
Inhibition of Cell Proliferation and Metastasis by Scutellarein Regulating PI3K/Akt/NF-κB Signaling through PTEN Activation in Hepatocellular Carcinoma

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
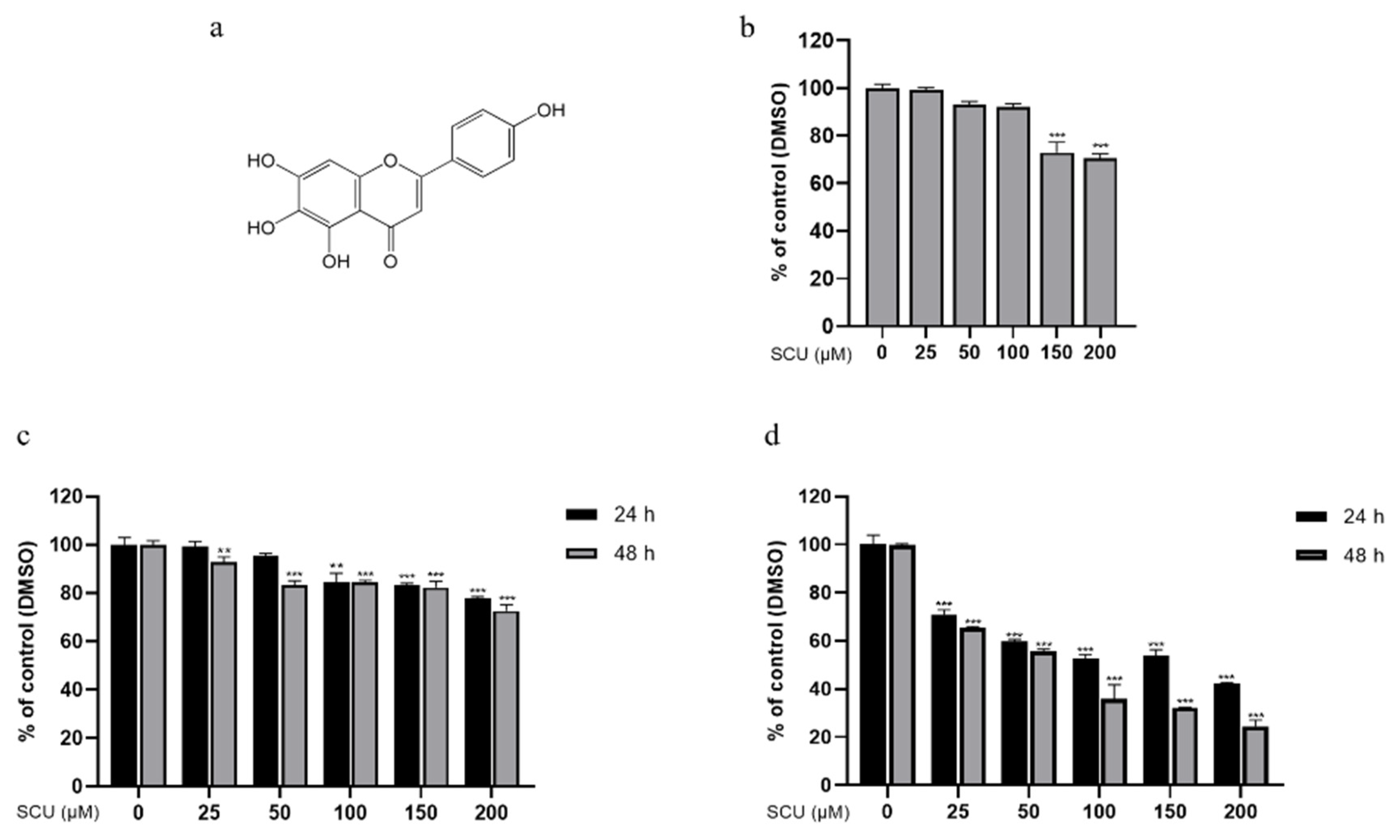
2.1. Scutellarein (SCU) Inhibits Cell Viability in Human Hepatocellular Carcinoma (HCC) Cell Lines and HaCaT Cells
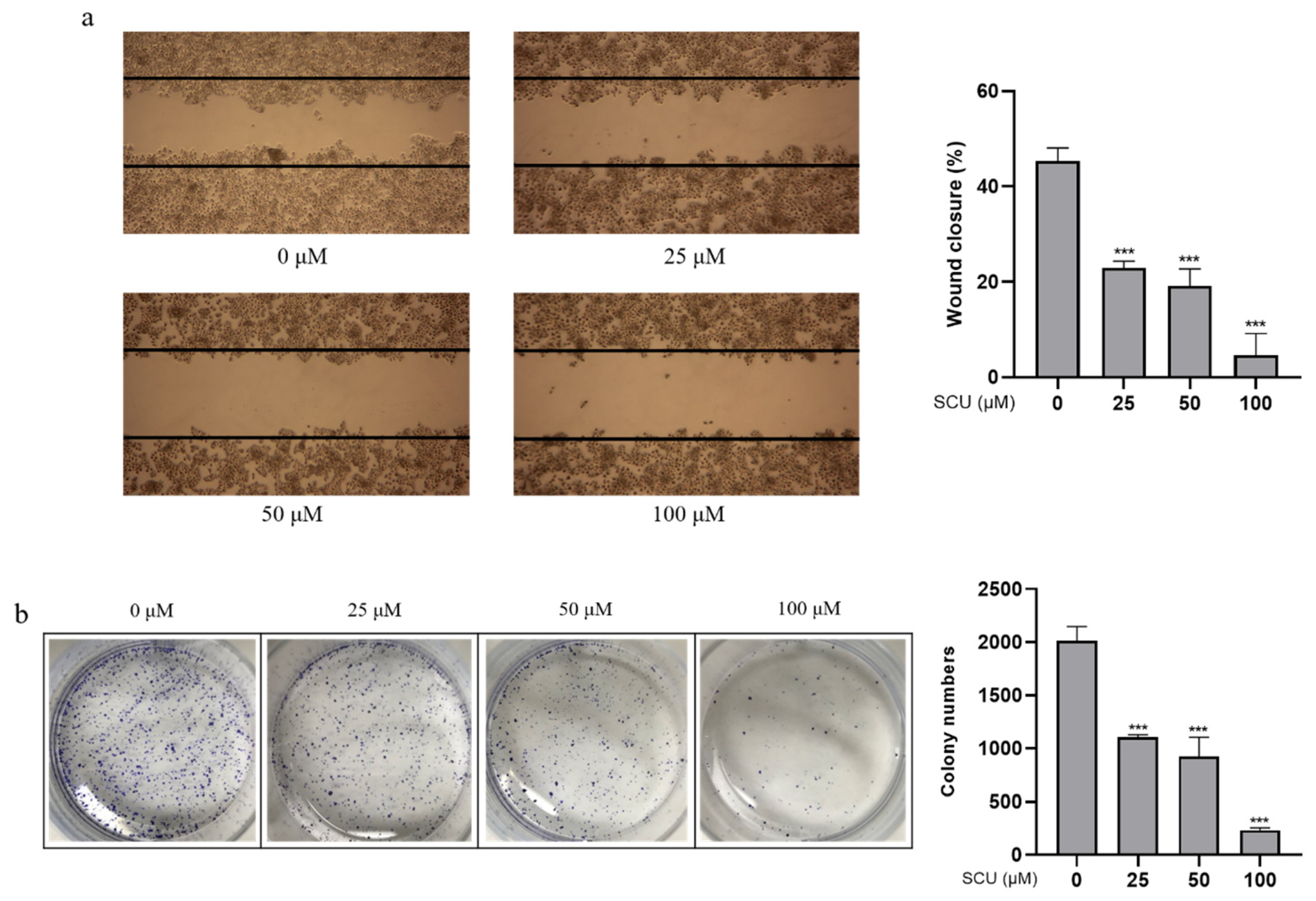
2.2. SCU Inhibits Cell Migration and Proliferation in HepG2 Cells
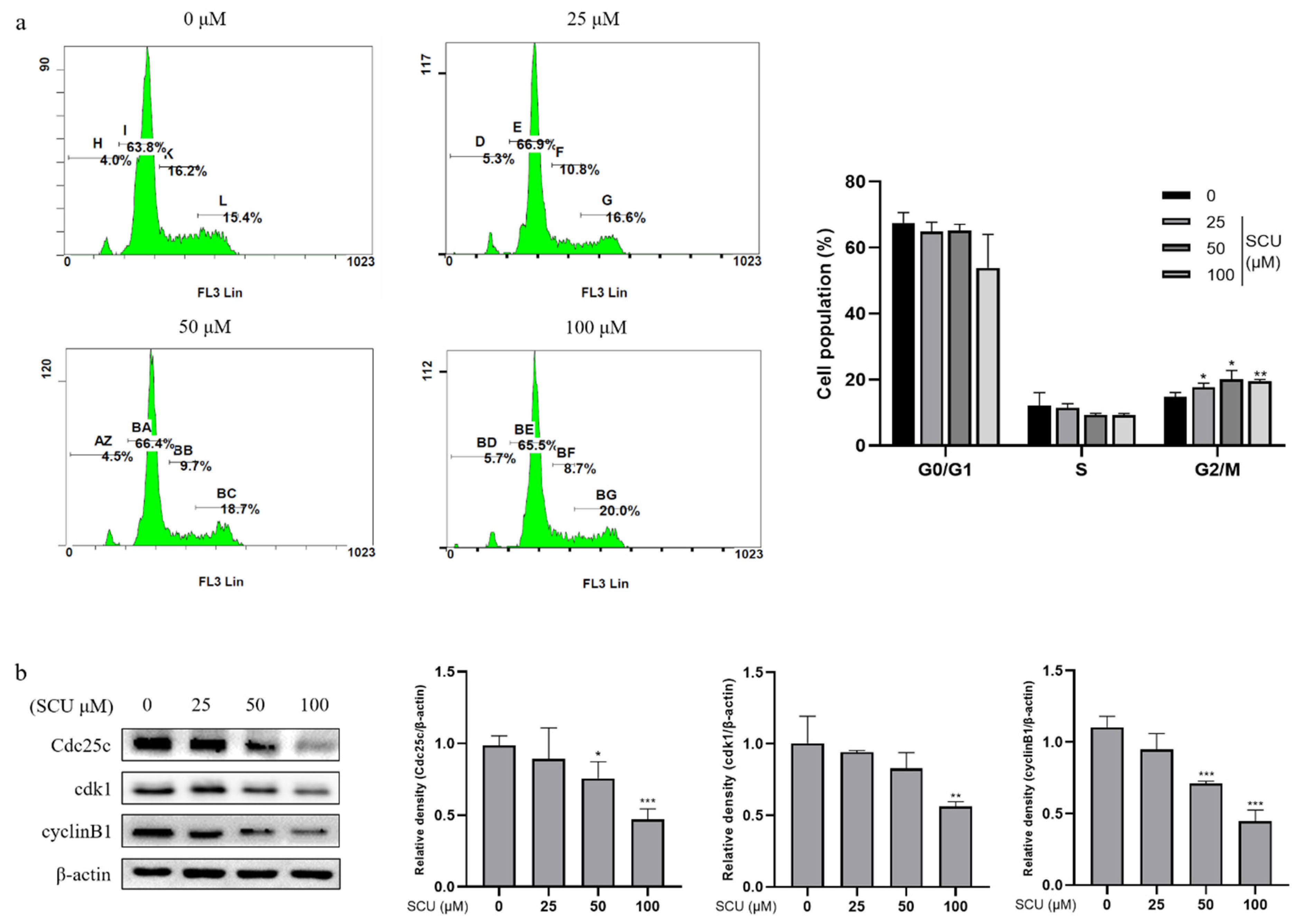
2.3. SCU Induces G2/M Phase Cell Cycle Arrest in HepG2 Cells
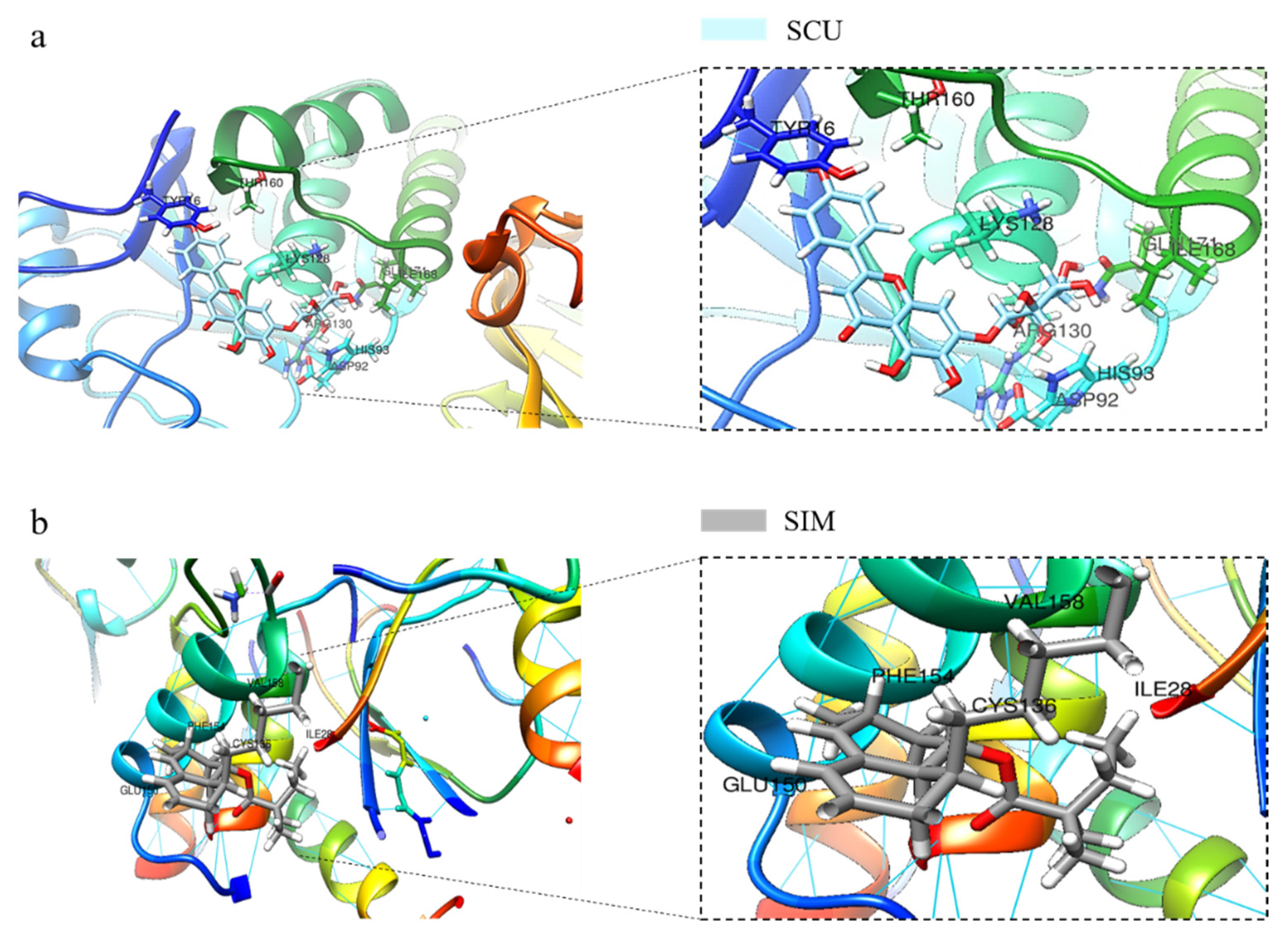
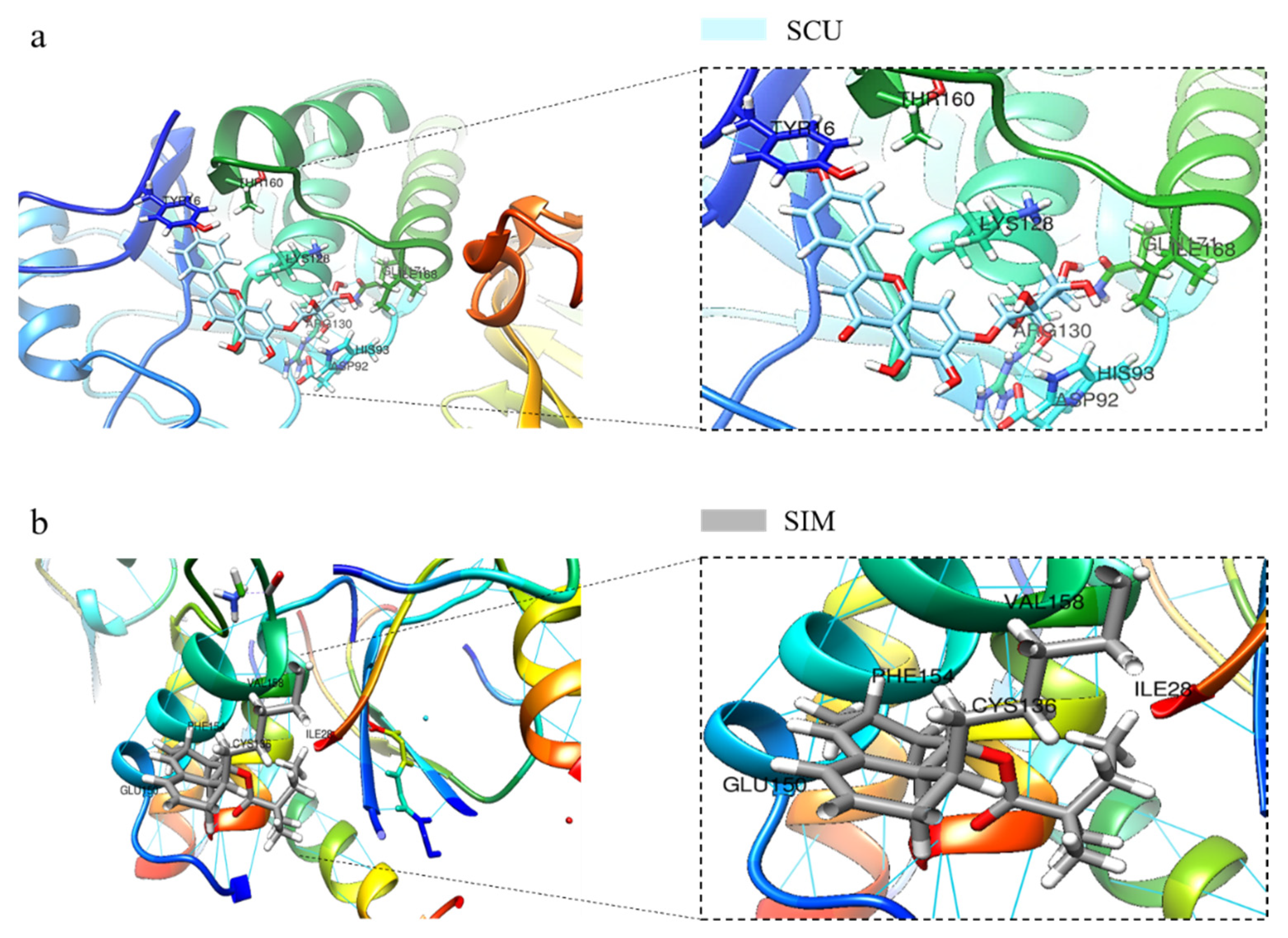
2.4. SCU Induces PTEN Expression in HepG2 Cells and Validation of SCU Binding with PTEN Using In Silico Molecular Docking Analysis
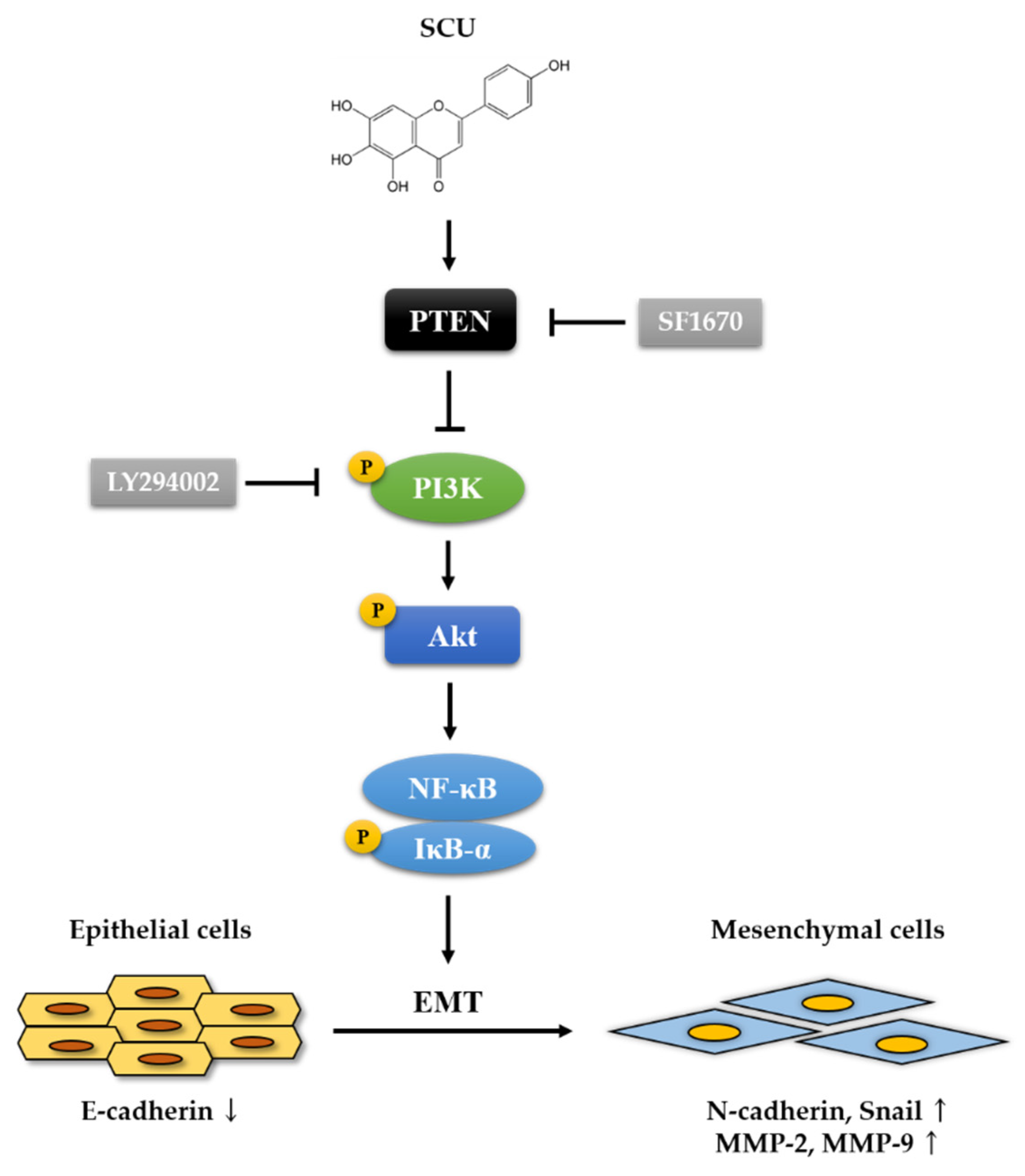
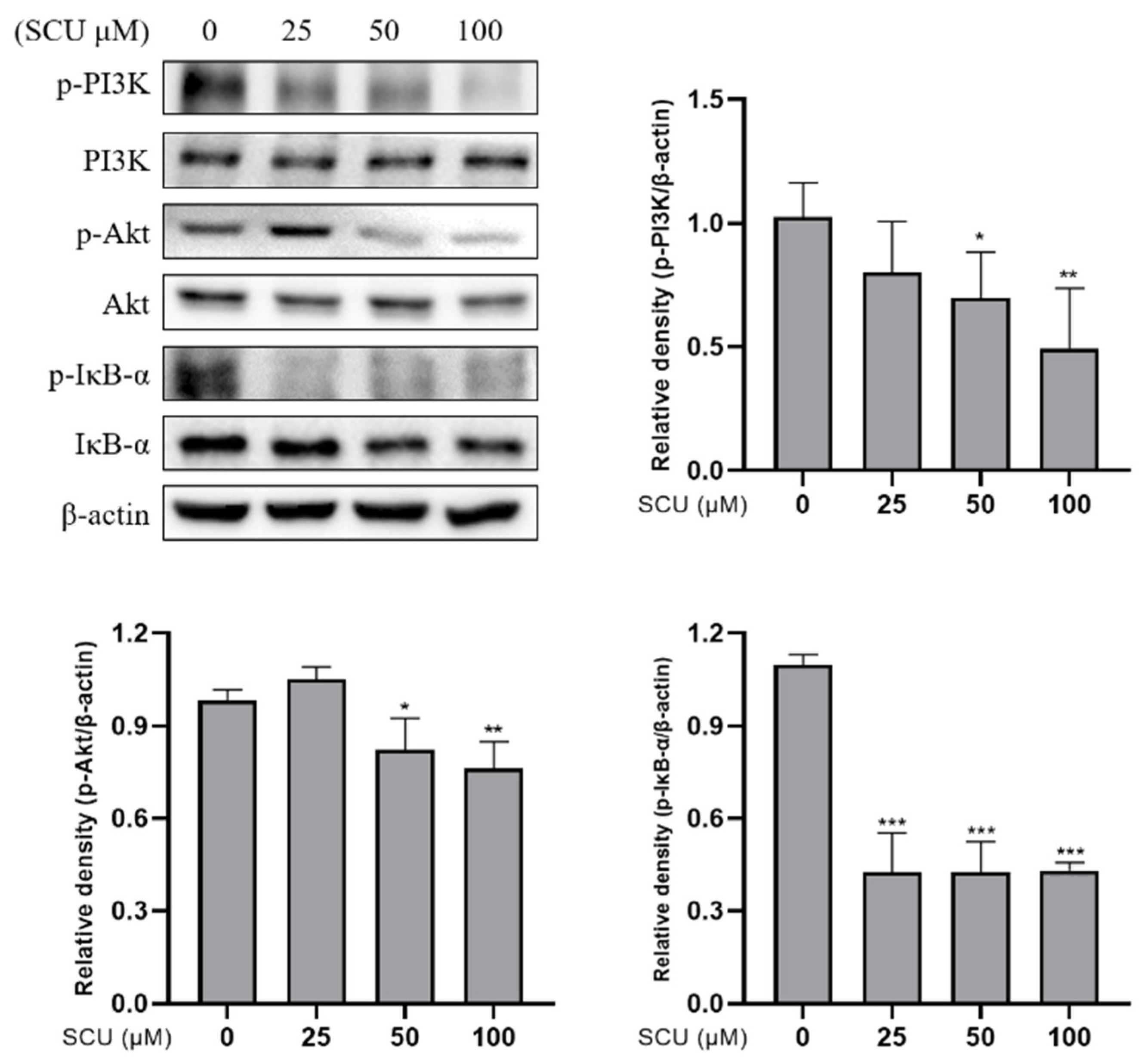
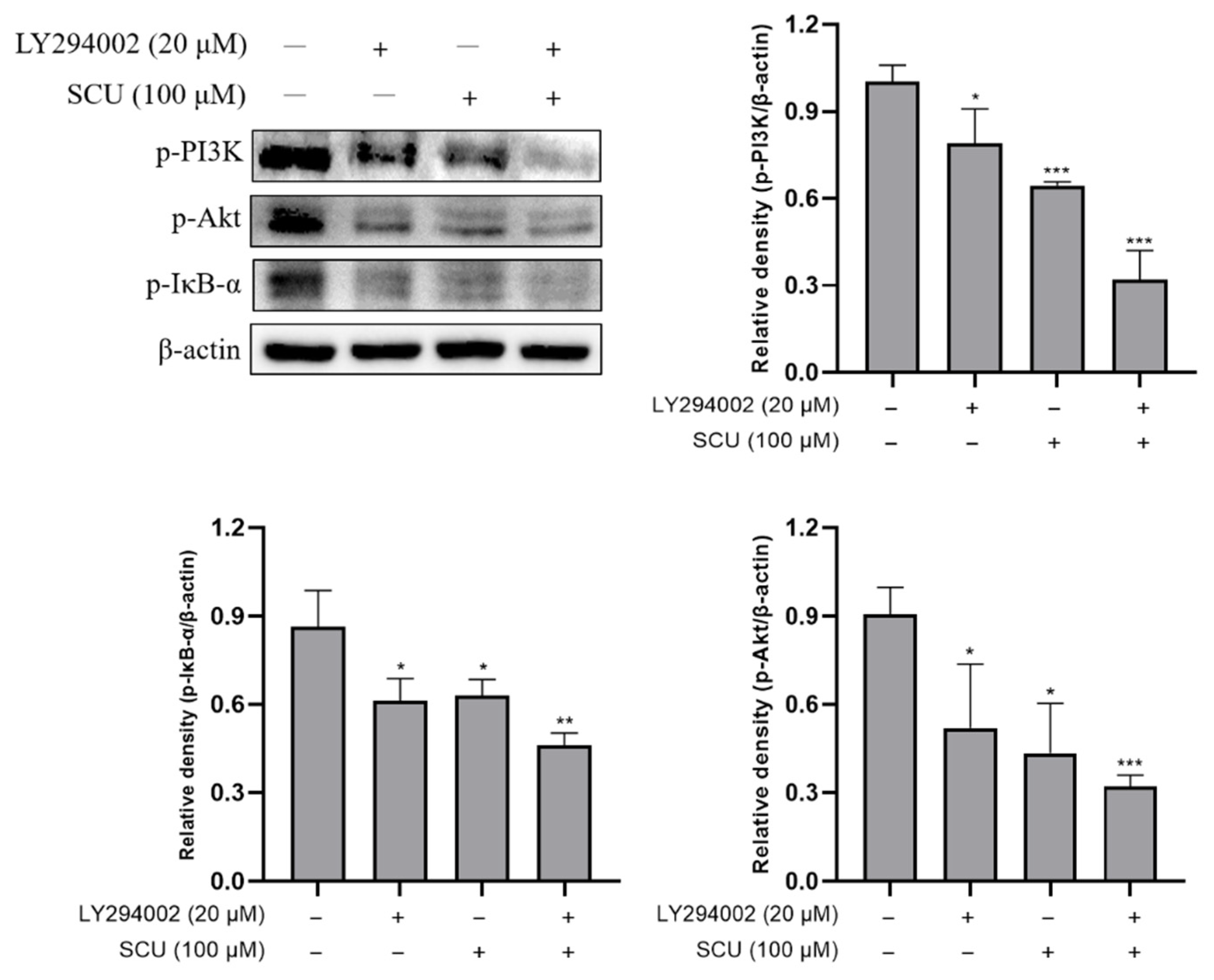
2.5. SCU Suppresses the PI3K/Akt/NF-κB Signaling Pathway in HepG2 Cells
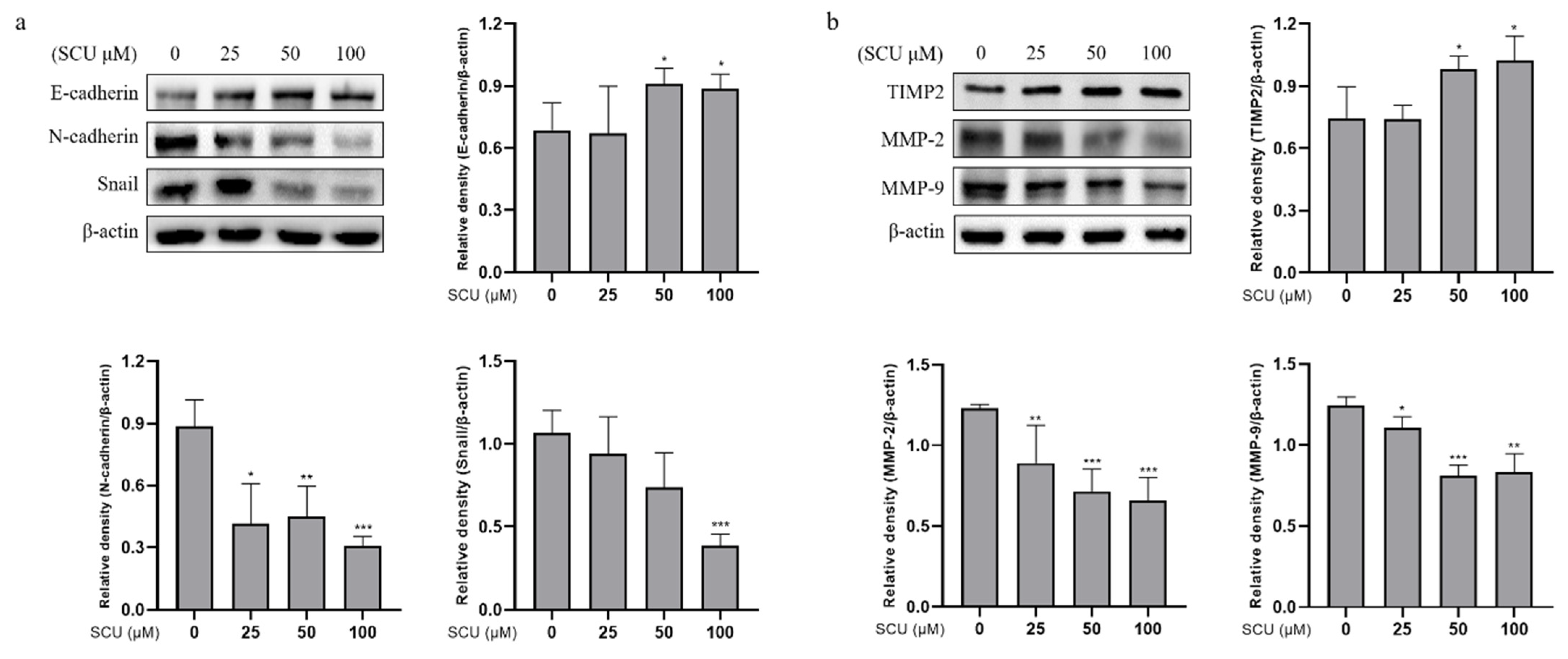
2.6. SCU Inhibits the Activity of EMT Markers and Migration-Related Proteins in HepG2 Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture and SCU Treatment
4.3. Cell Viability Assay
4.4. Wound Healing Assay
4.5. Colony Formation Assay
4.6. Analysis of Cell Cycle Distribution by Flow Cytometry
4.7. Western Blot Analysis
4.8. Inhibitor Assay
4.9. Immunocytochemistry
4.10. Molecular Docking Studies
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Altekruse, S.F.; McGlynn, K.A.; Reichman, M.E. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J. Clin. Oncol. 2009, 27, 1485–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, A.; Hoshida, Y.; Battiston, C.; Tovar, V.; Sia, D.; Alsinet, C.; Cornella, H.; Liberzon, A.; Kobayashi, M.; Kumada, H.; et al. Combining clinical, pathology, and gene expression data to predict recurrence of hepatocellular carcinoma. Gastroenterology 2011, 140, 1501–1512. [Google Scholar] [CrossRef] [Green Version]
- Dave, B.; Migliaccio, I.; Gutierrez, M.C.; Wu, M.F.; Chamness, G.C.; Wong, H.; Narasanna, A.; Chakrabarty, A.; Hilsenbeck, S.G.; Huang, J.; et al. Loss of phosphatase and tensin homolog or phosphoinositol-3 kinase activation and response to trastuzumab or lapatinib in human epidermal growth factor receptor 2-overexpressing locally advanced breast cancers. J. Clin. Oncol. 2011, 29, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.M.; Miller, T.W. Therapeutic targeting of cancers with loss of PTEN function. Curr. Drug Targets 2014, 15, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Dasari, V.R.; Kaur, K.; Velpula, K.K.; Gujrati, M.; Fassett, D.; Klopfenstein, J.D.; Dinh, D.H.; Rao, J.S. Upregulation of PTEN in glioma cells by cord blood mesenchymal stem cells inhibits migration via downregulation of the PI3K/Akt pathway. PLoS ONE 2010, 5, e10350. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef]
- Bouali, S.; Chretien, A.S.; Ramacci, C.; Rouyer, M.; Becuwe, P.; Merlin, J.L. PTEN expression controls cellular response to cetuximab by mediating PI3K/AKT and RAS/RAF/MAPK downstream signaling in KRAS wild-type, hormone refractory prostate cancer cells. Oncol. Rep. 2009, 21, 731–735. [Google Scholar]
- Lu, X.X.; Cao, L.Y.; Chen, X.; Xiao, J.; Zou, Y.; Chen, Q. PTEN Inhibits Cell Proliferation, Promotes Cell Apoptosis, and Induces Cell Cycle Arrest via Downregulating the PI3K/AKT/hTERT Pathway in Lung Adenocarcinoma A549 Cells. BioMed Res. Int. 2016, 2016, 2476842. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.B.; Gao, Z.M.; Huang, W.Y.; Lu, Z.B. The mechanism of epithelial-mesenchymal transition induced by TGF-beta1 in neuroblastoma cells. Int. J. Oncol. 2017, 50, 1623–1633. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef]
- Melnikova, V.O.; Bar-Eli, M. Transcriptional control of the melanoma malignant phenotype. Cancer Biol. Ther. 2008, 7, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Sethi, S.; Macoska, J.; Chen, W.; Sarkar, F.H. Molecular signature of epithelial-mesenchymal transition (EMT) in human prostate cancer bone metastasis. Am. J. Transl. Res. 2010, 3, 90–99. [Google Scholar] [PubMed]
- Gos, M.; Miloszewska, J.; Przybyszewska, M. Epithelial-mesenchymal transition in cancer progression. Postepy Biochem. 2009, 55, 121–128. [Google Scholar] [PubMed]
- Nguyen, P.T.; Kudo, Y.; Yoshida, M.; Iizuka, S.; Ogawa, I.; Takata, T. N-cadherin expression is correlated with metastasis of spindle cell carcinoma of head and neck region. J. Oral Pathol. Med. 2011, 40, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer 2010, 102, 639–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Li, S.; Deng, L.; Yang, S.; Li, M.; Long, S.; Chen, S.; Lin, F.; Xiao, L. Decreased MT1-MMP in gastric cancer suppressed cell migration and invasion via regulating MMPs and EMT. Tumour Biol. 2015, 36, 6883–6889. [Google Scholar] [CrossRef] [PubMed]
- Le Marchand, L. Cancer preventive effects of flavonoids—A review. Biomed. Pharmacother. 2002, 56, 296–301. [Google Scholar] [CrossRef]
- Shi, X.; Chen, G.; Liu, X.; Qiu, Y.; Yang, S.; Zhang, Y.; Fang, X.; Zhang, C.; Liu, X. Scutellarein inhibits cancer cell metastasis in vitro and attenuates the development of fibrosarcoma in vivo. Int. J. Mol. Med. 2015, 35, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Zhu, B.H.; Tang, S.B.; Wang, J.F.; Ren, J. Scutellarein inhibits hypoxia- and moderately-high glucose-induced proliferation and VEGF expression in human retinal endothelial cells. Acta Pharmacol. Sin. 2008, 29, 707–712. [Google Scholar] [CrossRef] [Green Version]
- Li, N.G.; Song, S.L.; Shen, M.Z.; Tang, Y.P.; Shi, Z.H.; Tang, H.; Shi, Q.P.; Fu, Y.F.; Duan, J.A. Mannich bases of scutellarein as thrombin-inhibitors: Design, synthesis, biological activity and solubility. Bioorg. Med. Chem. 2012, 20, 6919–6923. [Google Scholar] [CrossRef]
- Ha, S.E.; Kim, S.M.; Lee, H.J.; Vetrivel, P.; Venkatarame Gowda Saralamma, V.; Heo, J.D.; Kim, E.H.; Lee, S.J.; Kim, G.S. Scutellarein Induces Fas-Mediated Extrinsic Apoptosis and G2/M Cell Cycle Arrest in Hep3B Hepatocellular Carcinoma Cells. Nutrients 2019, 11, 263. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, J.; Zhong, S.; Li, J.; Du, W. Scutellarein inhibits the development of colon cancer via CDC4mediated RAGE ubiquitination. Int. J. Mol. Med. 2020, 45, 1059–1072. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [Green Version]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Efferth, T. Cancer therapy with natural products and medicinal plants. Planta Med. 2010, 76, 1035–1036. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.K.; Basu, S.; Sarkar, N.; Ghosh, A.C. Advances in cancer therapy with plant based natural products. Curr. Med. Chem. 2001, 8, 1467–1486. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, A.B.; Lopes, R.M.; Schwartsmann, G. Natural products in anticancer therapy. Curr. Opin. Pharmacol. 2001, 1, 364–369. [Google Scholar] [CrossRef]
- Nakamura, K.; Peng, Y.; Utsumi, F.; Tanaka, H.; Mizuno, M.; Toyokuni, S.; Hori, M.; Kikkawa, F.; Kajiyama, H. Novel Intraperitoneal Treatment with Non-Thermal Plasma-Activated Medium Inhibits Metastatic Potential of Ovarian Cancer Cells. Sci. Rep. 2017, 7, 6085. [Google Scholar] [CrossRef]
- Schulte-Hermann, R.; Bursch, W.; Low-Baselli, A.; Wagner, A.; Grasl-Kraupp, B. Apoptosis in the liver and its role in hepatocarcinogenesis. Cell Biol. Toxicol. 1997, 13, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, J.; Xiong, X.; Yuan, W.; Qin, S.; Cao, W.; Dai, L.; Xie, F.; Li, A.; Liu, Z. Icariin suppresses cell cycle transition and cell migration in ovarian cancer cells. Oncol. Rep. 2019, 41, 2321–2328. [Google Scholar] [CrossRef] [Green Version]
- Casella, M.L.; Parody, J.P.; Ceballos, M.P.; Quiroga, A.D.; Ronco, M.T.; Frances, D.E.; Monti, J.A.; Pisani, G.B.; Carnovale, C.E.; Carrillo, M.C.; et al. Quercetin prevents liver carcinogenesis by inducing cell cycle arrest, decreasing cell proliferation and enhancing apoptosis. Mol. Nutr. Food Res. 2014, 58, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Kao, G.D.; McKenna, W.G.; Maity, A.; Blank, K.; Muschel, R.J. Cyclin B1 availability is a rate-limiting component of the radiation-induced G2 delay in HeLa cells. Cancer Res. 1997, 57, 753–758. [Google Scholar] [PubMed]
- Perdiguero, E.; Nebreda, A.R. Regulation of Cdc25C activity during the meiotic G2/M transition. Cell Cycle 2004, 3, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; De Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 282–290. [Google Scholar] [CrossRef]
- Miller, T.W.; Perez-Torres, M.; Narasanna, A.; Guix, M.; Stal, O.; Perez-Tenorio, G.; Gonzalez-Angulo, A.M.; Hennessy, B.T.; Mills, G.B.; Kennedy, J.P.; et al. Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009, 69, 4192–4201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Shen, W.H. PTEN: A new guardian of the genome. Oncogene 2008, 27, 5443–5453. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Gu, W.; Zhang, Y.; Ji, Y.; Wen, Y.; Xu, X. Nicotine promotes cervical carcinoma cell line HeLa migration and invasion by activating PI3k/Akt/NF-kappaB pathway in vitro. Exp. Toxicol. Pathol. 2017, 69, 402–407. [Google Scholar] [CrossRef]
- Zhang, L.L.; Mu, G.G.; Ding, Q.S.; Li, Y.X.; Shi, Y.B.; Dai, J.F.; Yu, H.G. Phosphatase and Tensin Homolog (PTEN) Represses Colon Cancer Progression through Inhibiting Paxillin Transcription via PI3K/AKT/NF-kappaB Pathway. J. Biol. Chem. 2015, 290, 15018–15029. [Google Scholar] [CrossRef] [Green Version]
- Orlowski, R.Z.; Baldwin, A.S., Jr. NF-kappaB as a therapeutic target in cancer. Trends Mol. Med. 2002, 8, 385–389. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Ghani, R.; Serra, V.; Gyorffy, B.; Jurchott, K.; Solf, A.; Dietel, M.; Schafer, R. The PI3K inhibitor LY294002 blocks drug export from resistant colon carcinoma cells overexpressing MRP1. Oncogene 2006, 25, 1743–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.Y.; Wang, Q.; Shen, S.; Wei, X.L.; Li, G.X. Oridonin inhibits migration, invasion, adhesion and TGF-beta1-induced epithelial-mesenchymal transition of melanoma cells by inhibiting the activity of PI3K/Akt/GSK-3beta signaling pathway. Oncol. Lett. 2018, 15, 1362–1372. [Google Scholar] [CrossRef]
- Lee, Y.J.; Han, H.J. Troglitazone ameliorates high glucose-induced EMT and dysfunction of SGLTs through PI3K/Akt, GSK-3beta, Snail1, and beta-catenin in renal proximal tubule cells. J. Physiol. Ren. Physiol. 2010, 298, F1263–F1275. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Choi, E.J.; Cho, E.J.; Lee, Y.B.; Lee, J.H.; Yu, S.J.; Yoon, J.H.; Kim, Y.J. Inhibition of PI3K/Akt signaling suppresses epithelial-to-mesenchymal transition in hepatocellular carcinoma through the Snail/GSK-3/beta-catenin pathway. Clin. Mol. Hepatol. 2020, 26, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Yang, Q.; Lee, J.D. BMK1 kinase suppresses epithelial-mesenchymal transition through the Akt/GSK3beta signaling pathway. Cancer Res. 2012, 72, 1579–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nwomeh, B.C.; Liang, H.X.; Diegelmann, R.F.; Cohen, I.K.; Yager, D.R. Dynamics of the matrix metalloproteinases MMP-1 and MMP-8 in acute open human dermal wounds. Wound Repair Regen. 1998, 6, 127–134. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, S.E.; Kim, S.M.; Vetrivel, P.; Kim, H.H.; Bhosale, P.B.; Heo, J.D.; Lee, H.J.; Kim, G.S. Inhibition of Cell Proliferation and Metastasis by Scutellarein Regulating PI3K/Akt/NF-κB Signaling through PTEN Activation in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 8841. https://doi.org/10.3390/ijms22168841
Ha SE, Kim SM, Vetrivel P, Kim HH, Bhosale PB, Heo JD, Lee HJ, Kim GS. Inhibition of Cell Proliferation and Metastasis by Scutellarein Regulating PI3K/Akt/NF-κB Signaling through PTEN Activation in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2021; 22(16):8841. https://doi.org/10.3390/ijms22168841
Chicago/Turabian StyleHa, Sang Eun, Seong Min Kim, Preethi Vetrivel, Hun Hwan Kim, Pritam Bhagwan Bhosale, Jeong Doo Heo, Ho Jeong Lee, and Gon Sup Kim. 2021. "Inhibition of Cell Proliferation and Metastasis by Scutellarein Regulating PI3K/Akt/NF-κB Signaling through PTEN Activation in Hepatocellular Carcinoma" International Journal of Molecular Sciences 22, no. 16: 8841. https://doi.org/10.3390/ijms22168841
APA StyleHa, S. E., Kim, S. M., Vetrivel, P., Kim, H. H., Bhosale, P. B., Heo, J. D., Lee, H. J., & Kim, G. S. (2021). Inhibition of Cell Proliferation and Metastasis by Scutellarein Regulating PI3K/Akt/NF-κB Signaling through PTEN Activation in Hepatocellular Carcinoma. International Journal of Molecular Sciences, 22(16), 8841. https://doi.org/10.3390/ijms22168841