
Role of β-Adrenergic Receptors and Estrogen in Cardiac Repair after Myocardial Infarction: An Overview

 , ,
, ,
Abstract
:1. Introduction
2. Cardiac Repair after Myocardial Infarction
3. β-Adrenergic Receptors
4. β-Adrenergic Signaling in the Post-Infarction Inflammatory Response
5. β-Adrenergic Signaling in the Post-Infarction Reparative Phase
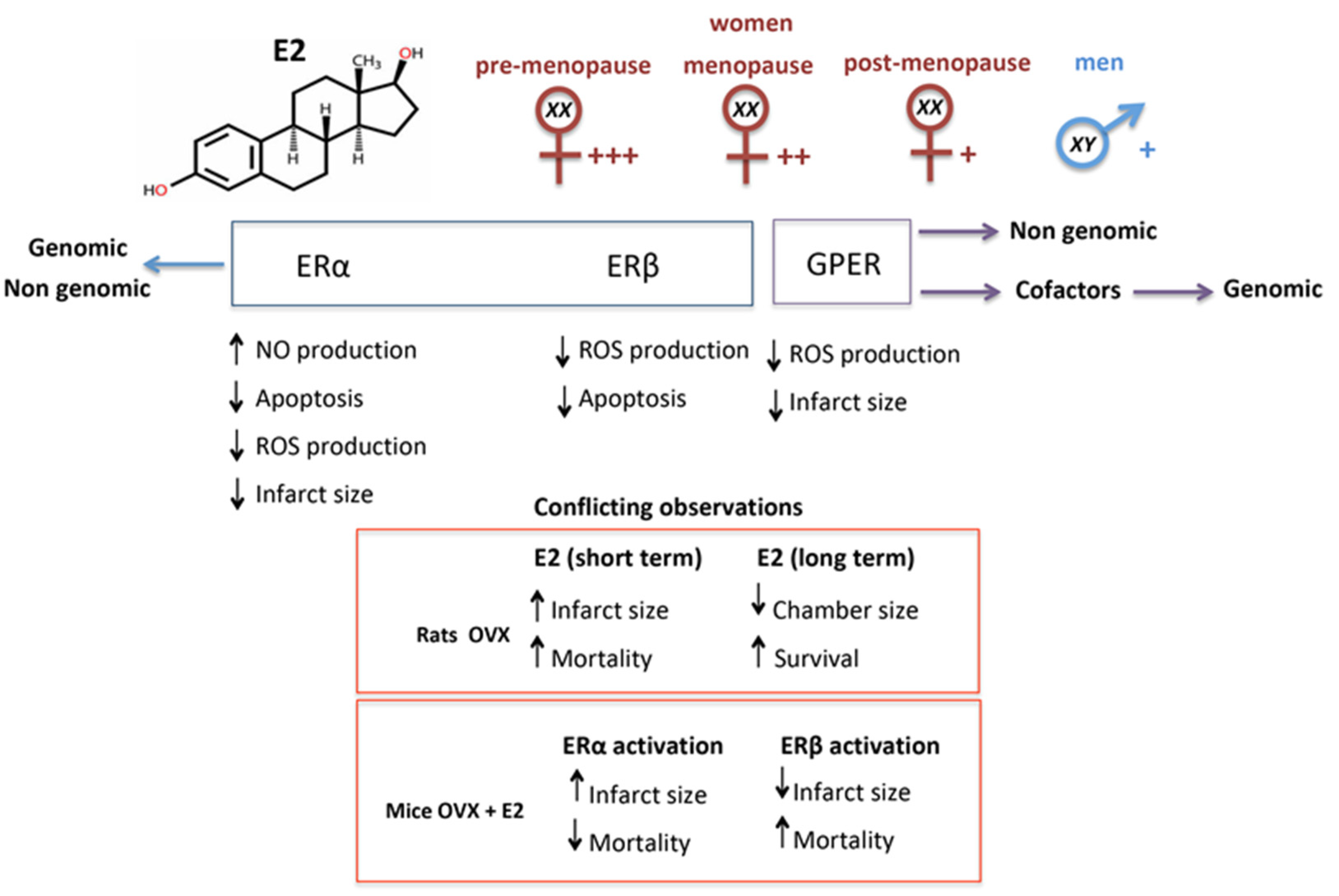
6. Estrogen and Estrogen Receptors
7. Estrogen in Cardiac Repair
8. Notes on the Role of Estrogen in Cardiac Regeneration
9. Interplay between ERs and β-ARs and Its Potential Therapeutic Value
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Voorhees, A.P.; DeLeon-Pennell, K.Y.; Ma, Y.; Halade, G.V.; Yabluchanskiy, A.; Iyer, R.P.; Flynn, E.; Cates, C.A.; Lindsey, M.L.; Han, H.C. Building a better infarct: Modulation of collagen cross-linking to increase infarct stiffness and reduce left ventricular dilation post-myocardial infarction. J. Mol. Cell Cardiol. 2015, 85, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Rathod, K.S.; Kapil, V.; Velmurugan, S.; Khambata, R.S.; Siddique, U.; Khan, S.; van Eijl, S.; Gee, L.C.; Bansal, J.; Pitrola, K.; et al. Accelerated resolution of inflammation underlies sex differences in inflammatory responses in humans. J. Clin. Invest. 2017, 127, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Pathophysiology of Myocardial Infarction. Compr. Physiol. 2015, 5, 1841–1875. [Google Scholar]
- Souders, C.A.; Bowers, S.L.; Baudino, T.A. Cardiac fibroblast: The renaissance cell. Circ. Res. 2009, 105, 1164–1176. [Google Scholar] [CrossRef]
- Yue, L.; Xie, J.; Nattel, S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc. Res. 2011, 89, 744–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakaya, M.; Watari, K.; Tajima, M.; Nakaya, T.; Matsuda, S.; Ohara, H.; Nishihara, H.; Yamaguchi, H.; Hashimoto, A.; Nishida, M.; et al. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J. Clin. Invest. 2017, 127, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Yekkala, K.; Borg, T.K.; Baudino, T.A. Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Ann. N. Y. Acad. Sci. 2006, 1080, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Dimitrov, S.; Cheng, T.; Redwine, L.; Pruitt, C.; Mills, P.J.; Ziegler, M.G.; Green, J.M.; Shaikh, F.; Wilson, K. Beta-adrenergic receptor mediated inflammation control by monocytes is associated with blood pressure and risk factors for cardiovascular disease. Brain Behav. Immun. 2015, 50, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Grisanti, L.A.; Gumpert, A.M.; Traynham, C.J.; Gorsky, J.E.; Repas, A.A.; Gao, E.; Carter, R.L.; Yu, D.; Calvert, J.W.; Garcia, A.P.; et al. Leukocyte-Expressed beta2-Adrenergic Receptors Are Essential for Survival After Acute Myocardial Injury. Circulation 2016, 134, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Tanner, M.A.; Thomas, T.P.; Maitz, C.A.; Grisanti, L.A. Beta2-Adrenergic Receptors Increase Cardiac Fibroblast Proliferation Through the Galphas/ERK1/2-Dependent Secretion of Interleukin-6. Int. J. Mol. Sci. 2020, 21, 8507. [Google Scholar] [CrossRef]
- Dixon, R.A.; Kobilka, B.K.; Strader, D.J.; Benovic, J.L.; Dohlman, H.G.; Frielle, T.; Bolanowski, M.A.; Bennett, C.D.; Rands, E.; Diehl, R.E.; et al. Cloning of the gene and cDNA for mammalian beta-adrenergic receptor and homology with rhodopsin. Nature 1986, 321, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- De Lucia, C.; Eguchi, A.; Koch, W.J. New Insights in Cardiac beta-Adrenergic Signaling During Heart Failure and Aging. Front. Pharmacol. 2018, 9, 904. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.A.; Sato, M.; Sarwar, M.; Hutchinson, D.S.; Summers, R.J. Ligand-directed signalling at beta-adrenoceptors. Br. J. Pharmacol. 2010, 159, 1022–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, N.D.; Sloan, E.K.; Bailey, M.T.; Arevalo, J.M.; Miller, G.E.; Chen, E.; Kobor, M.S.; Reader, B.F.; Sheridan, J.F.; Cole, S.W. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc. Natl. Acad. Sci. USA 2013, 110, 16574–16579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.H.; Gorouhi, F.; Ramirez, S.; Granick, J.L.; Byrne, B.A.; Soulika, A.M.; Simon, S.I.; Isseroff, R.R. Catecholamine stress alters neutrophil trafficking and impairs wound healing by beta2-adrenergic receptor-mediated upregulation of IL-6. J. Invest. Dermatol. 2014, 134, 809–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scanzano, A.; Schembri, L.; Rasini, E.; Luini, A.; Dallatorre, J.; Legnaro, M.; Bombelli, R.; Congiu, T.; Cosentino, M.; Marino, F. Adrenergic modulation of migration, CD11b and CD18 expression, ROS and interleukin-8 production by human polymorphonuclear leukocytes. Inflamm. Res. 2015, 64, 127–135. [Google Scholar] [CrossRef]
- Horn, N.A.; Anastase, D.M.; Hecker, K.E.; Baumert, J.H.; Robitzsch, T.; Rossaint, R. Epinephrine enhances platelet-neutrophil adhesion in whole blood in vitro. Anesth. Analg. 2005, 100, 520–526. [Google Scholar] [CrossRef]
- Padro, C.J.; Sanders, V.M. Neuroendocrine regulation of inflammation. Semin. Immunol. 2014, 26, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Effect of beta-agonists on inflammatory cells. J. Allergy Clin. Immunol. 1999, 104 Pt 2, S10–S17. [Google Scholar] [CrossRef]
- Elferink, J.G.; VanUffelen, B.E. The role of cyclic nucleotides in neutrophil migration. Gen. Pharmacol. 1996, 27, 387–393. [Google Scholar] [CrossRef]
- Straub, R.H.; Mayer, M.; Kreutz, M.; Leeb, S.; Scholmerich, J.; Falk, W. Neurotransmitters of the sympathetic nerve terminal are powerful chemoattractants for monocytes. J. Leukoc. Biol. 2000, 67, 553–558. [Google Scholar] [CrossRef]
- Johnson, M. Effects of beta2-agonists on resident and infiltrating inflammatory cells. J. Allergy Clin. Immunol. 2002, 110 (Suppl. S6), S282–S290. [Google Scholar] [CrossRef]
- Grisanti, L.A.; Traynham, C.J.; Repas, A.A.; Gao, E.; Koch, W.J.; Tilley, D.G. Beta2-Adrenergic receptor-dependent chemokine receptor 2 expression regulates leukocyte recruitment to the heart following acute injury. Proc. Natl. Acad. Sci. USA 2016, 113, 15126–15131. [Google Scholar] [CrossRef] [Green Version]
- Grisanti, L.A.; de Lucia, C.; Thomas, T.P.; Stark, A.; Strony, J.T.; Myers, V.D.; Beretta, R.; Yu, D.; Sardu, C.; Marfella, R.; et al. Prior beta-blocker treatment decreases leukocyte responsiveness to injury. JCI Insight 2019, 5, 9. [Google Scholar] [CrossRef]
- Grisanti, L.A.; Evanson, J.; Marchus, E.; Jorissen, H.; Woster, A.P.; DeKrey, W.; Sauter, E.R.; Combs, C.K.; Porter, J.E. Pro-inflammatory responses in human monocytes are beta1-adrenergic receptor subtype dependent. Mol. Immunol. 2010, 47, 1244–1254. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Prieto, J.; Villena-Gutierrez, R.; Gomez, M.; Bernardo, E.; Pun-Garcia, A.; Garcia-Lunar, I.; Crainiciuc, G.; Fernandez-Jimenez, R.; Sreeramkumar, V.; Bourio-Martinez, R.; et al. Neutrophil stunning by metoprolol reduces infarct size. Nat. Commun. 2017, 8, 14780. [Google Scholar] [CrossRef]
- Gao, X.M.; Dilley, R.J.; Samuel, C.S.; Percy, E.; Fullerton, M.J.; Dart, A.M.; Du, X.J. Lower risk of postinfarct rupture in mouse heart overexpressing beta 2-adrenergic receptors: Importance of collagen content. J. Cardiovasc. Pharmacol. 2002, 40, 632–640. [Google Scholar] [CrossRef]
- Ravindranathan, M.P.; Jenkins, B.; Haider, B.; Regan, T.J. Effects of beta-adrenergic inhibition on scar formation after myocardial infarction. Am. Heart J. 1984, 108, 25–30. [Google Scholar] [CrossRef]
- Fishbein, M.C.; Lei, L.Q.; Rubin, S.A. Long-term propranolol administration alters myocyte and ventricular geometry in rat hearts with and without infarction. Circulation 1988, 78, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Aranguiz-Urroz, P.; Canales, J.; Copaja, M.; Troncoso, R.; Vicencio, J.M.; Carrillo, C.; Lara, H.; Lavandero, S.; Diaz-Araya, G. Beta(2)-adrenergic receptor regulates cardiac fibroblast autophagy and collagen degradation. Biochim. Biophys. Acta 2011, 1812, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Araya, G.; Vivar, R.; Humeres, C.; Boza, P.; Bolivar, S.; Munoz, C. Cardiac fibroblasts as sentinel cells in cardiac tissue: Receptors, signaling pathways and cellular functions. Pharmcol. Res. 2015, 101, 30–40. [Google Scholar] [CrossRef]
- Turner, N.A.; Porter, K.E.; Smith, W.H.; White, H.L.; Ball, S.G.; Balmforth, A.J. Chronic beta2-adrenergic receptor stimulation increases proliferation of human cardiac fibroblasts via an autocrine mechanism. Cardiovasc. Res. 2003, 57, 784–792. [Google Scholar] [CrossRef] [Green Version]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmcol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef]
- Brown, R.D.; Ambler, S.K.; Mitchell, M.D.; Long, C.S. The cardiac fibroblast: Therapeutic target in myocardial remodeling and failure. Annu. Rev. Pharmcol. Toxicol. 2005, 45, 657–687. [Google Scholar] [CrossRef] [PubMed]
- Camelliti, P.; Borg, T.K.; Kohl, P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 2005, 65, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T. Fibrosis in hypertensive heart disease: Focus on cardiac fibroblasts. J. Hypertens. 2004, 22, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.R.; Mellor, K.M.; Wollermann, A.C.; Ip, W.T.; Reichelt, M.E.; Meachem, S.J.; Simpson, E.R.; Delbridge, L.M. Aromatase deficiency confers paradoxical postischemic cardioprotection. Endocrinology 2011, 152, 4937–4947. [Google Scholar] [CrossRef] [Green Version]
- Hammes, S.R.; Levin, E.R. Extranuclear steroid receptors: Nature and actions. Endocr. Rev. 2007, 28, 726–741. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. Estrogen biology: New insights into GPER function and clinical opportunities. Mol. Cell Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef]
- Grohe, C.; Kahlert, S.; Lobbert, K.; Stimpel, M.; Karas, R.H.; Vetter, H.; Neyses, L. Cardiac myocytes and fibroblasts contain functional estrogen receptors. FEBS Lett. 1997, 416, 107–112. [Google Scholar] [CrossRef]
- Taylor, A.H.; Al-Azzawi, F. Immunolocalisation of oestrogen receptor beta in human tissues. J. Mol. Endocrinol. 2000, 24, 145–155. [Google Scholar] [CrossRef]
- Mahmoodzadeh, S.; Eder, S.; Nordmeyer, J.; Ehler, E.; Huber, O.; Martus, P.; Weiske, J.; Pregla, R.; Hetzer, R.; Regitz-Zagrosek, V. Estrogen receptor alpha up-regulation and redistribution in human heart failure. FASEB J. 2006, 20, 926–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, S.; Sundberg, M.; Pristovsek, N.; Ibrahim, A.; Jonsson, P.; Katona, B.; Clausson, C.M.; Zieba, A.; Ramstrom, M.; Soderberg, O.; et al. Corrigendum: Insufficient antibody validation challenges oestrogen receptor beta research. Nat. Commun. 2017, 8, 16164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutson, D.D.; Gurrala, R.; Ogola, B.O.; Zimmerman, M.A.; Mostany, R.; Satou, R.; Lindsey, S.H. Estrogen receptor profiles across tissues from male and female Rattus norvegicus. Biol. Sex Differ. 2019, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safe, S.; Kim, K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J. Mol. Endocrinol. 2008, 41, 263–275. [Google Scholar] [CrossRef]
- Meyer, M.R.; Barton, M. Estrogens and Coronary Artery Disease: New Clinical Perspectives. Adv. Pharmacol. 2016, 77, 307–360. [Google Scholar]
- Patten, R.D.; Karas, R.H. Estrogen replacement and cardiomyocyte protection. Trends Cardiovasc. Med. 2006, 16, 69–75. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Maggiolini, M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol. Cell Endocrinol. 2009, 308, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Cavasin, M.A.; Yang, X.P.; Liu, Y.H.; Mehta, D.; Karumanchi, R.; Bulagannawar, M.; Carretero, O.A. Effects of ACE inhibitor, AT1 antagonist, and combined treatment in mice with heart failure. J. Cardiovasc. Pharmacol. 2000, 36, 472–480. [Google Scholar] [CrossRef]
- Van Eickels, M.; Patten, R.D.; Aronovitz, M.J.; Alsheikh-Ali, A.; Gostyla, K.; Celestin, F.; Grohe, C.; Mendelsohn, M.E.; Karas, R.H. 17-beta-estradiol increases cardiac remodeling and mortality in mice with myocardial infarction. J. Am. Coll. Cardiol. 2003, 41, 2084–2092. [Google Scholar] [CrossRef]
- De Almeida, S.A.; Claudio, E.R.G.; Mengal, V.; Brasil, G.A.; Merlo, E.; Podratz, P.L.; Graceli, J.B.; Gouvea, S.A.; de Abreu, G.R. Estrogen Therapy Worsens Cardiac Function and Remodeling and Reverses the Effects of Exercise Training After Myocardial Infarction in Ovariectomized Female Rats. Front. Physiol. 2018, 9, 1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.J.; Ornatsky, O.; Stewart, D.J.; Picard, P.; Dawood, F.; Wen, W.H.; Liu, P.P.; Webb, D.J.; Monge, J.C. Effects of estrogen replacement on infarct size, cardiac remodeling, and the endothelin system after myocardial infarction in ovariectomized rats. Circulation 2000, 102, 2983–2989. [Google Scholar] [CrossRef] [Green Version]
- Sivasinprasasn, S.; Palee, S.; Chattipakorn, K.; Jaiwongkum, T.; Apaijai, N.; Pratchayasakul, W.; Chattipakorn, S.C.; Chattipakorn, N. N-acetylcysteine with low-dose estrogen reduces cardiac ischemia-reperfusion injury. J. Endocrinol. 2019, 242, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Babiker, F.A.; Lips, D.J.; Delvaux, E.; Zandberg, P.; Janssen, B.J.; Prinzen, F.; van Eys, G.; Grohe, C.; Doevendans, P.A. Oestrogen modulates cardiac ischaemic remodelling through oestrogen receptor-specific mechanisms. Acta Physiol. 2007, 189, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabe-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Dawn, B.; Bolli, R. Increasing evidence that estrogen is an important modulator of bone marrow-mediated cardiac repair after acute infarction. Circulation 2006, 114, 2203–2205. [Google Scholar] [CrossRef] [Green Version]
- Broughton, K.M.; Wang, B.J.; Firouzi, F.; Khalafalla, F.; Dimmeler, S.; Fernandez-Aviles, F.; Sussman, M.A. Mechanisms of Cardiac Repair and Regeneration. Circ. Res. 2018, 122, 1151–1163. [Google Scholar] [CrossRef]
- Ghiroldi, A.; Piccoli, M.; Cirillo, F.; Monasky, M.M.; Ciconte, G.; Pappone, C.; Anastasia, L. Cell-Based Therapies for Cardiac Regeneration: A Comprehensive Review of Past and Ongoing Strategies. Int. J. Mol. Sci. 2018, 19, 3194. [Google Scholar] [CrossRef] [Green Version]
- Loffredo, F.S.; Steinhauser, M.L.; Gannon, J.; Lee, R.T. Bone marrow-derived cell therapy stimulates endogenous cardiomyocyte progenitors and promotes cardiac repair. Cell Stem Cell 2011, 8, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Hu, L.; Xu, Q.; Yuan, H.; Ba, L.; He, Y.; Che, H. Cytoprotective Role of Alpha-1 Antitrypsin in Vascular Endothelial Cell Under Hypoxia/Reoxygenation Condition. J. Cardiovasc. Pharmacol. 2015, 66, 96–107. [Google Scholar] [CrossRef]
- Kunz, G.A.; Liang, G.; Cuculi, F.; Gregg, D.; Vata, K.C.; Shaw, L.K.; Goldschmidt-Clermont, P.J.; Dong, C.; Taylor, D.A.; Peterson, E.D. Circulating endothelial progenitor cells predict coronary artery disease severity. Am. Heart J. 2006, 152, 190–195. [Google Scholar] [CrossRef]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Murakami, M.; Qi, X.; McAnally, J.; Porrello, E.R.; Mahmoud, A.I.; Tan, W.; Shelton, J.M.; et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13839–13844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, F.; Zhang, W.; Liu, J. Membrane estrogen receptor alpha is an important modulator of bone marrow C-Kit+ cells mediated cardiac repair after myocardial infarction. Int. J. Clin. Exp. Pathol. 2015, 8, 4284–4295. [Google Scholar] [PubMed]
- Strehlow, K.; Werner, N.; Berweiler, J.; Link, A.; Dirnagl, U.; Priller, J.; Laufs, K.; Ghaeni, L.; Milosevic, M.; Bohm, M.; et al. Estrogen increases bone marrow-derived endothelial progenitor cell production and diminishes neointima formation. Circulation 2003, 107, 3059–3065. [Google Scholar] [CrossRef] [Green Version]
- Hamada, H.; Kim, M.K.; Iwakura, A.; Ii, M.; Thorne, T.; Qin, G.; Asai, J.; Tsutsumi, Y.; Sekiguchi, H.; Silver, M.; et al. Estrogen receptors alpha and beta mediate contribution of bone marrow-derived endothelial progenitor cells to functional recovery after myocardial infarction. Circulation 2006, 114, 2261–2270. [Google Scholar] [CrossRef] [Green Version]
- Patten, R.D.; Pourati, I.; Aronovitz, M.J.; Baur, J.; Celestin, F.; Chen, X.; Michael, A.; Haq, S.; Nuedling, S.; Grohe, C.; et al. 17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circ. Res. 2004, 95, 692–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deschamps, A.M.; Murphy, E.; Sun, J. Estrogen receptor activation and cardioprotection in ischemia reperfusion injury. Trends Cardiovasc. Med. 2010, 20, 73–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, E.A.; Obeid, N.R.; Lucchesi, B.R. Activation of estrogen receptor-alpha protects the in vivo rabbit heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2039–H2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nova-Lamperti, E.; Zúñiga, F.; Ormazábal, V.; Escudero, C.; Aguayo, C. Vascular regeneration by endothelial progenitor cells in health and diseases. In Microcirculation Revisited—From Molecules to Clinical Practice; IntechOpen: London, UK, 2016. [Google Scholar]
- Gonzalez-Arenas, A.; Aguilar-Maldonado, B.; Avendano-Vazquez, S.E.; Garcia-Sainz, J.A. Estrogens cross-talk to alpha1b-adrenergic receptors. Mol. Pharmacol. 2006, 70, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.R.; Sharma, R. Cross-talk between beta-adrenergic stimulation and estrogen receptors: Isoproterenol inhibits 17beta-estradiol-induced gene transcription in A7r5 cells. J. Cardiovasc. Pharmacol. 2003, 42, 266–274. [Google Scholar] [CrossRef]
- Willis, B.C.; Salazar-Cantu, A.; Silva-Platas, C.; Fernandez-Sada, E.; Villegas, C.A.; Rios-Argaiz, E.; Gonzalez-Serrano, P.; Sanchez, L.A.; Guerrero-Beltran, C.E.; Garcia, N.; et al. Impaired oxidative metabolism and calcium mishandling underlie cardiac dysfunction in a rat model of post-acute isoproterenol-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H467–H477. [Google Scholar] [CrossRef] [Green Version]
- Riedel, K.; Deussen, A.J.; Tolkmitt, J.; Weber, S.; Schlinkert, P.; Zatschler, B.; Friebel, C.; Muller, B.; El-Armouche, A.; Morawietz, H.; et al. Estrogen determines sex differences in adrenergic vessel tone by regulation of endothelial beta-adrenoceptor expression. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H243–H254. [Google Scholar] [CrossRef]
- Wu, Q.; Zhao, Z.; Sun, H.; Hao, Y.L.; Yan, C.D.; Gu, S.L. Oestrogen changed cardiomyocyte contraction and beta-adrenoceptor expression in rat hearts subjected to ischaemia-reperfusion. Exp. Physiol. 2008, 93, 1034–1043. [Google Scholar] [CrossRef]
- Liu, C.G.; Xu, K.Q.; Xu, X.; Huang, J.J.; Xiao, J.C.; Zhang, J.P.; Song, H.P. 17Beta-oestradiol regulates the expression of Na+/K+-ATPase beta1-subunit, sarcoplasmic reticulum Ca2+-ATPase and carbonic anhydrase iv in H9C2 cells. Clin. Exp. Pharmacol. Physiol. 2007, 34, 998–1004. [Google Scholar] [CrossRef]
- Chu, S.H.; Goldspink, P.; Kowalski, J.; Beck, J.; Schwertz, D.W. Effect of estrogen on calcium-handling proteins, beta-adrenergic receptors, and function in rat heart. Life Sci. 2006, 79, 1257–1267. [Google Scholar] [CrossRef]
- Kang, S.; Liu, Y.; Sun, D.; Zhou, C.; Liu, A.; Xu, C.; Hao, Y.; Li, D.; Yan, C.; Sun, H. Chronic activation of the G protein-coupled receptor 30 with agonist G-1 attenuates heart failure. PLoS ONE 2012, 7, e48185. [Google Scholar] [CrossRef]
- Ma, Y.; Cheng, W.T.; Wu, S.; Wong, T.M. Oestrogen confers cardioprotection by suppressing Ca2+/calmodulin-dependent protein kinase II. Br. J. Pharmacol. 2009, 157, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Hill, B.J.; Muldrew, E. Oestrogen upregulates the sarcoplasmic reticulum Ca(2+) ATPase pump in coronary arteries. Clin. Exp. Pharmacol. Physiol. 2014, 41, 430–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitcomb, V.; Wauson, E.; Christian, D.; Clayton, S.; Giles, J.; Tran, Q.K. Regulation of beta adrenoceptor-mediated myocardial contraction and calcium dynamics by the G protein-coupled estrogen receptor 1. Biochem. Pharmacol. 2020, 171, 113727. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.S. Models of excitation-contraction coupling in cardiac ventricular myocytes. Methods Mol. Biol. 2012, 910, 309–335. [Google Scholar] [PubMed] [Green Version]
- Machuki, J.O.; Zhang, H.Y.; Geng, J.; Fu, L.; Adzika, G.K.; Wu, L.; Shang, W.; Wu, J.; Kexue, L.; Zhao, Z.; et al. Estrogen regulation of cardiac cAMP-L-type Ca(2+) channel pathway modulates sex differences in basal contraction and responses to beta2AR-mediated stress in left ventricular apical myocytes. Cell Commun. Signal. 2019, 17, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenfeld, J.; Cleveland, J.C., Jr.; Kao, D.P.; White, M.; Wichman, S.; Bristow, J.C.; Peterson, V.; Rodegheri-Brito, J.; Korst, A.; Blain-Nelson, P.; et al. Sex-related differences in age-associated downregulation of human ventricular myocardial beta1-adrenergic receptors. J. Heart Lung Transpl. 2016, 35, 352–361. [Google Scholar] [CrossRef]
- Cao, X.; Zhou, C.; Chong, J.; Fu, L.; Zhang, L.; Sun, D.; Hou, H.; Zhang, Y.; Li, D.; Sun, H. Estrogen resisted stress-induced cardiomyopathy through increasing the activity of beta(2)AR-Galphas signal pathway in female rats. Int. J. Cardiol. 2015, 187, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Li, W.L.; Xiang, W.; Ping, Y. Activation of novel estrogen receptor GPER results in inhibition of cardiocyte apoptosis and cardioprotection. Mol. Med. Rep. 2015, 12, 2425–2430. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Liu, A.; Sun, H.; Sun, Y.; Wang, G.; Gao, L.; Hao, Y.; Yan, C. Beta2-Adrenoceptor confers cardioprotection against hypoxia in isolated ventricular myocytes and the effects depend on estrogenic environment. J. Recept. Signal. Transduct. Res. 2010, 30, 255–261. [Google Scholar] [CrossRef]
- Machuki, J.O.; Zhang, H.Y.; Harding, S.E.; Sun, H. Molecular pathways of oestrogen receptors and beta-adrenergic receptors in cardiac cells: Recognition of their similarities, interactions and therapeutic value. Acta Physiol. 2018, 222, e12978. [Google Scholar] [CrossRef]
- Chambliss, K.L.; Yuhanna, I.S.; Anderson, R.G.; Mendelsohn, M.E.; Shaul, P.W. ERbeta has nongenomic action in caveolae. Mol. Endocrinol. 2002, 16, 938–946. [Google Scholar] [PubMed] [Green Version]
- Ke, X.; Shu, X.R.; Wu, F.; Hu, Q.S.; Deng, B.Q.; Wang, J.F.; Nie, R.Q. Overexpression of the beta2AR gene improves function and re-endothelialization capacity of EPCs after arterial injury in nude mice. Stem Cell Res. Ther. 2016, 7, 73. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Biondi-Zoccai, G.; Abbate, A.; D’Ascenzo, F.; Castagno, D.; Van Tassell, B.; Mukherjee, D.; Lichstein, E. Benefits of beta blockers in patients with heart failure and reduced ejection fraction: Network meta-analysis. BMJ 2013, 346, f55. [Google Scholar] [CrossRef] [Green Version]
- Hunt, S.A.; Abraham, W.T.; Chin, M.H.; Feldman, A.M.; Francis, G.S.; Ganiats, T.G.; Jessup, M.; Konstam, M.A.; Mancini, D.M.; Michl, K.; et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: Developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009, 119, e391–e479. [Google Scholar] [PubMed] [Green Version]
- Giannakopoulos, G.; Noble, S. Should We Be Using Upstream Beta-Blocker Therapy for Acute Myocardial Infarction? Curr Cardiol. Rep. 2021, 23, 66. [Google Scholar] [CrossRef] [PubMed]
- Ahmet, I.; Morrell, C.; Lakatta, E.G.; Talan, M.I. Therapeutic efficacy of a combination of a beta1-adrenoreceptor (AR) blocker and beta2-AR agonist in a rat model of postmyocardial infarction dilated heart failure exceeds that of a beta1-AR blocker plus angiotensin-converting enzyme inhibitor. J. Pharmacol. Exp. Ther. 2009, 331, 178–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.B.; Lu, Q.; Nguyen, M.N.; Su, Y.; Ziemann, M.; Wang, L.N.; Kiriazis, H.; Puthalakath, H.; Sadoshima, J.; Hu, H.Y.; et al. Stimulation of beta-adrenoceptors up-regulates cardiac expression of galectin-3 and BIM through the Hippo signalling pathway. Br. J. Pharmacol. 2019, 176, 2465–2481. [Google Scholar] [CrossRef]
- Hu, Q.; Guo, Y.; Zhang, T.; Feng, J.; Wang, J.; Dong, X.; Chen, Y.; Nie, R.; Feng, Z.; Huang, Y.; et al. Importance of beta2AR elevation for re-endothelialization capacity mediated by late endothelial progenitor cells in hypertensive patients. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H867–H880. [Google Scholar] [CrossRef]
- Luzier, A.B.; Killian, A.; Wilton, J.H.; Wilson, M.F.; Forrest, A.; Kazierad, D.J. Gender-related effects on metoprolol pharmacokinetics and pharmacodynamics in healthy volunteers. Clin. Pharmacol. Ther. 1999, 66, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, D.A.; Gal, J.; Gerber, J.G.; Nies, A.S. Age and gender influence the stereoselective pharmacokinetics of propranolol. J. Pharmacol. Exp. Ther. 1992, 261, 1181–1186. [Google Scholar] [PubMed]
- Thurmann, P.A.; Haack, S.; Werner, U.; Szymanski, J.; Haase, G.; Drewelow, B.; Reimann, I.R.; Hippius, M.; Siegmund, W.; May, K.; et al. Tolerability of beta-blockers metabolized via cytochrome P450 2D6 is sex-dependent. Clin. Pharmacol. Ther. 2006, 80, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Seeland, U.; Regitz-Zagrosek, V. Sex and gender differences in cardiovascular drug therapy. Handb. Exp. Pharmacol. 2012, 214, 211–236. [Google Scholar]
- Vizgirda, V.M.; Wahler, G.M.; Sondgeroth, K.L.; Ziolo, M.T.; Schwertz, D.W. Mechanisms of sex differences in rat cardiac myocyte response to beta-adrenergic stimulation. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H256–H263. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| β-AR Agonists | β-AR Antagonists |
|---|---|
| Adrenaline non-selective β-AR physiological ligand | Propranolol non-selective β1 and β2-AR |
| Noradrenaline non-selective β-AR physiological ligand | Metoprolol selective β1-AR |
| Isoprotere nolnon-selective β-AR | Bisoprolol selective β1-AR |
| Dobutamine selective β1-AR | Carvedilol non-selective β1 and β2-AR |
| Salbutamol selective β2-AR | Atenolol selective β1-AR |
| Clenbuterol selective β2-AR | Practolol selective β1-AR |
| Salmeterol selective β2-AR | Sotalol non-selective β1 and β2-AR |
| Receptor | Effects | Mechanisms | Refs. |
|---|---|---|---|
| β2-AR | Impaired scar formation followed by cardiac rupture and death in β2-AR KO mice subjected to MI | Decreased migration of leukocytes to the injured heart | [10] |
| β2-AR | Chronic β-blocker treatment with ICI 118,551 or carvedilol increases cardiac rupture events after MI | Splenic leukocyte accumulation and decreased migration of leukocytes to the injured heart | [27] |
| β1-AR | Treatment with metoprolol, a β1-blocker, reduces infarct size in animal models of cardiac ischemia | Inhibition of neutrophil-driven inflammatory responses | [29] |
| β2-AR | Transgenic mice overexpressing the β2-AR have lower incidence of cardiac rupture following MI | Increased production of collagen | [30] |
| β2-AR | β2-AR activation opposes the expansion of infarct area | Increased production of collagen and fibroblast proliferation | [11] |
| β-ARs | Chronic treatment with nonselective β-blockers causes cardiac cavity enlargement and scar thinning | Reduced scar amount | [31,32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matarrese, P.; Maccari, S.; Vona, R.; Gambardella, L.; Stati, T.; Marano, G. Role of β-Adrenergic Receptors and Estrogen in Cardiac Repair after Myocardial Infarction: An Overview. Int. J. Mol. Sci. 2021, 22, 8957. https://doi.org/10.3390/ijms22168957
Matarrese P, Maccari S, Vona R, Gambardella L, Stati T, Marano G. Role of β-Adrenergic Receptors and Estrogen in Cardiac Repair after Myocardial Infarction: An Overview. International Journal of Molecular Sciences. 2021; 22(16):8957. https://doi.org/10.3390/ijms22168957
Chicago/Turabian StyleMatarrese, Paola, Sonia Maccari, Rosa Vona, Lucrezia Gambardella, Tonino Stati, and Giuseppe Marano. 2021. "Role of β-Adrenergic Receptors and Estrogen in Cardiac Repair after Myocardial Infarction: An Overview" International Journal of Molecular Sciences 22, no. 16: 8957. https://doi.org/10.3390/ijms22168957
APA StyleMatarrese, P., Maccari, S., Vona, R., Gambardella, L., Stati, T., & Marano, G. (2021). Role of β-Adrenergic Receptors and Estrogen in Cardiac Repair after Myocardial Infarction: An Overview. International Journal of Molecular Sciences, 22(16), 8957. https://doi.org/10.3390/ijms22168957