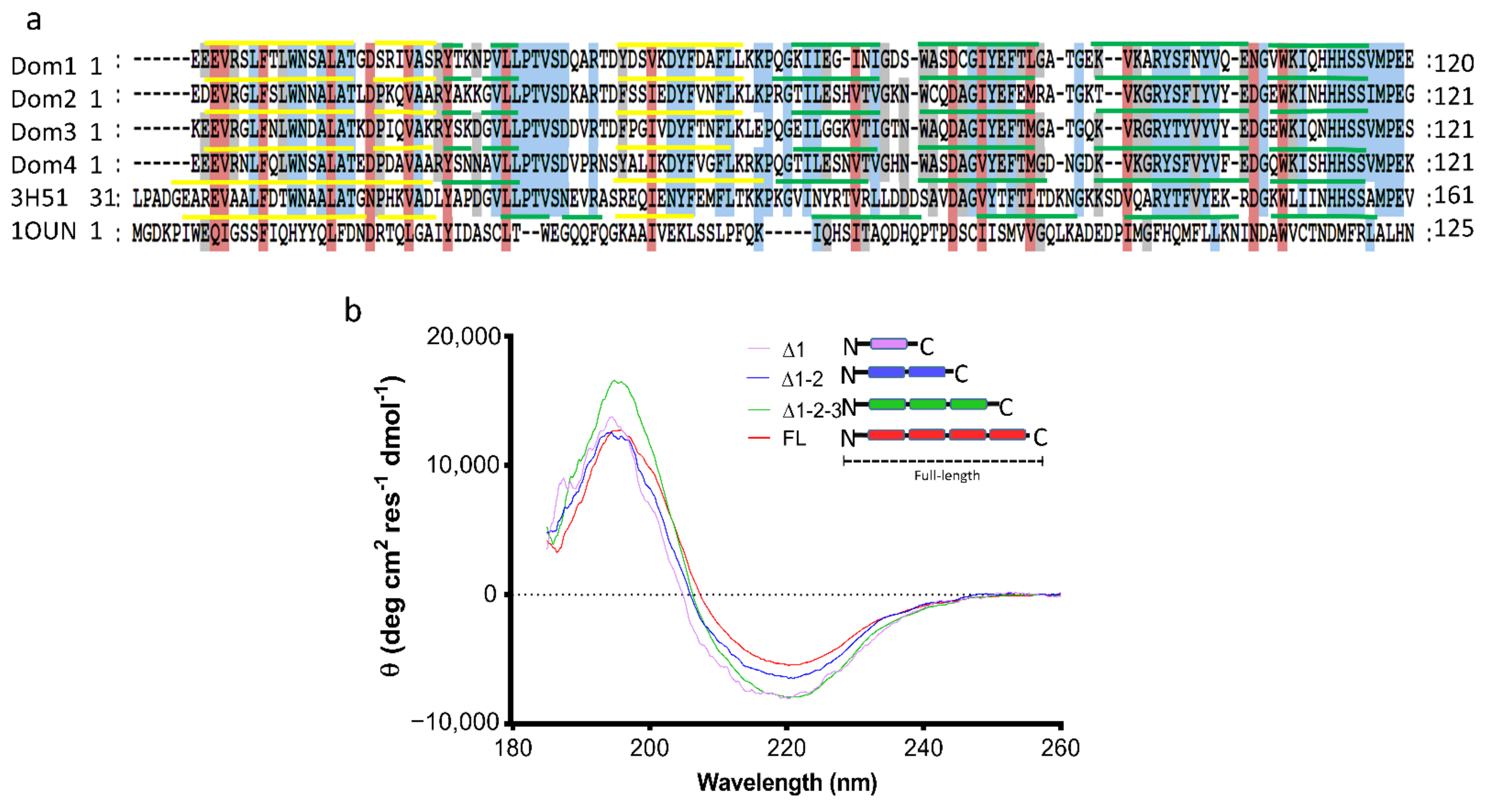
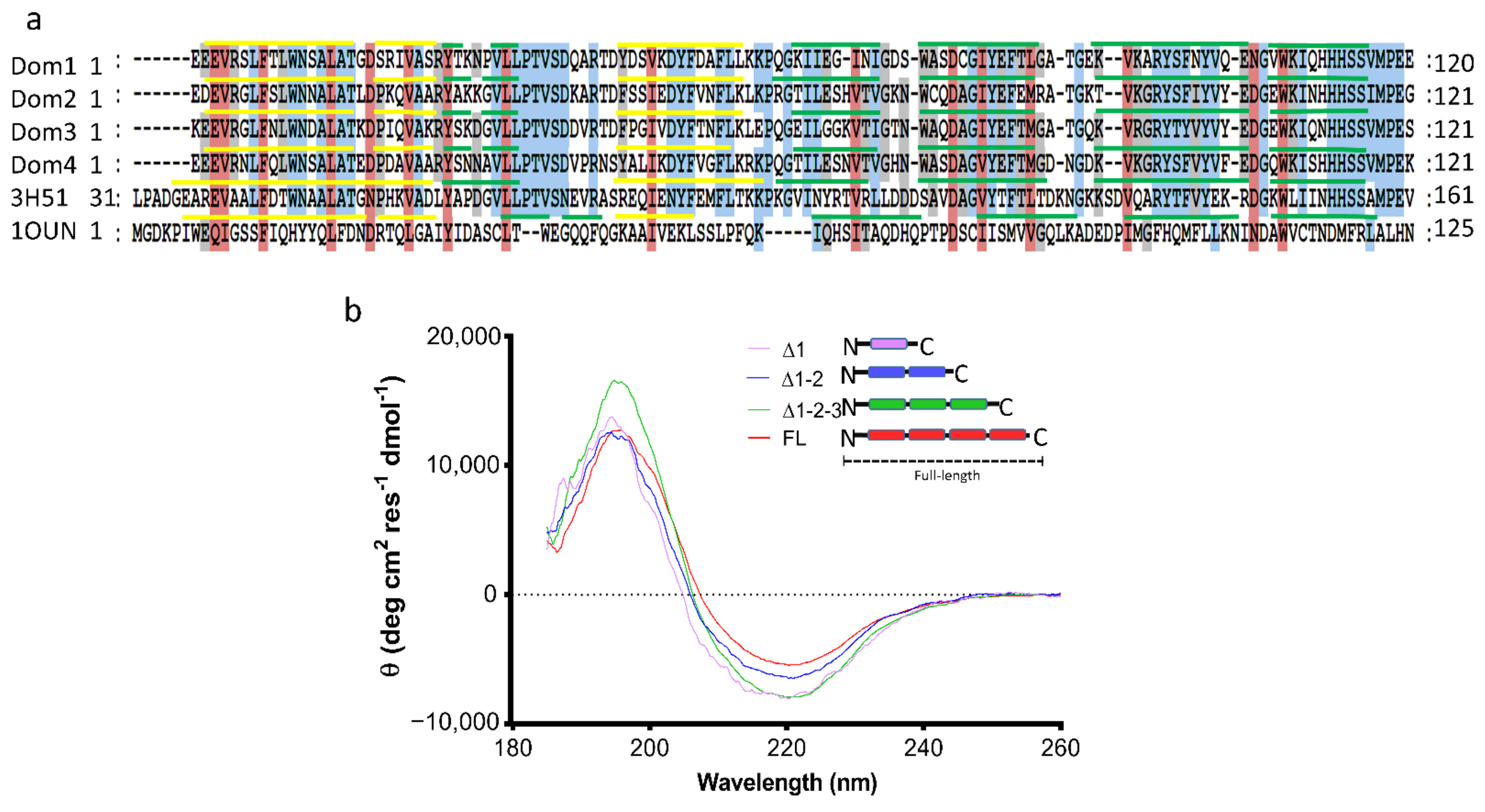
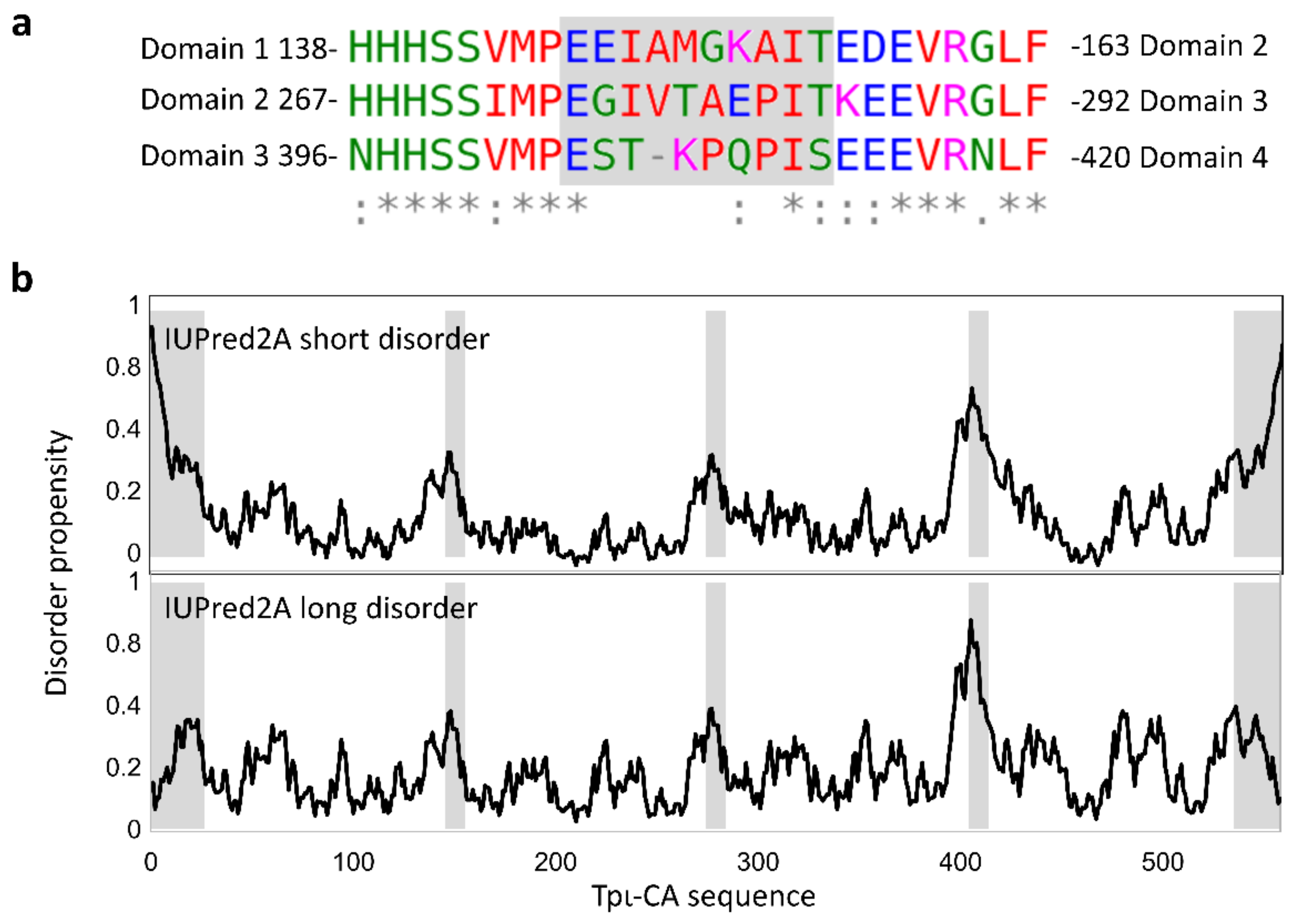
2.2. Characterization of the Secondary Structure of tpι-CA and Its Domain Variants
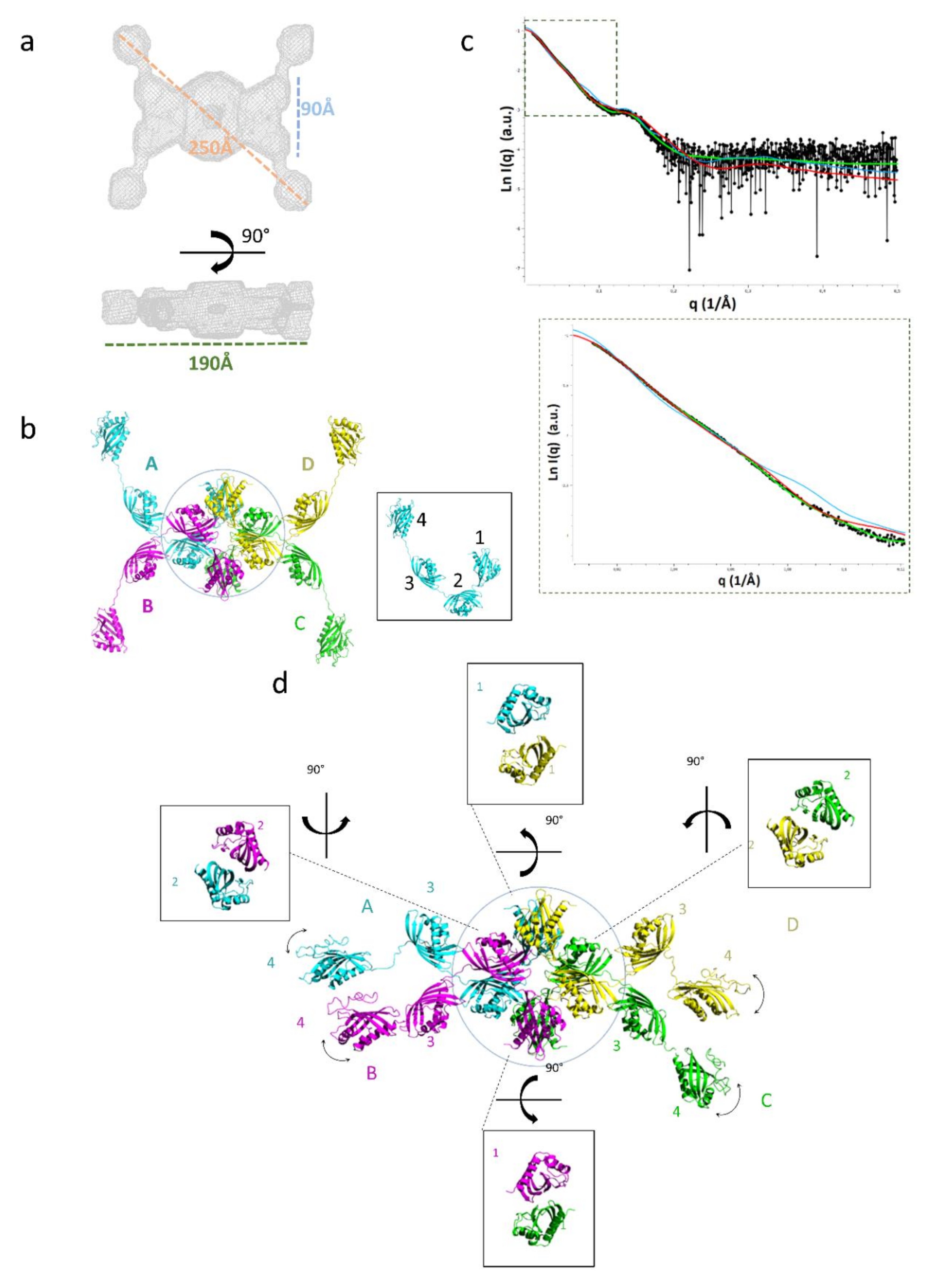
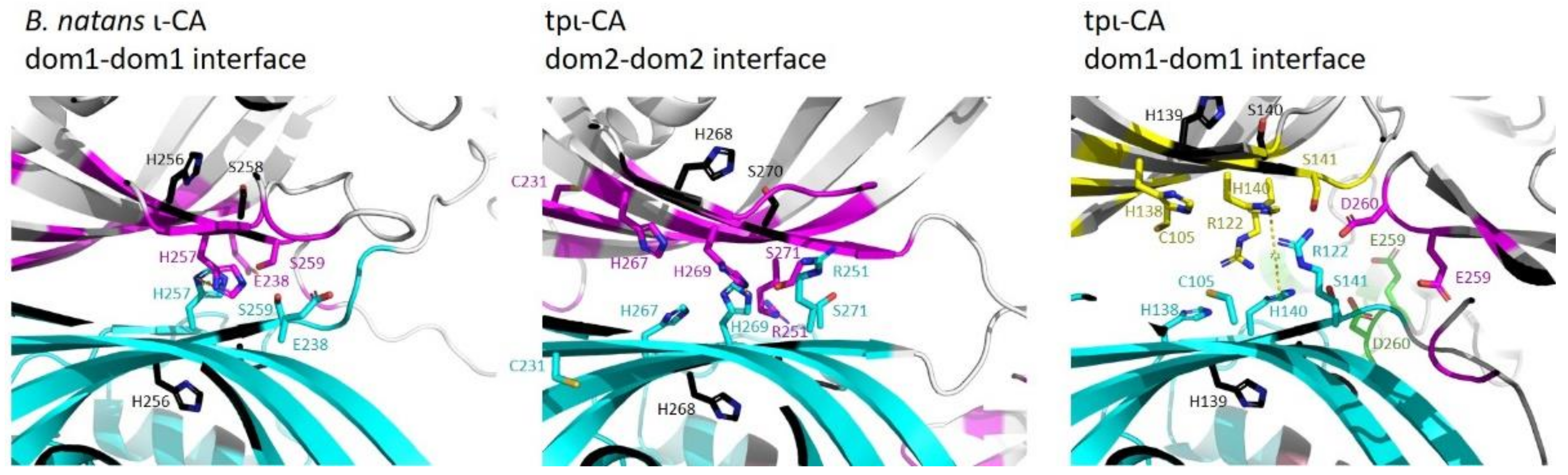
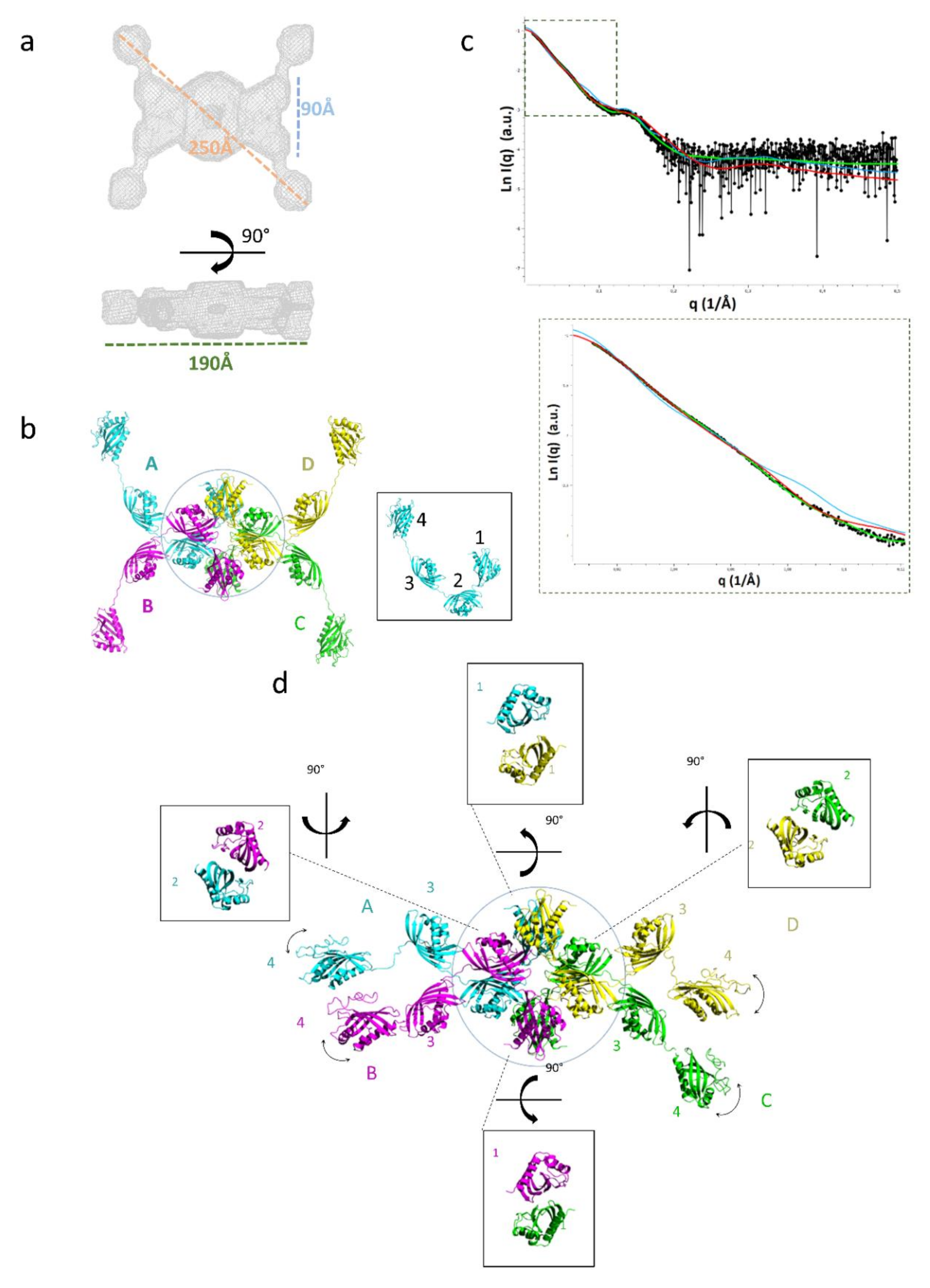
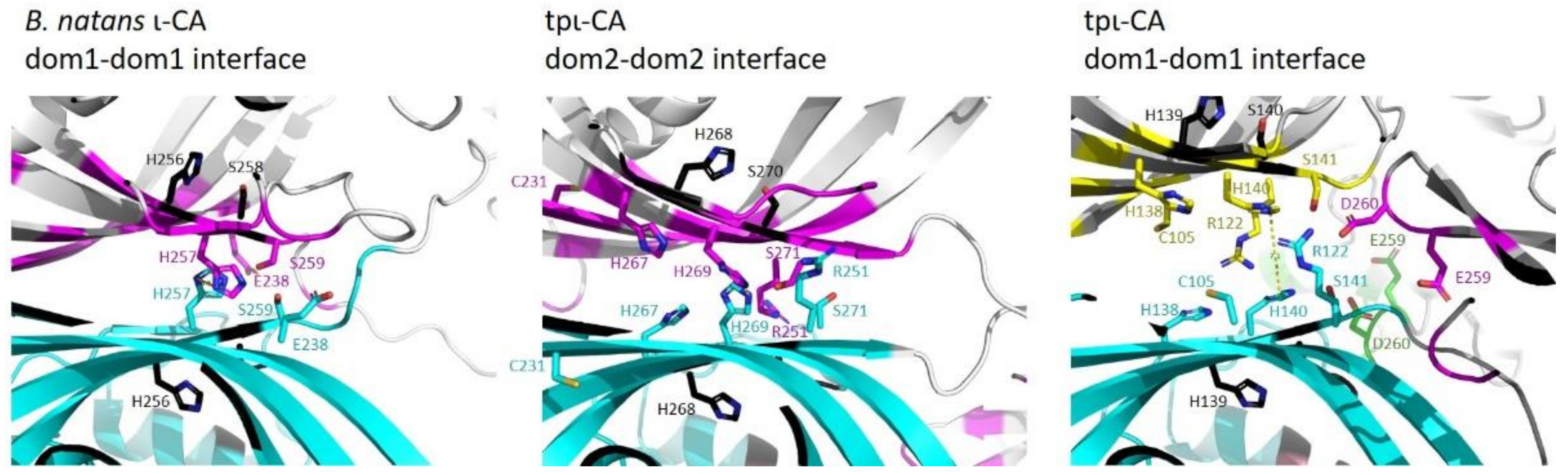
2.3. Domain Organization in Tetrameric tpι-CA
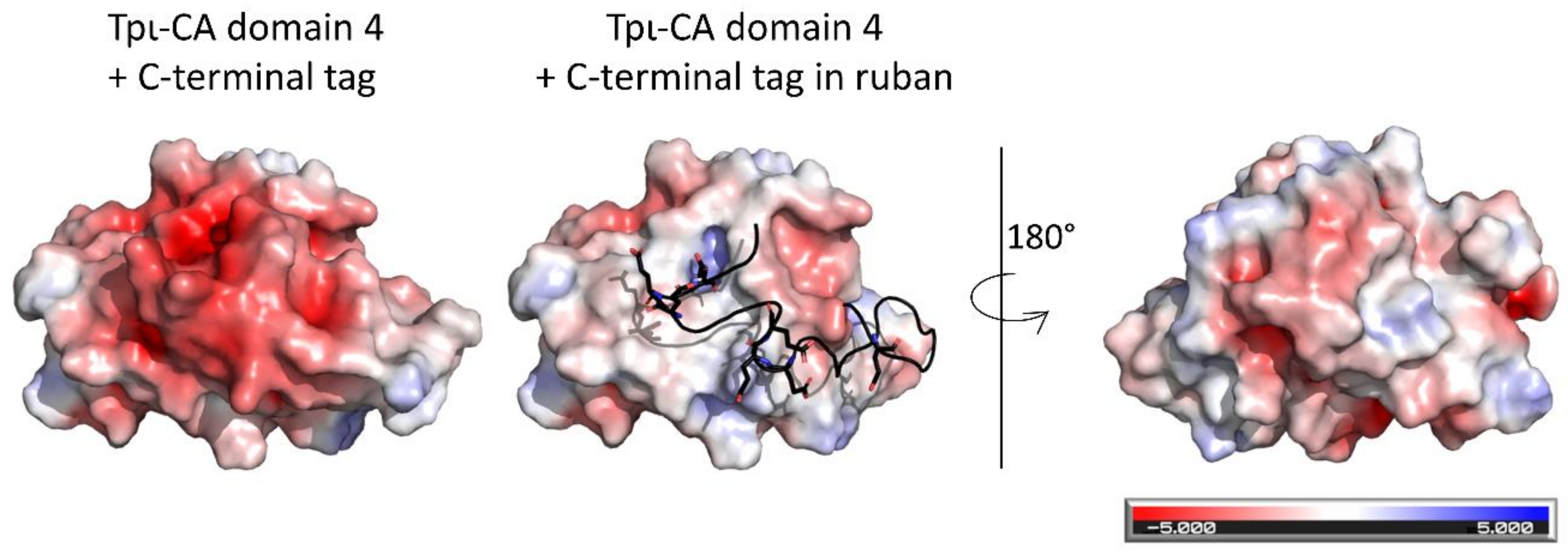
2.4. Flexibility in the Protruding Arms Brought by the Linkers
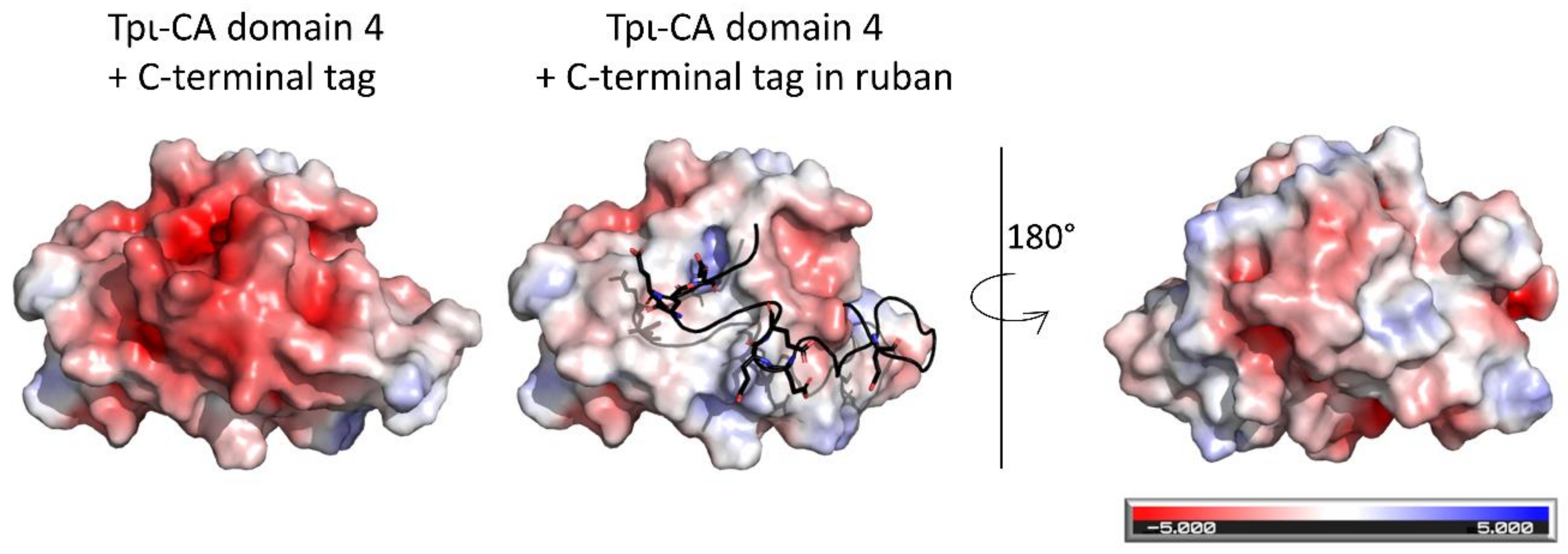
2.5. Experimental Validation of the “Drone-Like” Structural Model
3. Discussion
4. Conclusion
5. Materials and Methods
5.1. Protein Expression and Purification
5.2. Size Exclusion Chromatography (SEC)
5.3. Congo Red Assay
5.4. Protein Cross-Linking
5.5. Protein Analysis
5.6. Analytical Ultracentrifugation (AUC)
5.7. Electrospray Mass Spectrometry (ESI-MS)
5.8. Circular Dichroism (CD)
5.9. Dynamic Light Scattering (DLS)
5.10. Size Exclusion Chromatography Coupled to Small-Angle X-Ray Scattering (SEC-SAXS)
5.11. Diffusion-Ordered Spectroscopy Nuclear Magnetic Resonance (DOSY-NMR)
5.12. D-Modeling
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lionetto, M.G.; Caricato, R.; Giordano, M.E.; Schettino, T. The complex relationship between metals and carbonic anhydrase: New insights and perspectives. Int. J. Mol. Sci. 2016, 17, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, E.L.; Maberly, S.C.; Gontero, B. Insights on the functions and ecophysiological relevance of the diverse carbonic anhydrases in microalgae. Int. J. Mol. Sci. 2020, 21, 2922. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, S.; Vullo, D.; Fisher, G.M.; Andrews, K.T.; Poulsen, S.A.; Capasso, C.; Supuran, C.T. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum—The η-carbonic anhydrases. Bioorgan. Med. Chem. Lett. 2014, 24, 4389–4396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Feng, L.; Jeffrey, P.D.; Shi, Y.; Morel, F.M.M.M. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature 2008, 452, 56–61. [Google Scholar] [CrossRef]
- Kikutani, S.; Nakajima, K.; Nagasato, C.; Tsuji, Y.; Miyatake, A.; Matsuda, Y. Thylakoid luminal θ-carbonic anhydrase critical for growth and photosynthesis in the marine diatom Phaeodactylum tricornutum. Proc. Natl. Acad. Sci. USA 2016, 113, 9828–9833. [Google Scholar] [CrossRef] [Green Version]
- Jensen, E.L.; Clement, R.; Kosta, A.; Maberly, S.C.; Gontero, B. A new widespread subclass of carbonic anhydrase in marine phytoplankton. ISME J. 2019, 13, 2094–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef]
- DiMario, R.J.; Machingura, M.C.; Waldrop, G.L.; Moroney, J.V. The many types of carbonic anhydrases in photosynthetic organisms. Plant Sci. 2018, 268, 11–17. [Google Scholar] [CrossRef]
- Tsuji, Y.; Nakajima, K.; Matsuda, Y. Molecular aspects of the biophysical CO2-concentrating mechanism and its regulation in marine diatoms. J. Exp. Bot. 2017, 68, 3763–3772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacAuley, S.R.; Zimmerman, S.A.; Apolinario, E.E.; Evilia, C.; Hou, Y.M.; Ferry, J.G.; Sowers, K.R. The archetype γ-class carbonic anhydrase (cam) contains iron when synthesized in vivo. Biochemistry 2009, 48, 817–819. [Google Scholar] [CrossRef]
- DiMario, R.J.; Clayton, H.; Mukherjee, A.; Ludwig, M.; Moroney, J.V. Plant Carbonic Anhydrases: Structures, Locations, Evolution, and Physiological Roles. Mol. Plant 2017, 10, 30–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, R.; Lignon, S.; Mansuelle, P.; Jensen, E.; Pophillat, M.; Lebrun, R.; Denis, Y.; Puppo, C.; Maberly, S.C.; Gontero, B. Responses of the marine diatom Thalassiosira pseudonana to changes in CO2 concentration: A proteomic approach. Sci. Rep. 2017, 7, 42333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, M.; Beardall, J.; Raven, J.A. CO2 concentrating mechanisms in algae: Mechanisms, environmental modulation, and evolution. Annu. Rev. Plant Biol. 2005, 56, 99–131. [Google Scholar] [CrossRef] [Green Version]
- Del Prete, S.; Nocentini, A.; Supuran, C.T.; Capasso, C. Bacterial ι-carbonic anhydrase: A new active class of carbonic anhydrase identified in the genome of the Gram-negative bacterium Burkholderia territorii. J. Enzym. Inhib. Med. Chem. 2020, 35, 1060–1068. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, Y.; Senda, M.; Fukuda, K.; Yu, H.Y.; Ishida, M.; Taira, M.; Kinbara, K.; Senda, T. Characterization of a novel type of carbonic anhydrase that acts without metal cofactors. BMC Biol. 2021, 19, 1–15. [Google Scholar] [CrossRef]
- Eberhardt, R.Y.; Chang, Y.; Bateman, A.; Murzin, A.G.; Axelrod, H.L.; Hwang, W.C.; Aravind, L. Filling out the structural map of the NTF2-like superfamily. BMC Bioinform. 2013, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, O.S.; Deindl, S.; Comolli, L.R.; Hoelz, A.; Downing, K.H.; Nairn, A.C.; Kuriyan, J. Oligomerization states of the association domain and the holoenyzme of Ca2+/CaM kinase II. FEBS J. 2006, 273, 682–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, M.; Kent, H.M.; Mccoy, A.J. Structural basis for molecular recognition between nuclear transport factor 2 (NTF2) and the GDP-bound form of the Ras-family GTPase Ran. J. Mol. Biol. 1998, 277, 635–646. [Google Scholar] [CrossRef]
- Vognsen, T.; Kristensen, O. Crystal structure of the Rasputin NTF2-like domain from Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2012, 420, 188–192. [Google Scholar] [CrossRef]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, 668–673. [Google Scholar] [CrossRef] [Green Version]
- Mészáros, B.; Erdös, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Petoukhov, M.V.; Franke, D.; Shkumatov, A.V.; Tria, G.; Kikhney, A.G.; Gajda, M.; Gorba, C.; Mertens, H.D.T.; Konarev, P.V.; Svergun, D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012, 45, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Ortega, A.; Amorós, D.; De La Torre, J.G. Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 2011, 101, 892–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liljas, A.; Laurberg, M. A wheel invented three times. EMBO Rep. 2000, 1, 16–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahaka, M.; Amara, S.; Wattanakul, J.; Gedi, M.A.; Aldai, N.; Parsiegla, G.; Lecomte, J.; Christeller, J.T.; Gray, D.; Gontero, B.; et al. The digestion of galactolipids and its ubiquitous function in Nature for the uptake of the essential a-linolenic acid. Food Funct. 2020, 11, 6710–6744. [Google Scholar] [CrossRef]
- Satoh, D.; Hiraoka, Y.; Colman, B.; Matsuda, Y. Physiological and molecular biological characterization of intracellular carbonic anhydrase from the marine diatom Phaeodactylum tricornutum. Plant. Physiol. 2001, 126, 1459–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitao, Y.; Matsuda, Y. Formation of macromolecular complexes of carbonic anhydrases in the chloroplast of a marine diatom by the action of the C-terminal helix. Biochem. J. 2009, 688, 681–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maberly, S.C.; Gontero, B.; Puppo, C.; Villain, A.; Severi, I.; Giordano, M. Inorganic carbon uptake in a freshwater diatom, Asterionella formosa (Bacillariophyceae): From ecology to genomics. Phycologia 2021, 1–12. [Google Scholar] [CrossRef]
- Udayalaxmi, S.; Gangula, M.R. Investigation of manganese metal coordination in proteins: A comprehensive PDB analysis and quantum mechanical study. Struct. Chem. 2020, 31, 1057–1064. [Google Scholar]
- Zheng, H.; Chruszcz, M.; Lasota, P.; Lebioda, L.; Minor, W. Data mining of metal ion environments present in protein structures. J. Inorg. Biochem. 2008, 102, 1765–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antimonova, O.I.; Grudinina, N.A.; Egorov, V.V.; Polyakov, D.S. Interaction of the Dye Congo Red with Fibrils of Lysozyme, Beta2-Microglobulin, and Transthyretin. Cell Tissue Biol. 2016, 10, 468–475. [Google Scholar] [CrossRef]
- Laue, T.M.; Shah, B.D.; Ridgeway, T.M.; Pelletier, S.L. Computer-aided interpretation of analytical sedimentation data for proteins. In Analytical Ultracentrifugation in Biochemistry and Polymer Science; Harding, S.E., Rowe, A.J., Horton, J.C., Eds.; Royal Society of Chemistry: Cambridge, UK, 1992; pp. 90–125. ISBN 0851863450. [Google Scholar]
- Schuck, P.; Rossmanith, P. Determination of the sedimentation coefficient distribution by least-squares boundary modeling. Biopolymers 2000, 54, 328–341. [Google Scholar] [CrossRef]
- Franke, D.; Petoukhov, M.V.; Konarev, P.V.; Panjkovich, A.; Tuukkanen, A.; Mertens, H.D.T.; Kikhney, A.G.; Hajizadeh, N.R.; Franklin, J.M.; Jeffries, C.M.; et al. ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017, 50, 1212–1225. [Google Scholar] [CrossRef] [Green Version]
- Piiadov, V.; de Araújo, E.A.; Neto, M.O.; Craievich, A.F.; Polikarpov, I. SAXSMoW 2.0: Online calculator of the molecular weight of proteins in dilute solution from experimental SAXS data measured on a relative scale. Protein Sci. 2019, 28, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Charge State | m/z | Delta m/z | |
|---|---|---|---|
| Theoretical | Experimental * | ||
| 28 | 9285.00 | 9288.00 | 3.00 |
| 29 | 8964.86 | 8970.22 | 5.36 |
| 30 | 8666.07 | 8670.32 | 4.25 |
| 31 | 8386.55 | 8390.12 | 3.57 |
| 32 | 8124.50 | 8127.45 | 2.95 |
| Deduced multimeric mass (Da) | Oligomer state | Deduced monomer mass (theoretical) (Da) | Error (ppm) |
| 260,066.20 | 4 | 65,016.55 (64,988) | 439 |
| Tp-ιCA Domain-Repeat Variant | Experimental CD (DichroWeb Analysis) | Predicted Secondary Structure (PSIPred) | ||||
|---|---|---|---|---|---|---|
| β-Strand | α-Helix | Coil | β-Strand | α-Helix | Coil | |
| FL | 0.39 | 0.07 | 0.54 | 0.32 | 0.19 | 0.49 |
| ∆1-2-3 | 0.35 | 0.18 | 0.48 | 0.33 | 0.17 | 0.50 |
| ∆1-2 | 0.37 | 0.08 | 0.55 | 0.34 | 0.17 | 0.49 |
| ∆1 | 0.31 | 0.19 | 0.50 | 0.32 | 0.16 | 0.52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jensen, E.L.; Receveur-Brechot, V.; Hachemane, M.; Wils, L.; Barbier, P.; Parsiegla, G.; Gontero, B.; Launay, H. Structural Contour Map of the Iota Carbonic Anhydrase from the Diatom Thalassiosira pseudonana Using a Multiprong Approach. Int. J. Mol. Sci. 2021, 22, 8723. https://doi.org/10.3390/ijms22168723
Jensen EL, Receveur-Brechot V, Hachemane M, Wils L, Barbier P, Parsiegla G, Gontero B, Launay H. Structural Contour Map of the Iota Carbonic Anhydrase from the Diatom Thalassiosira pseudonana Using a Multiprong Approach. International Journal of Molecular Sciences. 2021; 22(16):8723. https://doi.org/10.3390/ijms22168723
Chicago/Turabian StyleJensen, Erik L., Véronique Receveur-Brechot, Mohand Hachemane, Laura Wils, Pascale Barbier, Goetz Parsiegla, Brigitte Gontero, and Hélène Launay. 2021. "Structural Contour Map of the Iota Carbonic Anhydrase from the Diatom Thalassiosira pseudonana Using a Multiprong Approach" International Journal of Molecular Sciences 22, no. 16: 8723. https://doi.org/10.3390/ijms22168723
APA StyleJensen, E. L., Receveur-Brechot, V., Hachemane, M., Wils, L., Barbier, P., Parsiegla, G., Gontero, B., & Launay, H. (2021). Structural Contour Map of the Iota Carbonic Anhydrase from the Diatom Thalassiosira pseudonana Using a Multiprong Approach. International Journal of Molecular Sciences, 22(16), 8723. https://doi.org/10.3390/ijms22168723