
Parallel Reporter Assays Identify Altered Regulatory Role of rs684232 in Leading to Prostate Cancer Predisposition


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
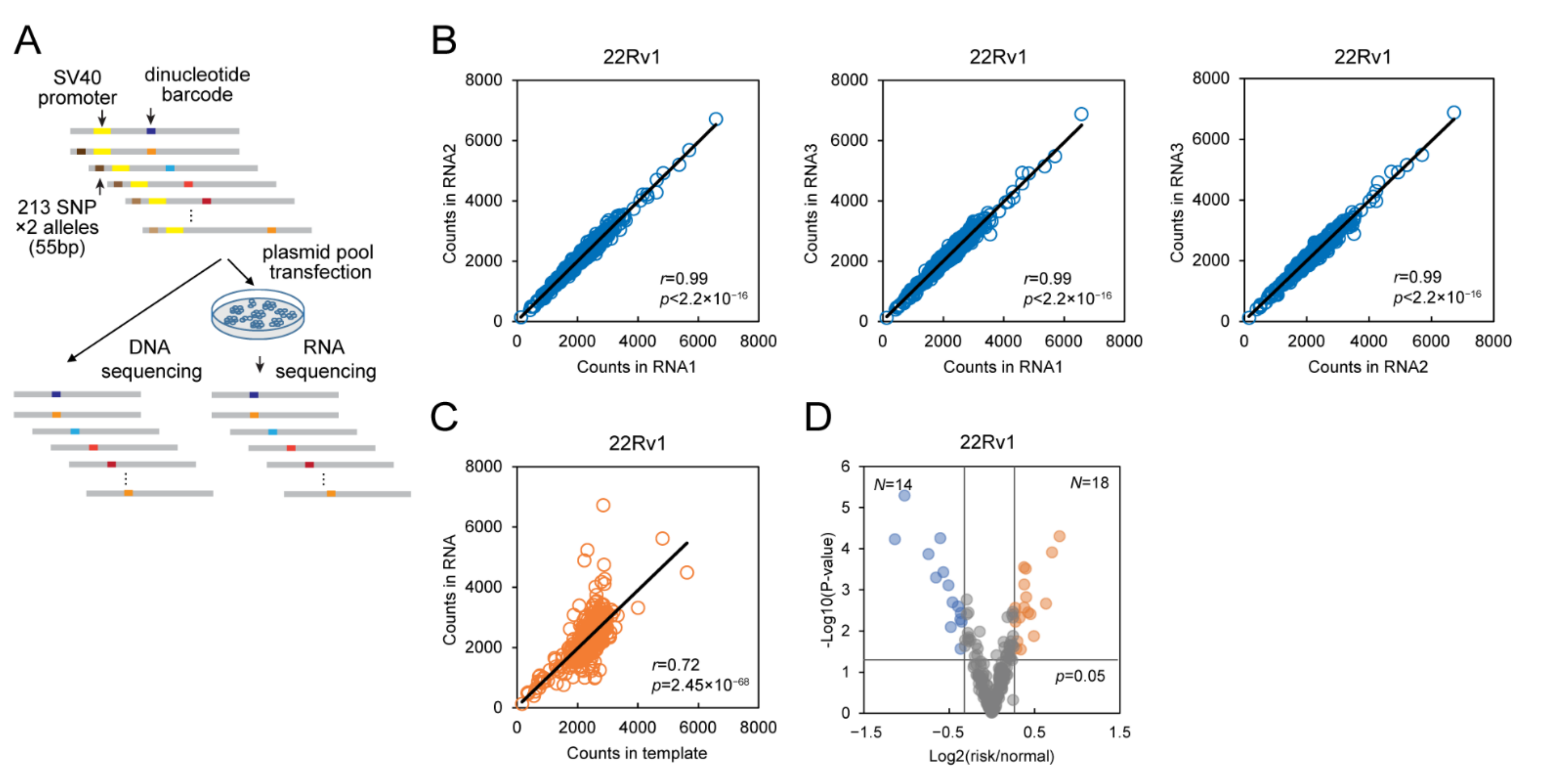
2.1. DiR Assay Discovers Regulatory SNPs in Prostate Cancer Cells
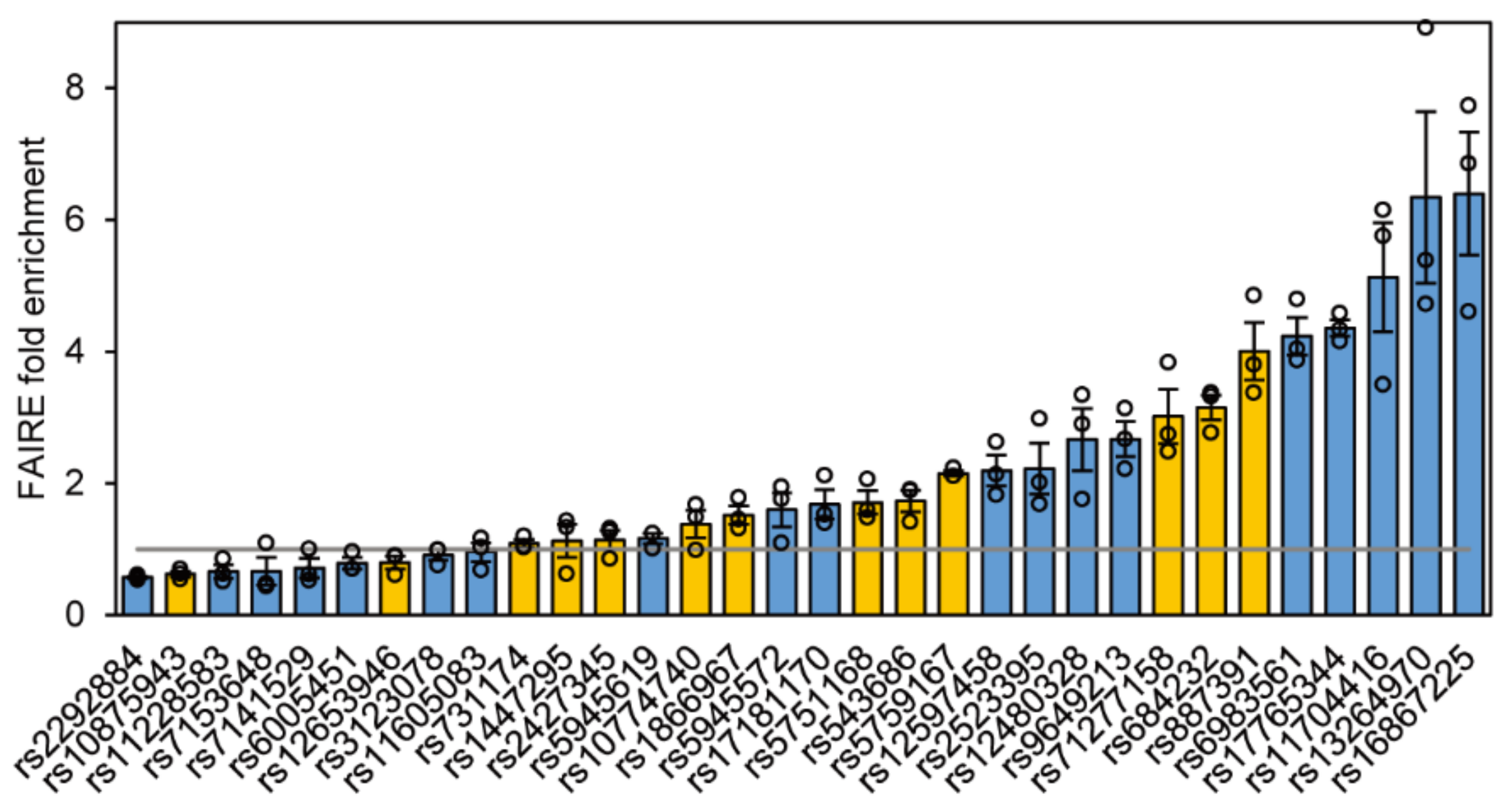
2.2. Chromatin Status of 32 Functional SNPs
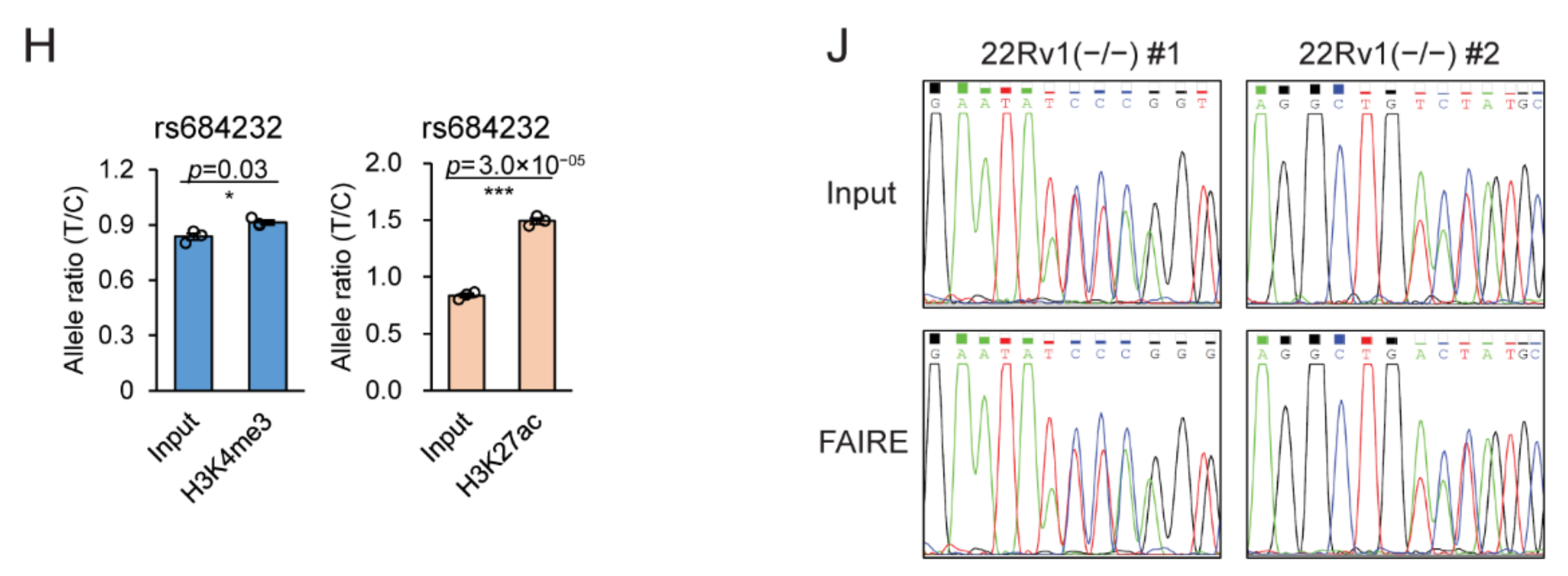
2.3. Allele-Specific Activity of rs684232 and rs887391
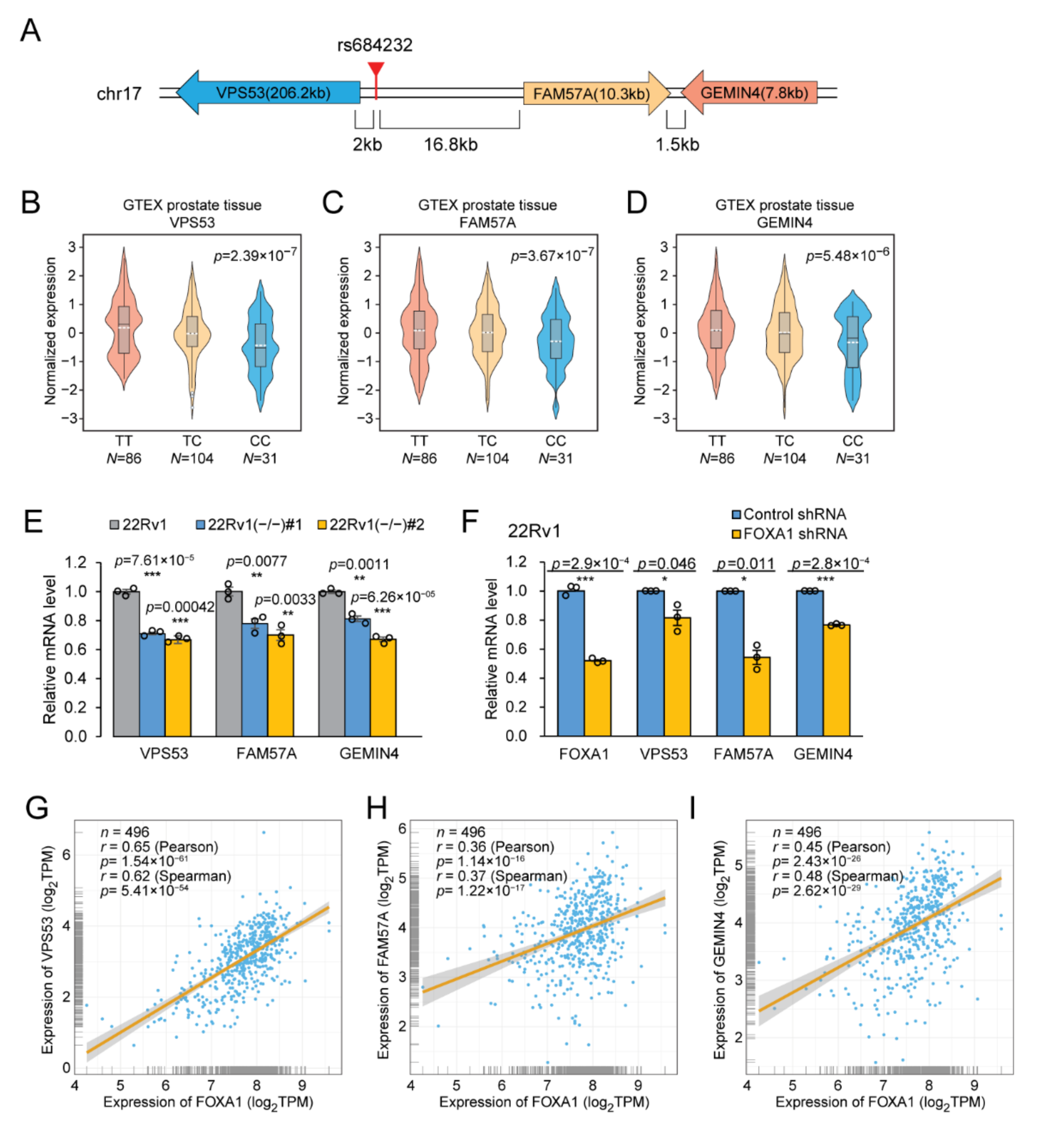
2.4. The Gene Regulatory Function of SNP rs684232
2.5. rs684232 Affects FOXA1 Chromatin Binding
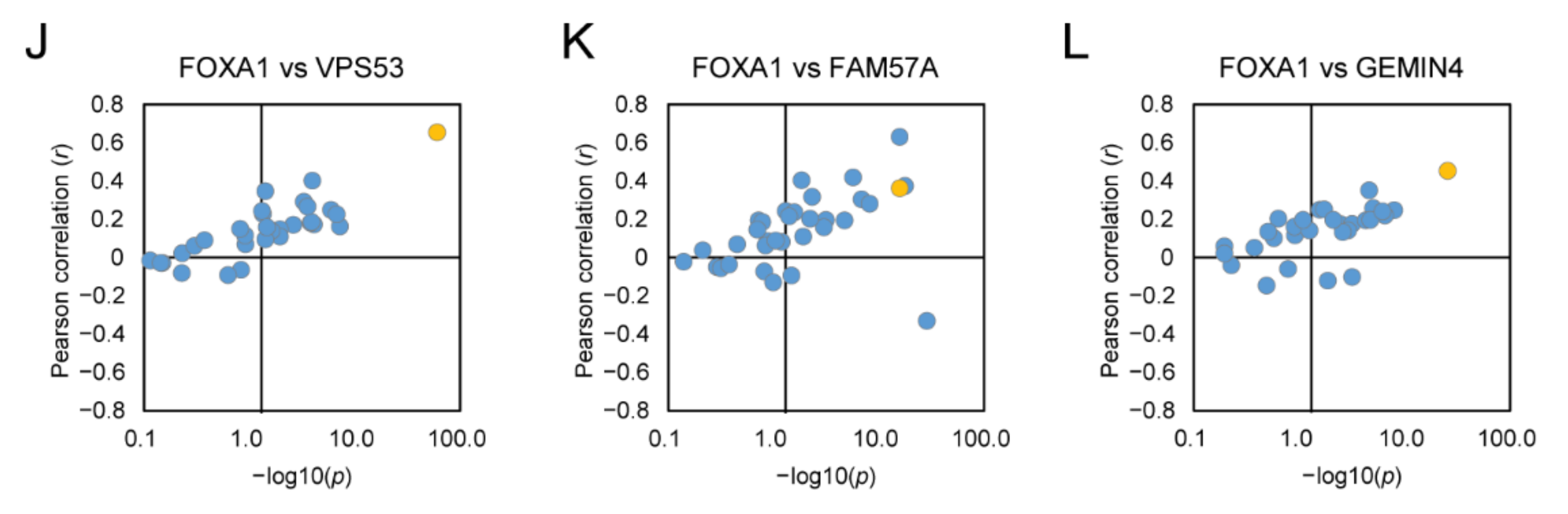
2.6. rs684232 Regulates Gene Expression of VPS53, FAM57A, and GEMIN4 through FOXA1
2.7. VPS53, FAM57A, and GEMIN4 Knockdown Impedes Cancerous Phenotypes
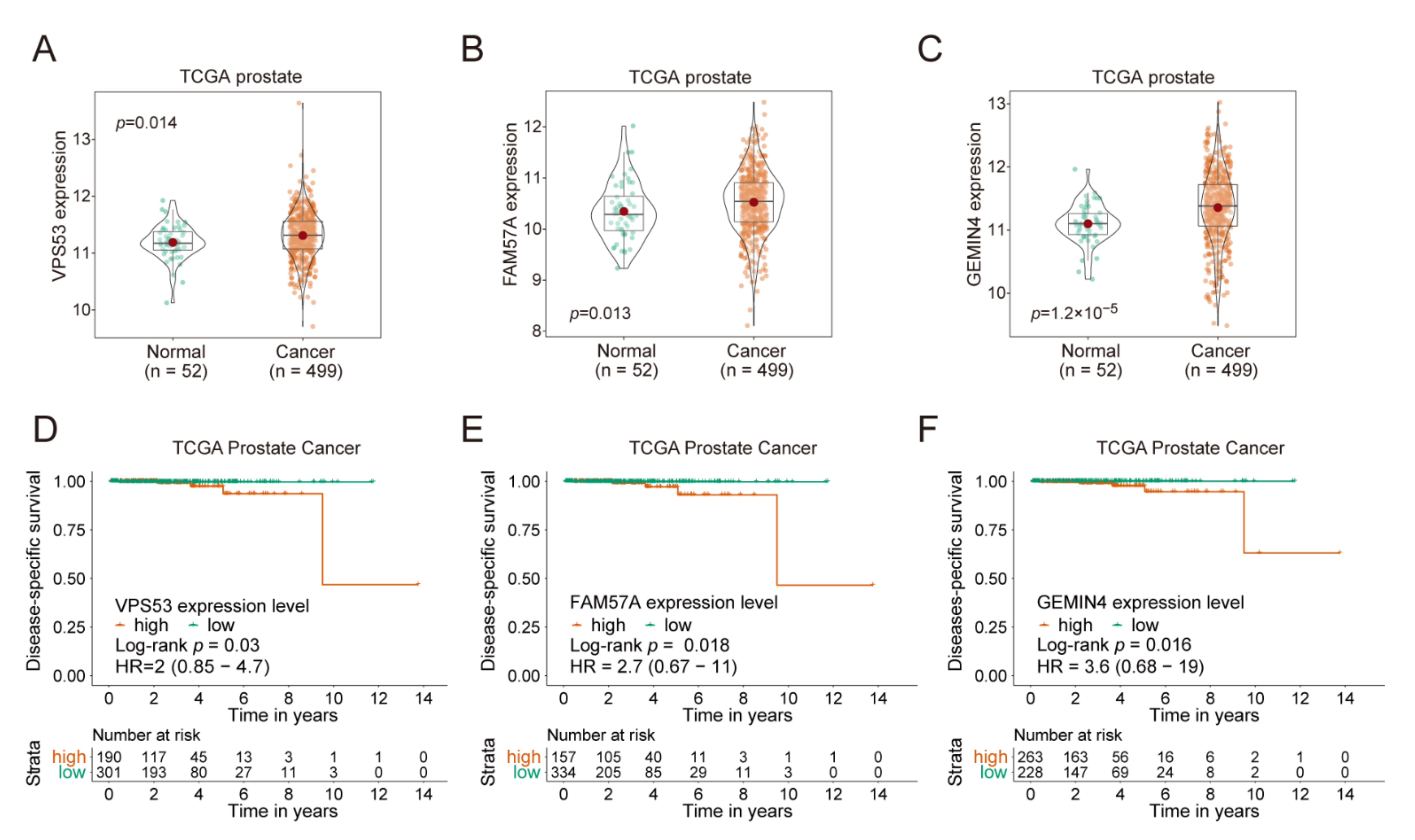
2.8. VPS53, FAM57A, and GEMIN4 Affect Cancer Progression
3. Discussion
4. Materials and Methods
4.1. Construction of the DiR Reporter Pool for Prostate Cancer Risk SNPs
4.2. Cell Culture
4.3. Cell Transfection
4.4. RNA Isolation and Reverse Transcription
4.5. Quantitative PCR
4.6. DiR-Seq Library Preparation for Illumina Sequencing
4.7. NGS Data Processing
4.8. Luciferase Reporter Assays
4.9. Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE)
4.10. Chromatin Immunoprecipitation (ChIP)
4.11. Lentiviral Constructs, Lentivirus Production, and Infection
4.12. rs684232 Knockout Using CRISPR/Cas9
4.13. Genome Editing Efficiency Determination and Single-Cell Clone Screening
4.14. Cell Viability and Proliferation Assays
4.15. Colony-Forming Assay
4.16. Wound Healing Assays
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sud, A.; Kinnersley, B.; Houlston, R.S. Genome-wide association studies of cancer: Current insights and future perspectives. Nat. Rev. Cancer 2017, 17, 692–704. [Google Scholar] [CrossRef]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2020, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Olama, A.A.; Kote-Jarai, Z.; Berndt, S.I.; Conti, D.V.; Schumacher, F.; Han, Y.; Benlloch, S.; Hazelett, D.J.; Wang, Z.M.; Saunders, E.; et al. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat. Genet. 2014, 46, 1103–1109. [Google Scholar] [CrossRef] [Green Version]
- Eeles, R.; Goh, C.; Castro, E.; Bancroft, E.; Guy, M.; Al Olama, A.A.; Easton, D.; Kote-Jarai, Z. The genetic epidemiology of prostate cancer and its clinical implications. Nat. Rev. Urol. 2014, 11, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Eeles, R.A.; Al Olama, A.A.; Benlloch, S.; Saunders, E.J.; Leongamornlert, D.A.; Tymrakiewicz, M.; Ghoussaini, M.; Luccarini, C.; Dennis, J.; Jugurnauth-Little, S.; et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat. Genet. 2013, 45, 385–391. [Google Scholar] [CrossRef]
- Schumacher, F.R.; Al Olama, A.A.; Berndt, S.I.; Benlloch, S.; Ahmed, M.; Saunders, E.J.; Dadaev, T.; Leongamornlert, D.; Anokian, E.; Cieza-Borrella, C.; et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 2019, 50, 928–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virlogeux, V.; Graff, R.E.; Hoffmann, T.J.; Witte, J.S. Replication and Heritability of Prostate Cancer Risk Variants: Impact of Population-Specific Factors. Cancer Epidemiol. Prev. Biomark. 2015, 24, 938–943. [Google Scholar] [CrossRef] [Green Version]
- Benafif, S.; Kote-Jarai, Z.; Eeles, R.A.; Consortium, P. A Review of Prostate Cancer Genome-Wide Association Studies (GWAS). Cancer Epidemiol. Prev. Biomark. 2018, 27, 845–857. [Google Scholar] [CrossRef] [Green Version]
- Khurana, E.; Fu, Y.; Chakravarty, D.; Demichelis, F.; Rubin, M.A.; Gerstein, M. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016, 17, 93–108. [Google Scholar] [CrossRef]
- Farashi, S.; Kryza, T.; Clements, J.; Batra, J. Post-GWAS in prostate cancer: From genetic association to biological contribution. Nat. Rev. Cancer 2019, 19, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.D.; Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat. Biotechnol. 2012, 30, 1095–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Whitington, T.; Gao, P.; Lindberg, J.F.; Yang, Y.; Sun, J.; Vaisanen, M.R.; Szulkin, R.; Annala, M.; Yan, J.; et al. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nat. Genet. 2014, 46, 126–135. [Google Scholar] [CrossRef]
- Gao, P.; Xia, J.H.; Sipeky, C.; Dong, X.M.; Zhang, Q.; Yang, Y.; Zhang, P.; Cruz, S.P.; Zhang, K.; Zhu, J.; et al. Biology and Clinical Implications of the 19q13 Aggressive Prostate Cancer Susceptibility Locus. Cell 2018, 174, 576–589.e18. [Google Scholar] [CrossRef] [Green Version]
- Takeda, D.Y.; Spisak, S.; Seo, J.H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szallasi, Z.; et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018, 174, 422–432.e13. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Tian, J.; Zhao, Q.; Yang, N.; Ying, P.; Peng, X.; Zou, D.; Zhu, Y.; Zhong, R.; Gao, Y.; et al. Functional characterization of a low-frequency V1937I variant in FASN associated with susceptibility to esophageal squamous cell carcinoma. Arch. Toxicol. 2020, 94, 2039–2046. [Google Scholar] [CrossRef]
- Yu, C.Y.; Han, J.X.; Zhang, J.; Jiang, P.; Shen, C.; Guo, F.; Tang, J.; Yan, T.; Tian, X.; Zhu, X.; et al. A 16q22.1 variant confers susceptibility to colorectal cancer as a distal regulator of ZFP90. Oncogene 2020, 39, 1347–1360. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Seo, J.H.; Stranger, B.; McKenna, A.; Pe’er, I.; Laframboise, T.; Brown, M.; Tyekucheva, S.; Freedman, M.L. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell 2013, 152, 633–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldridge, D.A.; Wood, A.C.; Weichert-Leahey, N.; Crimmins, I.; Sussman, R.; Winter, C.; McDaniel, L.D.; Diamond, M.; Hart, L.S.; Zhu, S.; et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature 2015, 528, 418–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Ren, N.; Huang, Q. rs10514231 Leads to Breast Cancer Predisposition by Altering ATP6AP1L Gene Expression. Cancers 2021, 13, 3752. [Google Scholar] [CrossRef]
- Li, B.; Huang, Q.; Wei, G.H. The Role of HOX Transcription Factors in Cancer Predisposition and Progression. Cancers 2019, 11, 528. [Google Scholar] [CrossRef] [Green Version]
- Brandao, A.; Paulo, P.; Teixeira, M.R. Hereditary Predisposition to Prostate Cancer: From Genetics to Clinical Implications. Int. J. Mol. Sci. 2020, 21, 5036. [Google Scholar] [CrossRef] [PubMed]
- Tak, Y.G.; Farnham, P.J. Making sense of GWAS: Using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenet. Chromatin 2015, 8, 57. [Google Scholar] [CrossRef] [Green Version]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [Green Version]
- Patwardhan, R.P.; Lee, C.; Litvin, O.; Young, D.L.; Pe’er, D.; Shendure, J. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol. 2009, 27, 1173–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patwardhan, R.P.; Hiatt, J.B.; Witten, D.M.; Kim, M.J.; Smith, R.P.; May, D.; Lee, C.; Andrie, J.M.; Lee, S.I.; Cooper, G.M.; et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol. 2012, 30, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.P.; Taher, L.; Patwardhan, R.P.; Kim, M.J.; Inoue, F.; Shendure, J.; Ovcharenko, I.; Ahituv, N. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat. Genet. 2013, 45, 1021–1028. [Google Scholar] [CrossRef] [Green Version]
- Melnikov, A.; Murugan, A.; Zhang, X.; Tesileanu, T.; Wang, L.; Rogov, P.; Feizi, S.; Gnirke, A.; Callan, C.G., Jr.; Kinney, J.B.; et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012, 30, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Arnold, C.D.; Gerlach, D.; Stelzer, C.; Boryn, L.M.; Rath, M.; Stark, A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 2013, 339, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Xia, J.H.; Zhu, J.; Gao, P.; Tian, Y.J.; Du, M.; Guo, Y.C.; Suleman, S.; Zhang, Q.; Kohli, M.; et al. High-throughput screening of prostate cancer risk loci by single nucleotide polymorphisms sequencing. Nat. Commun. 2018, 9, 2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhille, L.; Griffon, A.; Maqbool, M.A.; Zacarias-Cabeza, J.; Dao, L.T.; Fernandez, N.; Ballester, B.; Andrau, J.C.; Spicuglia, S. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nat. Commun. 2015, 6, 6905. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Liu, Y.; Zhang, Q.; Wu, J.; Liang, J.; Yu, S.; Wei, G.H.; White, K.P.; Wang, X. Systematic identification of regulatory variants associated with cancer risk. Genome Biol. 2017, 18, 194. [Google Scholar] [CrossRef] [Green Version]
- Ren, N.; Li, B.; Liu, Q.; Yang, L.; Liu, X.; Huang, Q. A dinucleotide tag-based parallel reporter gene assay method. bioRxiv 2021. [Google Scholar] [CrossRef]
- Al Olama, A.A.; Dadaev, T.; Hazelett, D.J.; Li, Q.; Leongamornlert, D.; Saunders, E.J.; Stephens, S.; Cieza-Borrella, C.; Whitmore, I.; Benlloch Garcia, S.; et al. Multiple novel prostate cancer susceptibility signals identified by fine-mapping of known risk loci among Europeans. Hum. Mol. Genet. 2015, 24, 5589–5602. [Google Scholar] [CrossRef] [PubMed]
- Dadaev, T.; Saunders, E.J.; Newcombe, P.J.; Anokian, E.; Leongamornlert, D.A.; Brook, M.N.; Cieza-Borrella, C.; Mijuskovic, M.; Wakerell, S.; Olama, A.A.A.; et al. Fine-mapping of prostate cancer susceptibility loci in a large meta-analysis identifies candidate causal variants. Nat. Commun. 2018, 9, 2256. [Google Scholar] [CrossRef]
- Levo, M.; Segal, E. In pursuit of design principles of regulatory sequences. Nat. Rev. Genet. 2014, 15, 453–468. [Google Scholar] [CrossRef]
- Huang, Q.; Gong, C.; Li, J.; Zhuo, Z.; Chen, Y.; Wang, J.; Hua, Z.C. Distance and helical phase dependence of synergistic transcription activation in cis-regulatory module. PLoS ONE 2012, 7, e31198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, J.M.; Giresi, P.G.; Davis, I.J.; Lieb, J.D. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc. 2012, 7, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Hua, J.T.; Ahmed, M.; Guo, H.; Zhang, Y.; Chen, S.; Soares, F.; Lu, J.; Zhou, S.; Wang, M.; Li, H.; et al. Risk SNP-Mediated Promoter-Enhancer Switching Drives Prostate Cancer through lncRNA PCAT19. Cell 2018, 174, 564–575.e18. [Google Scholar] [CrossRef] [Green Version]
- Igolkina, A.A.; Zinkevich, A.; Karandasheva, K.O.; Popov, A.A.; Selifanova, M.V.; Nikolaeva, D.; Tkachev, V.; Penzar, D.; Nikitin, D.M.; Buzdin, A. H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 Histone Tags Suggest Distinct Regulatory Evolution of Open and Condensed Chromatin Landmarks. Cells 2019, 8, 1034. [Google Scholar] [CrossRef] [Green Version]
- Whitington, T.; Gao, P.; Song, W.; Ross-Adams, H.; Lamb, A.D.; Yang, Y.; Svezia, I.; Klevebring, D.; Mills, I.G.; Karlsson, R.; et al. Gene regulatory mechanisms underpinning prostate cancer susceptibility. Nat. Genet. 2016, 48, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Larson, N.B.; McDonnell, S.; French, A.J.; Fogarty, Z.; Cheville, J.; Middha, S.; Riska, S.; Baheti, S.; Nair, A.A.; Wang, L.; et al. Comprehensively evaluating cis-regulatory variation in the human prostate transcriptome by using gene-level allele-specific expression. Am. J. Hum. Genet. 2015, 96, 869–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houlahan, K.E.; Shiah, Y.J.; Gusev, A.; Yuan, J.; Ahmed, M.; Shetty, A.; Ramanand, S.G.; Yao, C.Q.; Bell, C.; O’Connor, E.; et al. Genome-wide germline correlates of the epigenetic landscape of prostate cancer. Nat. Med. 2019, 25, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Ramanand, S.G.; Chen, Y.; Yuan, J.; Daescu, K.; Lambros, M.B.; Houlahan, K.E.; Carreira, S.; Yuan, W.; Baek, G.; Sharp, A.; et al. The landscape of RNA polymerase II-associated chromatin interactions in prostate cancer. J. Clin. Investig. 2020, 130, 3987–4005. [Google Scholar] [CrossRef]
- Ward, L.D.; Kellis, M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012, 40, D930–D934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fachal, L.; Dunning, A.M. From candidate gene studies to GWAS and post-GWAS analyses in breast cancer. Curr. Opin. Genet. Dev. 2015, 30, 32–41. [Google Scholar] [CrossRef]
- Chen, H.; Yu, H.; Wang, J.; Zhang, Z.; Gao, Z.; Chen, Z.; Lu, Y.; Liu, W.; Jiang, D.; Zheng, S.L.; et al. Systematic enrichment analysis of potentially functional regions for 103 prostate cancer risk-associated loci. Prostate 2015, 75, 1264–1276. [Google Scholar] [CrossRef] [PubMed]
- Sahu, B.; Laakso, M.; Ovaska, K.; Mirtti, T.; Lundin, J.; Rannikko, A.; Sankila, A.; Turunen, J.P.; Lundin, M.; Konsti, J.; et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011, 30, 3962–3976. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhardt, J.; Montani, M.; Wild, P.; Beer, M.; Huber, F.; Hermanns, T.; Muntener, M.; Kristiansen, G. FOXA1 Promotes Tumor Progression in Prostate Cancer and Represents a Novel Hallmark of Castration-Resistant Prostate Cancer. Am. J. Pathol. 2012, 180, 848–861. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.H.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Sahu, B.; Laakso, M.; Pihlajamaa, P.; Ovaska, K.; Sinielnikov, I.; Hautaniemi, S.; Janne, O.A. FoxA1 Specifies Unique Androgen and Glucocorticoid Receptor Binding Events in Prostate Cancer Cells. Cancer Res. 2013, 73, 1570–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, N.; Zhang, J.F.; Rao, M.A.; Case, T.C.; Mirosevich, J.; Wang, Y.Q.; Jin, R.J.; Gupta, A.; Rennie, P.S.; Matusik, R.J. The role of hepatocyte nuclear factor-3 alpha (forkhead box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol. Endocrinol. 2003, 17, 1484–1507. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, M.M.; Li, F.; Takeda, D.Y.; Lenci, R.; Chonkar, A.; Chabot, M.; Cejas, P.; Vazquez, F.; Cook, J.; Shivdasani, R.A.; et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet. 2015, 47, 1346–1351. [Google Scholar] [CrossRef]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinform. 2012, 13, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.; Anders, S.; Lawrence, M.; Aboyoun, P.; Pages, H.; Gentleman, R. ShortRead: A bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 2009, 25, 2607–2608. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Ren, N.; Yang, L.; Liu, J.; Huang, Q. A qPCR method for genome editing efficiency determination and single-cell clone screening in human cells. Sci. Rep. 2019, 9, 18877. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, N.; Liu, Q.; Yan, L.; Huang, Q. Parallel Reporter Assays Identify Altered Regulatory Role of rs684232 in Leading to Prostate Cancer Predisposition. Int. J. Mol. Sci. 2021, 22, 8792. https://doi.org/10.3390/ijms22168792
Ren N, Liu Q, Yan L, Huang Q. Parallel Reporter Assays Identify Altered Regulatory Role of rs684232 in Leading to Prostate Cancer Predisposition. International Journal of Molecular Sciences. 2021; 22(16):8792. https://doi.org/10.3390/ijms22168792
Chicago/Turabian StyleRen, Naixia, Qingqing Liu, Lingjie Yan, and Qilai Huang. 2021. "Parallel Reporter Assays Identify Altered Regulatory Role of rs684232 in Leading to Prostate Cancer Predisposition" International Journal of Molecular Sciences 22, no. 16: 8792. https://doi.org/10.3390/ijms22168792