
Transglutaminase 6 Is Colocalized and Interacts with Mutant Huntingtin in Huntington Disease Rodent Animal Models

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Regional Distribution of TG6 in BACHD Mice
2.2. TG6 Enzymatic Activity Is Prominent in BACHD Mice
2.3. Regional Colocalization of TG6 and the TG-Enzymatic Reaction Products in the Brain of tgHD Rats
2.4. Distribution of Mutant Huntingtin Pathology in tgHD Rats
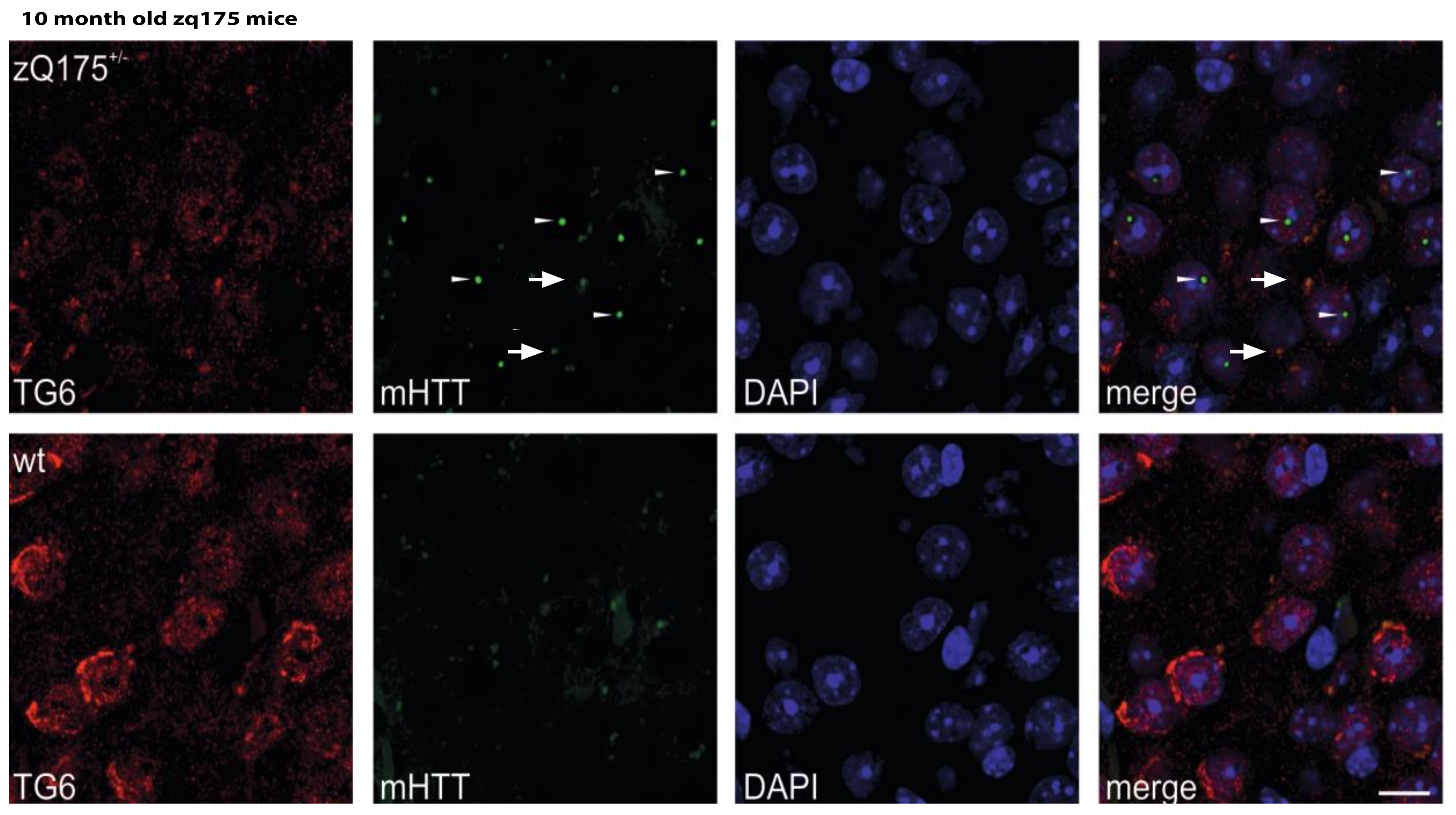
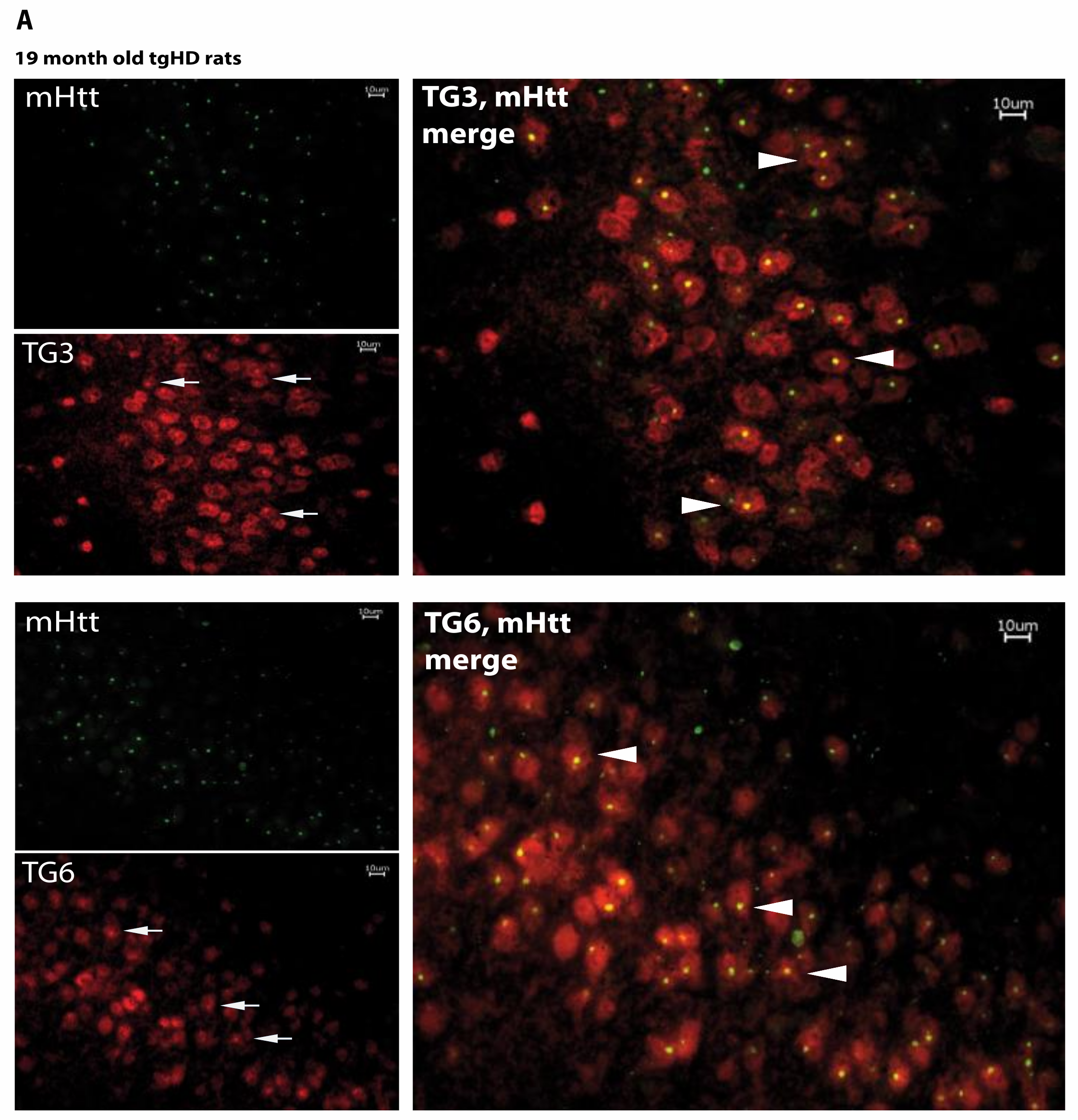
2.5. Huntingtin Aggregates Colocalize with TG6 in Different Rodent HD Transgenic Animal Models
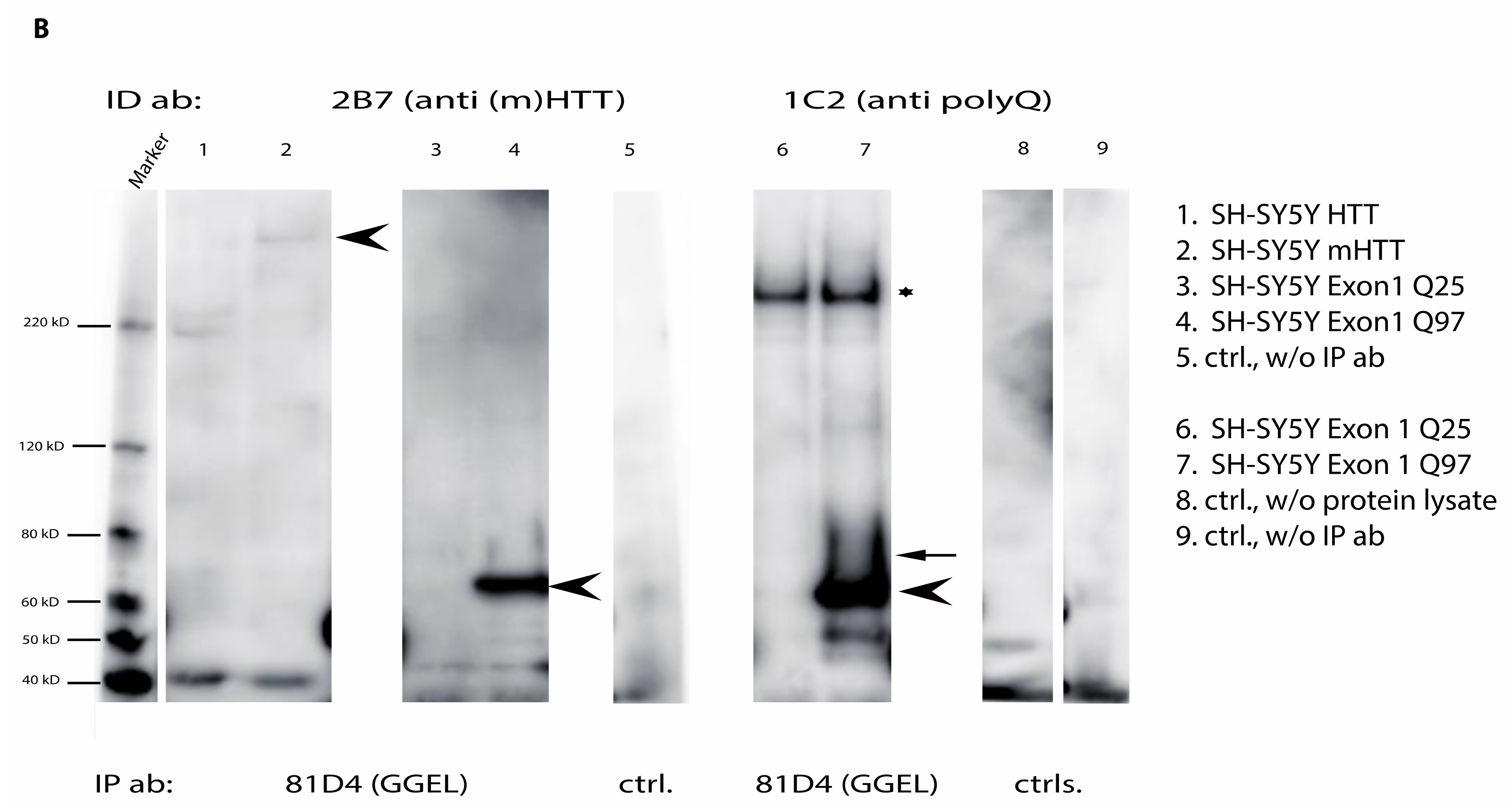
2.6. Mutant Huntingtin Contains Transglutaminase-Catalyzed Crosslinks
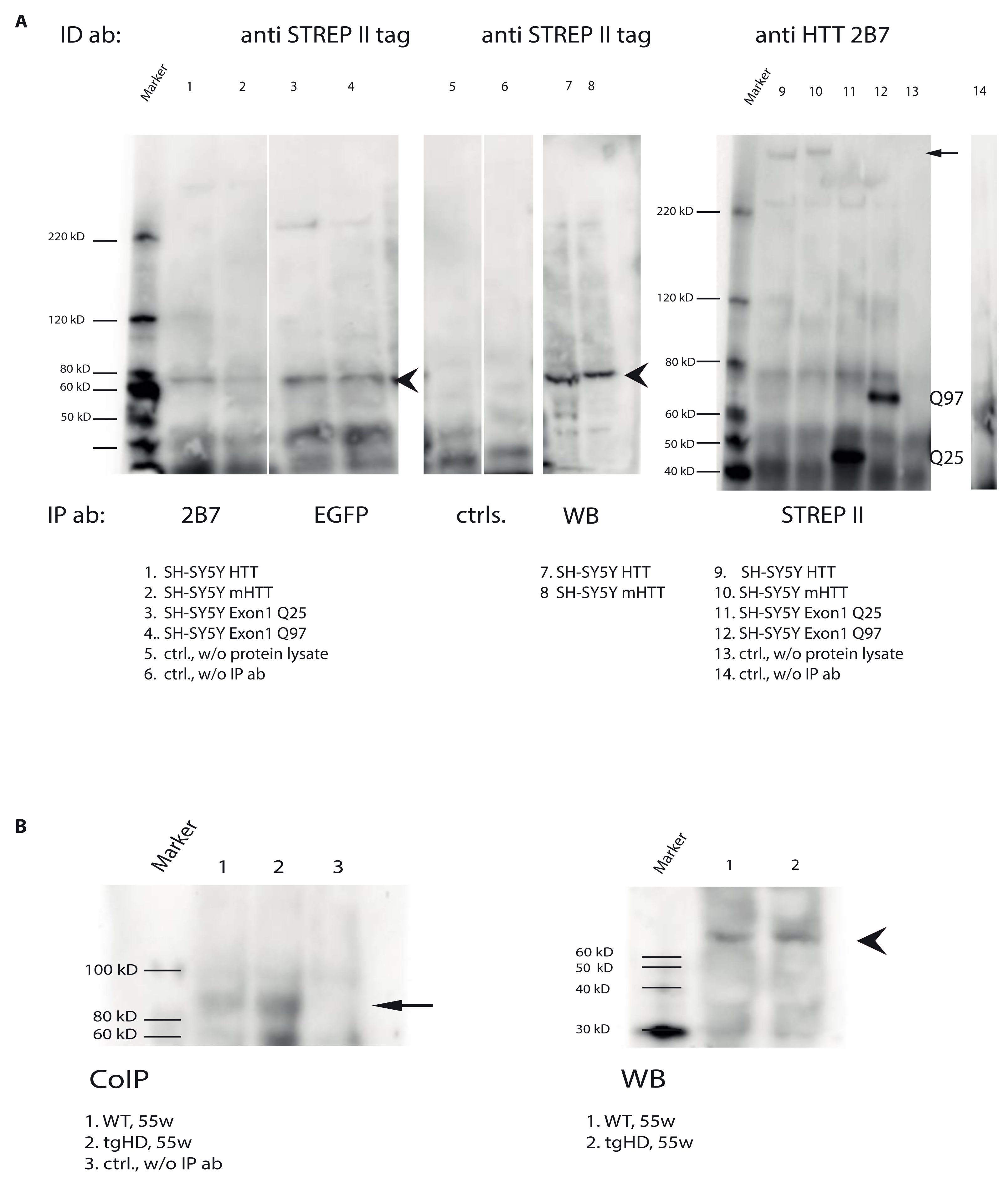
2.7. Direct Interaction of TG6 and Huntingtin
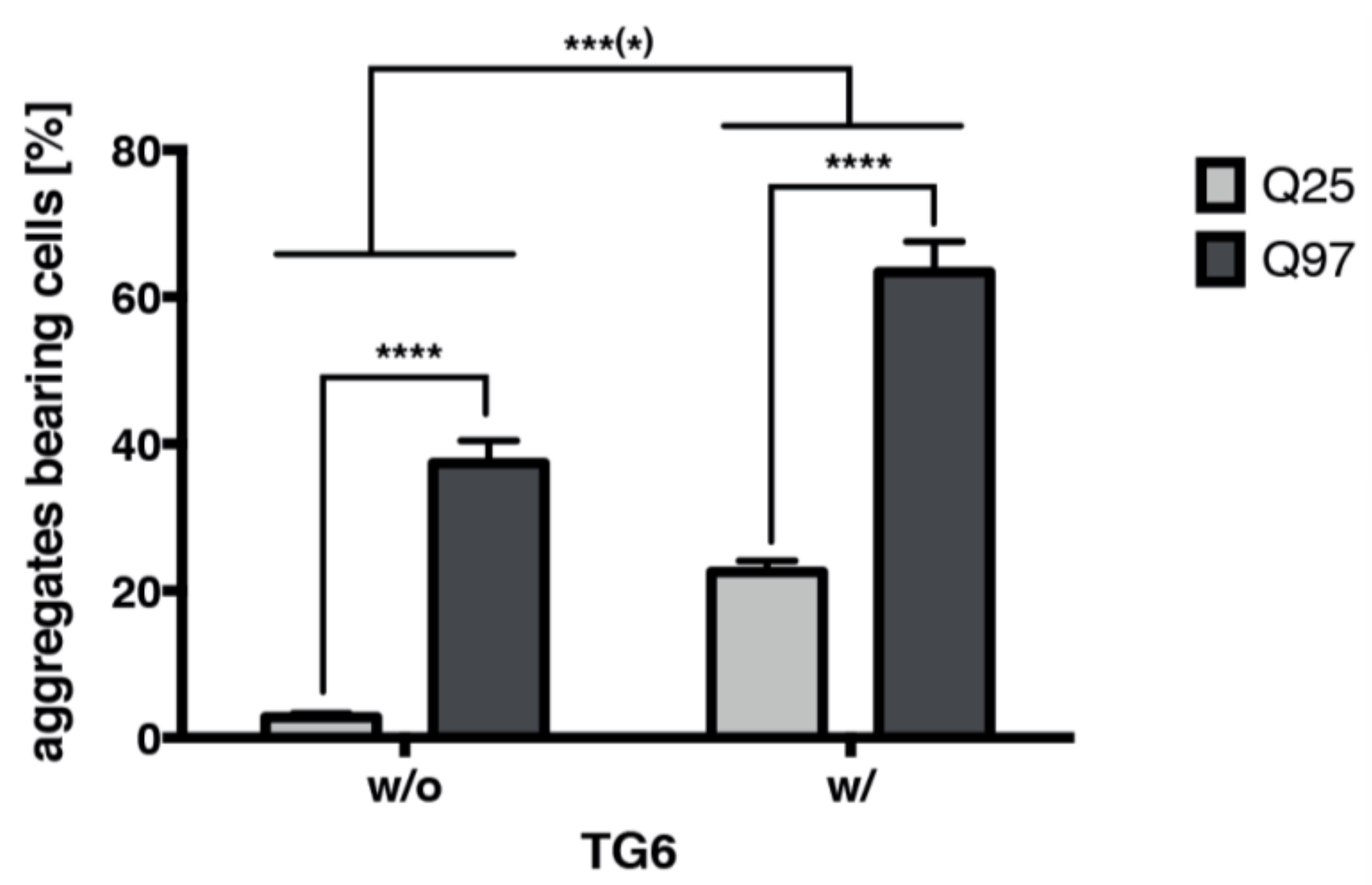
2.8. TG6 Overexpression Induces Accumulation of Mutant Huntingtin Exon 1 Aggregates
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. Plasmid Constructs
4.3. Cell Lines and Cell Culture
4.4. Quantification of Mutant Huntingtin Exon 1 Aggregates and Statistical Analysis
4.4.1. Glutamine-Donor Synthetic Peptides
4.4.2. In Situ TG Activity Assay on Brain Section
4.5. Immunohistochemistry and Immunofluorescence
4.6. Sample Preparation of Brain Tissues or Transfected Cells for Immunoblotting
4.7. Co-Immunoprecipitation
4.8. SDS-Gel Electrophoresis and Western Blot
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huntington’s Disease Collaborative Research. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s Disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Huntington’s disease. Handb. Exp. Pharmacol. 2014, 220, 357–409. [Google Scholar]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Gutekunst, C.A.; Li, S.H.; Yi, H.; Mulroy, J.S.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.J.; Hersch, S.M.; Li, X.J. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J. Neurosci. 1999, 19, 2522–2534. [Google Scholar] [CrossRef]
- Arrasate, M.; Finkbeiner, S. Protein aggregates in Huntington’s disease. Exp. Neurol. 2012, 238, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Saudou, F.; Finkbeiner, S.; Devys, D.; Greenberg, M.E. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 1998, 95, 55–66. [Google Scholar] [CrossRef]
- Esposito, C.; Caputo, I. Mammalian transglutaminases—Identification of substrates as a key to physiological function and physiopathological relevance. FEBS J. 2005, 272, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K. Mammalian transglutaminases: A family portrait. In Transglutaminases: Family of Enzymes with Diverse Functions; Mehta, K., Eckert, R., Eds.; Karger Medical and Scientific Publishers: Basel, Switzerland, 2005; pp. 1–18. [Google Scholar]
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003, 4, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Jeitner, T.M.; Steinert, P.M. Transglutaminase in disease. Neurochem. Int. 2002, 40, 85–103. [Google Scholar] [CrossRef]
- Bailey, C.D.C.; Graham, R.M.; Nanda, N.; Davies, P.J.A.; Johnson, G.V. Validity of mouse models for the study of tissue transglutaminase in neurodegenerative diseases. Mol. Cell. Neurosci. 2004, 25, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Iismaa, S.E.; Mearns, B.M.; Lorand, L.; Graham, R.M. Transglutaminases and Disease: Lessons From Genetically Engineered Mouse Models and Inherited Disorders. Physiol. Rev. 2009, 89, 991–1023. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, N.; Matsuki, M.; Yamanishi, K.; Yasuno, H. Transglutaminase 1 is essential to embryonic development of skin barrier. J. Dermatol. Sci. 2000, 23, 215. [Google Scholar]
- Matsuki, M.; Kuramoto, N.; Yamanishi, K.; Yasuno, H.; Takizawa, T.; Takizawa, T. Barrier defects leading into ichthyosiform skin phenotype in transglutaminase 1 deficiency. J. Dermatol. Sci. 2000, 23, 199. [Google Scholar]
- Fesus, L.; Piacentini, M. Transglutaminase 2: An enigmatic enzyme with diverse functions. Trends Biochem. Sci. 2002, 27, 534–539. [Google Scholar] [CrossRef]
- McConoughey, S.J.; Basso, M.; Niatsetskaya, Z.V.; Sleiman, S.F.; Smimova, N.A.; Langley, B.C.; Mahishi, L.; Cooper, A.J.L.; Antonyak, M.A.; Cerione, R.A.; et al. Inhibition of transglutaminase 2 mitigates transcriptional dysregulation in models of Huntington’s disease. EMBO Mol. Med. 2010, 2, 349–370. [Google Scholar] [CrossRef]
- D'Eletto, M.; Farrace, M.G.; Falasca, L.; Reali, V.; Oliverio, S.; Melino, G.; Griffin, M.; Fimia, G.M.; Piacentini, M. Transglutaminase 2 is involved in autophagosome maturation. Autophagy 2009, 5, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- D'Eletto, M.; Farrace, M.G.; Rossin, F.; Strappazzon, F.; Di Giacomo, G.; Cecconi, F.; Melino, G.; Sepe, S.; Moreno, S.; Fimia, G.M.; et al. Type 2 transglutaminase is involved in the autophagy-dependent clearance of ubiquitinated proteins. Cell Death Differ. 2012, 19, 1228–1238. [Google Scholar] [CrossRef]
- Dieterich, W.; Ehnis, T.; Bauer, M.; Donner, P.; Volta, U.; Riecken, E.O.; Schuppan, D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 1997, 3, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Ahvazi, B.; Boeshans, K.; Jang, S.I.; Kalinin, A.E.; Steinert, P.M. Transglutaminases in skin biology. Minerva Biotecnol. 2002, 14, 165–169. [Google Scholar]
- Kalinin, A.E.; Kajava, A.V.; Steinert, P.M. Epithelial barrier function: Assembly and structural features of the cornified cell envelope. Bioessays 2002, 24, 789–800. [Google Scholar] [CrossRef]
- John, S.; Thiebach, L.; Frie, C.; Mokkapati, S.; Bechtel, M.; Nischt, R.; Rosser-Davies, S.; Paulsson, M.; Smyth, N. Epidermal Transglutaminase (TGase 3) Is Required for Proper Hair Development, but Not the Formation of the Epidermal Barrier. PLoS ONE 2012, 7, e34252. [Google Scholar]
- Thomas, H.; Beck, K.; Adamczyk, M.; Aeschlimann, P.; Langley, M.; Oita, R.C.; Thiebach, L.; Hils, M.; Aeschlimann, D. Transglutaminase 6: A protein associated with central nervous system development and motor function. Amino Acids 2013, 44, 161–177. [Google Scholar] [CrossRef]
- Guan, W.J.; Liu, X.J.; Tang, B.S.; Liu, Y.T.; Zhou, Y.; Jiang, H.; Shen, L.; Xia, K.; Wang, J.-L. Gluten ataxia of sporadic and hereditary cerebellar ataxia in patients from mainland China. Neurol. India 2013, 61, 226–230. [Google Scholar] [PubMed]
- Guan, W.-J.; Wang, J.-L.; Liu, Y.-T.; Ma, Y.-T.; Zhou, Y.; Jiang, H.; Shen, L.; Guo, J.-F.; Xia, K.; Li, J.-D.; et al. Spinocerebellar ataxia type 35 (SCA35)-associated transglutaminase 6 mutants sensitize cells to apoptosis. Biochem. Biophys. Res. Commun. 2013, 430, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Xia, K.D.; Ma, Y.T.; Liu, Y.T.; Shi, Y.T.; Jiang, H.; Shen, L.; Xia, K.; Li, J.D.; Tang, B.S.; et al. Transglutaminase 6 interacts with polyQ proteins and promotes the formation of polyQ aggregates. Biochem. Biophys. Res. Commun. 2013, 437, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lu, Y.; Peng, F.; Yu, H.L.; Wu, J.Y.; Tan, Y.; Zhao, Y.X. TGM6 variants in Parkinson's disease: Clinical findings and functional evidence. J. Integr. Neurosci. 2020, 19, 51–64. [Google Scholar] [CrossRef]
- Aeschlimann, D.; Thomazy, V. Protein crosslinking in assembly and remodelling of extracellular matrices: The role of transglutaminases. Connect. Tissue Res. 2000, 41, 1–27. [Google Scholar] [CrossRef]
- Zeron, M.M.; Chen, N.S.; Moshaver, A.; Lee, A.T.C.; Wellington, C.L.; Hayden, M.R.; Raymond, L.A. Mutant huntingtin enhances excitotoxic cell death. Mol. Cell. Neurosci. 2001, 17, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Zeron, M.M.; Fernandes, H.B.; Krebs, C.; Shehadeh, J.; Wellington, C.L.; Leavitt, B.R.; Baimbridge, K.G.; Hayden, M.R.; Raymond, L.A. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington’s disease. Mol. Cell. Neurosci. 2004, 25, 469–479. [Google Scholar] [CrossRef]
- Kiraly, R.; Demeny, M.A.; Fesues, L. Protein transamidation by transglutaminase 2 in cells: A disputed Ca2+-dependent action of a multifunctional protein. FEBS J. 2011, 278, 4717–4739. [Google Scholar] [CrossRef] [PubMed]
- Jeitner, T.M.; Pinto, J.T.; Krasnikov, B.F.; Horswill, M.; Cooper, A.J.L. Transglutaminases and neurodegeneration. J. Neurochem. 2009, 109, 160–166. [Google Scholar] [CrossRef]
- Muma, N.A. Transglutaminase is linked to neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 258–263. [Google Scholar] [CrossRef]
- Hoffner, G.; Djian, P. Transglutaminase and diseases of the central nervous system. Front. Biosci. 2005, 10, 3078–3092. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zainelli, G.M.; Dudek, N.L.; Ross, C.A.; Kim, S.Y.; Muma, N.A. Mutant huntingtin protein—A substrate for transglutaminase 1, 2, and 3. J. Neuropathol. Exp. Neurol. 2005, 64, 58–65. [Google Scholar] [CrossRef]
- Caccamo, D.; Curro, M.; Condello, S.; Ferlazzo, N.; Ientile, R. Critical role of transglutaminase and other stress proteins during neurodegenerative processes. Amino Acids 2010, 38, 653–658. [Google Scholar] [CrossRef]
- Wolf, J.; Jager, C.; Lachmann, I.; Schonknecht, P.; Morawski, M.; Arendt, T.; Mothes, T. Tissue transglutaminase is not a biochemical marker for Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Jager, C.; Morawski, M.; Lachmann, I.; Schonknecht, P.; Mothes, T.; Arendt, T. Tissue transglutaminase in Alzheimer’s disease—Facts and fiction: A reply to “Tissue transglutaminase is a biochemical marker for Alzheimer’s disease”. Neurobiol. Aging 2014, 35, e5–e9. [Google Scholar] [CrossRef]
- Kumar, A.; Kneynsberg, A.; Tucholski, J.; Perry, G.; van Groen, T.; Detloff, P.J.; Lesort, M. Tissue transglutaminase overexpression does not modify the disease phenotype of the R6/2 mouse model of Huntington’s disease. Exp. Neurol. 2012, 237, 78–89. [Google Scholar] [CrossRef][Green Version]
- Wilhelmus, M.M.; Drukarch, B. Tissue transglutaminase is a biochemical marker for Alzheimer’s disease. Neurobiol. Aging 2014, 35, E3–E4. [Google Scholar] [CrossRef]
- Von Horsten, S.; Schmitt, I.; Nguyen, H.P.; Holzmann, C.; Schmidt, T.; Walther, T.; Bader, M.; Pabst, R.; Kobbe, P.; Krotova, J.; et al. Transgenic rat model of Huntington’s disease. Hum. Mol. Genet. 2003, 12, 617–624. [Google Scholar] [CrossRef]
- Gray, M.; Shirasaki, D.I.; Cepeda, C.; Andre, V.M.; Wilburn, B.; Lu, X.-H.; Tao, J.; Yamazaki, I.; Li, S.-H.; Sun, Y.E.; et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 2008, 28, 6182–6195. [Google Scholar] [CrossRef]
- Esposito, C.; Paparo, F.; Caputo, I.; Porta, R.; Salvati, V.M.; Mazzarella, G.; Auricchio, S.; Troncone, R. Expression and enzymatic activity of small intestinal tissue transglutaminase in celiac disease. Am. J. Gastroenterol. 2003, 98, 1813–1820. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Krebs, A.; Canneva, F.; Schnepf, R.; Dobner, J.; Dieterich, W.; von Horsten, S. In situ enzymatic activity of transglutaminase isoforms on brain tissue sections of rodents: A new approach to monitor differences in post-translational protein modifications during neurodegeneration. Brain Res. 2015, 1631, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Bhide, P.G.; Day, M.; Sapp, E.; Schwarz, C.; Sheth, A.; Kim, J.; Young, A.B.; Penney, J.; Golden, J.; Aronin, N.; et al. Expression of normal and mutant huntingtin in the developing brain. J. Neurosci. 1996, 16, 5523–5535. [Google Scholar] [CrossRef]
- Petrasch-Parwez, E.; Nguyen, H.-P.; Loebbecke-Schumacher, M.; Habbes, H.-W.; Wieczorek, S.; Riess, O.; Andres, K.-H.; Dermietzel, R.; Von Hoersten, S. Cellular and subcellular localization of Huntington aggregates in the brain of a rat transgenic for Huntington disease. J. Comp. Neurol. 2007, 501, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Southwell, A.L.; Smith-Dijak, A.; Kay, C.; Sepers, M.; Villanueva, E.B.; Parsons, M.P.; Xie, Y.; Anderson, L.; Felczak, B.; Waltl, S.; et al. An enhanced Q175 knock-in mouse model of Huntington disease with higher mutant huntingtin levels and accelerated disease phenotypes. Hum. Mol. Genet. 2016, 25, 3654–3675. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Vignoli, B.; Ramesh, N.; Polanco, M.J.; Coutelier, M.; Stephen, C.D.; Canossa, M.; Monin, M.L.; Aeschlimann, P.; Turberville, S.; et al. Mutations in TGM6 induce the unfolded protein response in SCA35. Hum. Mol. Genet. 2017, 26, 3749–3762. [Google Scholar] [CrossRef]
- Heng, M.Y.; Detloff, P.J.; Albin, R.L. Rodent genetic models of Huntington disease. Neurobiol. Dis. 2008, 32, 1–9. [Google Scholar] [CrossRef]
- Lee, C.Y.; Cantle, J.P.; Yang, X.W. Genetic manipulations of mutant huntingtin in mice: New insights into Huntington’s disease pathogenesis. FEBS J. 2013, 280, 4382–4394. [Google Scholar] [CrossRef]
- Ardan, T.; Baxa, M.; Levinska, B.; Sedlackova, M.; Nguyen, T.D.; Klima, J.; Juhas, S.; Juhasova, J.; Smatlikova, P.; Vochozkova, P.; et al. Transgenic minipig model of Huntington’s disease exhibiting gradually progressing neurodegeneration. Dis. Models Mech. 2019, 13, dmm041319. [Google Scholar] [CrossRef] [PubMed]
- Vidinska, D.; Vochozkova, P.; Smatlikova, P.; Ardan, T.; Klima, J.; Juhas, S.; Juhasova, J.; Bohuslavova, B.; Baxa, M.; Valekova, I.; et al. Gradual Phenotype Development in Huntington Disease Transgenic Minipig Model at 24 Months of Age. Neurodegener. Dis. 2018, 18, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Baxa, M.; Hruska-Plochan, M.; Juhas, S.; Vodicka, P.; Pavlok, A.; Juhasova, J.; Miyanohara, A.; Nejime, T.; Klima, J.; Macakova, M.; et al. A transgenic minipig model of Huntington’s Disease. J. Huntingt. Dis. 2013, 2, 47–68. [Google Scholar] [CrossRef] [PubMed]
- Holm, I.E.; Alstrup, A.K.; Luo, Y. Genetically modified pig models for neurodegenerative disorders. J. Pathol. 2016, 238, 267–287. [Google Scholar] [CrossRef] [PubMed]
- Sathasivam, K.; Woodman, B.; Mahal, A.; Bertaux, F.; Wanker, E.E.; Shima, D.T.; Bates, G.P. Centrosome disorganization in fibroblast cultures derived from R6/2 Huntington’s disease (HD) transgenic mice and HD patients. Hum. Mol. Genet. 2001, 10, 2425–2435. [Google Scholar] [CrossRef] [PubMed]
- Carty, N.; Berson, N.; Tillack, K.; Thiede, C.; Scholz, D.; Kottig, K.; Sedaghat, Y.; Gabrysiak, C.; Yohrling, G.; von der Kammer, H.; et al. Characterization of HTT inclusion size, location, and timing in the zQ175 mouse model of Huntington’s disease: An in vivo high-content imaging study. PLoS ONE 2015, 10, e0123527. [Google Scholar] [CrossRef]
- Abe, H.; Watanabe, M.; Yamakuni, T.; Kuwano, R.; Takahashi, Y.; Kondo, H. Localization of gene expression of calbindin in the brain of adult rats. Neurosci. Lett. 1992, 138, 211–215. [Google Scholar] [CrossRef]
- Cao, C.; Temel, Y.; Blokland, A.; Ozen, H.; Steinbusch, H.W.M.; Vlamings, R.; Nguyen, H.P.; von Horsten, S.; Schmitz, C.; Visser-Vandewalle, V. Progressive deterioration of reaction time performance and choreiform symptoms in a new Huntington’s disease transgenic ratmodel. Behav. Brain Res. 2006, 170, 257–261. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Kobbe, P.; Rahne, H.; Worpel, T.; Jager, B.; Stephan, M.; Pabst, R.; Holzmann, C.; Riess, O.; Korr, H.; et al. Behavioral abnormalities precede neuropathological markers in rats transgenic for Huntington’s disease. Hum. Mol. Genet. 2006, 15, 3177–3194. [Google Scholar] [CrossRef][Green Version]
- Menalled, L.B.; Kudwa, A.E.; Miller, S.; Fitzpatrick, J.; Watson-Johnson, J.; Keating, N.; Ruiz, M.; Mushlin, R.; Alosio, W.; McConnell, K.; et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PLoS ONE 2012, 7, e49838. [Google Scholar] [CrossRef]
- Mastroberardino, P.; Iannicola, C.; Nardacci, R.; Bernassola, F.; De Laurenzi, V.; Melino, G.; Moreno, S.; Pavone, F.; Oliverio, S.; Fesus, L.; et al. ‘Tissue’ transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington’s disease. Cell Death Differ. 2002, 9, 873–880. [Google Scholar] [CrossRef]
- Kim, H.C.; Lewis, M.S.; Gorman, J.J.; Park, S.C.; Girard, J.E.; Folk, J.E.; Chung, S.I. Protransglutaminase E from guinea pig skin. Isolation and partial characterization. J. Biol. Chem. 1990, 265, 21971–21978. [Google Scholar] [CrossRef]
- Dieterich, W.; Esslinger, B.; Trapp, D.; Hahn, E.; Huff, T.; Seilmeier, W.; Wieser, H.; Schuppan, D. Cross linking to tissue transglutaminase and collagen favours gliadin toxicity in coeliac disease. Gut 2006, 55, 478–484. [Google Scholar] [CrossRef]
- Dieterich, W.; Laag, E.; Bruckner-Tuderman, L.; Reunala, T.; Karpati, S.; Zagoni, T.; Riecken, E.O.; Schuppan, D. Antibodies to tissue transglutaminase as serologic markers in patients with dermatitis herpetiformis. J. Investig. Dermatol. 1999, 113, 133–136. [Google Scholar] [CrossRef]
- Ichinose, A.; Aoki, N. Reversible cross-linking of alpha 2-plasmin inhibitor to fibrinogen by fibrin-stabilizing factor. Biochim. Biophys. Acta 1982, 706, 158–164. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Bogdanov, M.B.; Matson, W.R.; Daikhin, Y.; Yudkoff, M.; Folk, J.E.; Steinman, L.; Browne, S.E.; Beal, M.F.; Blass, J.P.; et al. N-epsilon-(gamma-L-glutamyl)-L-lysine (GGEL) is increased in cerebrospinal fluid of patients with Huntington’s disease. J. Neurochem. 2001, 79, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Vermes, I.; Steur, E.N.; Jirikowski, G.F.; Haanen, C. Elevated concentration of cerebrospinal fluid tissue transglutaminase in Parkinson's disease indicating apoptosis. Mov. Disord. 2004, 19, 1252–1254. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, R.M.; Aschoff, A.; Niederwieser, G.; Heuberger, C.; Jirikowski, G. Cerebrospinal fluid tissue transglutaminase as a biochemical marker for Alzheimer’s disease. Neurobiol. Dis. 2002, 11, 106–110. [Google Scholar] [CrossRef][Green Version]
- Selkoe, D.J.; Abraham, C.; Ihara, Y. Crosslinking of alzheimer paired helical filaments—Analogies to transglutaminase-catalyzed filament crosslinking in vitro. J. Neuropathol. Exp. Neurol. 1982, 41, 354. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Abraham, C.; Ihara, Y. Brain transglutaminase—In vitro crosslinking of human neurofilament proteins into insoluble polymers. Proc. Natl. Acad. Sci. USA 1982, 79, 6070–6074. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-T.; Tang, B.-S.; Lan, W.; Song, N.-N.; Huang, Y.; Zhang, L.; Guan, W.-J.; Shi, Y.-T.; Shen, L.; Jiang, H.; et al. Distribution of transglutaminase 6 in the central nervous system of adult mice. Anat. Rec. 2013, 296, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Liu, M.; Li, X.; Liu, J.; Li, H. Origin of the Autophagosome Membrane in Mammals. BioMed Res. Int. 2018, 2018, 1012789. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in neurodegenerative diseases: Pathogenesis and therapy. Brain Pathol. 2018, 28, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Kesidou, E.; Lagoudaki, R.; Touloumi, O.; Poulatsidou, K.N.; Simeonidou, C. Autophagy and neurodegenerative disorders. Neural Regen. Res. 2013, 8, 2275–2283. [Google Scholar] [PubMed]
- Fujikake, N.; Shin, M.; Shimizu, S. Association Between Autophagy and Neurodegenerative Diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef]
- Cooper, A.J.; Jeitner, T.M.; Gentile, V.; Blass, J.P. Cross linking of polyglutamine domains catalyzed by tissue transglutaminase is greatly favored with pathological-length repeats: Does transglutaminase activity play a role in (CAG)(n)/Q(n)-expansion diseases? Neurochem. Int. 2002, 40, 53–67. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Battaile, K.; Cooper, A.J.L. Gamma-Glutamylamines and neurodegenerative diseases. Amino Acids 2013, 44, 129–142. [Google Scholar] [CrossRef][Green Version]
- Marambaud, P.; Dreses-Werringloer, U.; Vingtdeux, V. Calcium signaling in neurodegeneration. Mol. Neurodegener. 2009, 4, 20. [Google Scholar] [CrossRef]
- Zundorf, G.; Reiser, G. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef]
- Tang, T.S.; Slow, E.; Lupu, V.; Stavrovskaya, I.G.; Sugimori, M.; Llinas, R.; Kristal, B.S.; Hayden, M.R.; Bezprozvanny, I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 2602–2607. [Google Scholar] [CrossRef]
- Caccamo, D.; Campisi, A.; Curro, M.; Bramanti, V.; Tringali, M.; Li Volti, G.; Vanella, A.; Ientile, R. Antioxidant treatment inhibited glutamate-evoked NF-kappaB activation in primary astroglial cell cultures. Neurotoxicology 2005, 26, 915–921. [Google Scholar] [CrossRef]
- Lai, T.S.; Tucker, T.; Burke, J.R.; Strittmatter, W.J.; Greenberg, C.S. Effect of tissue transglutaminase on the solubility of proteins containing expanded polyglutamine repeats. J. Neurochem. 2004, 88, 1253–1260. [Google Scholar] [CrossRef]
- Landles, C.; Sathasivam, K.; Weiss, A.; Woodman, B.; Moffitt, H.; Finkbeiner, S.; Sun, B.; Gafni, J.; Ellerby, L.M.; Trottier, Y.; et al. Proteolysis of Mutant Huntingtin Produces an Exon 1 Fragment That Accumulates as an Aggregated Protein in Neuronal Nuclei in Huntington Disease. J. Biol. Chem. 2010, 285, 8808–8823. [Google Scholar] [CrossRef]
- Landles, C.; Milton, R.E.; Ali, N.; Flomen, R.; Flower, M.; Schindler, F.; Gomez-Paredes, C.; Bondulich, M.K.; Osborne, G.F.; Goodwin, D.; et al. Subcellular Localization and Formation of Huntingtin Aggregates Correlates with Symptom Onset and Progression in a Huntington’s Disease Model. Brain Commun. 2020, 2, fcaa066. [Google Scholar] [CrossRef] [PubMed]
- Hohenadl, C.; Mann, K.; Mayer, U.; Timpl, R.; Paulsson, M.; Aeschlimann, D. Two adjacent N-terminal glutamines of BM-40 (osteonectin, SPARC) act as amine acceptor sites in transglutaminaseC-catalyzed modification. J. Biol. Chem. 1995, 270, 23415–23420. [Google Scholar] [CrossRef] [PubMed]
- Adamcak, A.; Otten, B. Rodent therapeutics. Vet. Clin. N. Am. Exot. Anim. Pract. 2000, 3, 221–237. [Google Scholar] [CrossRef]
- Steffan, J.S.; Agrawal, N.; Pallos, J.; Rockabrand, E.; Trotman, L.C.; Slepko, N.; Illes, K.; Lukacsovich, T.; Zhu, Y.Z.; Cattaneo, E.; et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science 2004, 304, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Sakai, K.; Yanagi, T.; Fukushima, S.; Ihn, H.; Hitomi, K.; Shimizu, H. Synthetic Peptide K5 Efficiently Detects in situ Transglutaminase1 Activity; Its Application for Diagnosis of Lamellar Ichthyosis. J. Investig. Dermatol. 2010, 130, S21. [Google Scholar]
- Hitomi, K.; Kitamura, M.; Alea, M.P.; Ceylan, I.; Thomas, V.; El Alaoui, S. A specific colorimetric assay for measuring transglutaminase 1 and factor XIII activities. Anal. Biochem. 2009, 394, 281–283. [Google Scholar] [CrossRef]
- Sugimura, Y.; Kitamura, M.; Hosono, M.; Shibata, H.; Maki, M.; Hitomi, K. Characterization of Highly Reactive Sequences for Transglutaminase 2 and Factor XIIIa. In Animal Cell Technology: Basic & Applied Aspects; Springer: Dordrecht, The Netherlands, 2009; pp. 325–331. [Google Scholar]
- Fukui, M.; Kuramoto, K.; Yamasaki, R.; Shimizu, Y.; Itoh, M.; Kawamoto, T.; Hitomi, K. Identification of a highly reactive substrate peptide for transglutaminase 6 and its use in detecting transglutaminase activity in the skin epidermis. FEBS J. 2013, 280, 1420–1429. [Google Scholar] [CrossRef]
- Paganetti, P.; Weiss, A.; Trapp, M.; Hammerl, I.; Bleckmann, D.; Bodner, R.A.; Coven-Easter, S.; Housman, D.E.; Parker, C.N. Development of a method for the high-throughput quantification of cellular proteins. ChemBioChem 2009, 10, 1678–1688. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schulze-Krebs, A.; Canneva, F.; Stemick, J.; Plank, A.-C.; Harrer, J.; Bates, G.P.; Aeschlimann, D.; Steffan, J.S.; von Hörsten, S. Transglutaminase 6 Is Colocalized and Interacts with Mutant Huntingtin in Huntington Disease Rodent Animal Models. Int. J. Mol. Sci. 2021, 22, 8914. https://doi.org/10.3390/ijms22168914
Schulze-Krebs A, Canneva F, Stemick J, Plank A-C, Harrer J, Bates GP, Aeschlimann D, Steffan JS, von Hörsten S. Transglutaminase 6 Is Colocalized and Interacts with Mutant Huntingtin in Huntington Disease Rodent Animal Models. International Journal of Molecular Sciences. 2021; 22(16):8914. https://doi.org/10.3390/ijms22168914
Chicago/Turabian StyleSchulze-Krebs, Anja, Fabio Canneva, Judith Stemick, Anne-Christine Plank, Julia Harrer, Gillian P. Bates, Daniel Aeschlimann, Joan S. Steffan, and Stephan von Hörsten. 2021. "Transglutaminase 6 Is Colocalized and Interacts with Mutant Huntingtin in Huntington Disease Rodent Animal Models" International Journal of Molecular Sciences 22, no. 16: 8914. https://doi.org/10.3390/ijms22168914
APA StyleSchulze-Krebs, A., Canneva, F., Stemick, J., Plank, A.-C., Harrer, J., Bates, G. P., Aeschlimann, D., Steffan, J. S., & von Hörsten, S. (2021). Transglutaminase 6 Is Colocalized and Interacts with Mutant Huntingtin in Huntington Disease Rodent Animal Models. International Journal of Molecular Sciences, 22(16), 8914. https://doi.org/10.3390/ijms22168914