
Drug Discovery of Spinal Muscular Atrophy (SMA) from the Computational Perspective: A Comprehensive Review


Abstract
1. Introduction
2. Spinal Muscular Atrophy (SMA)
2.1. Disease Etiology
2.2. Clinical Classification of SMA Subtype
2.3. SMN Protein
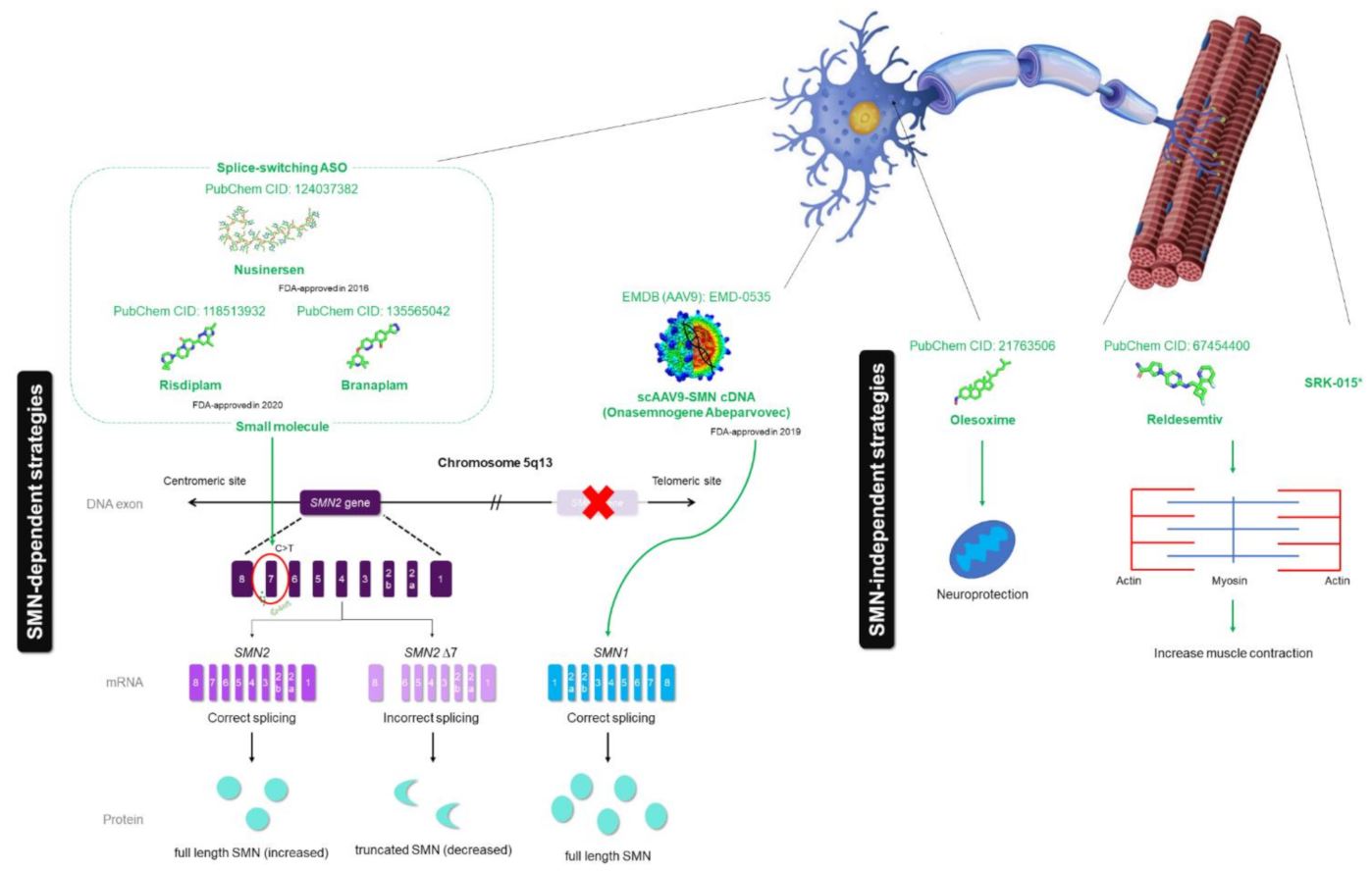
3. Current Drug of SMA
3.1. Current Drug—Early Success
3.2. Existing Drug—Clinical Trial Stage
4. Computer-Aided Drug Design (CADD)—The Open Window of Therapeutic Agents
4.1. In Silico Drug Repurposing
4.2. Network-Driven Drug Discovery (NDD)
4.3. AI-Assisted Drug Discovery (AID)
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SMA | spinal muscular atrophy |
| SMN1 | survival motor neuron 1 |
| FDA | U.S. Food and Drug Administration |
| NMD | neuromuscular disease |
| AI | artificial intelligence |
| ML | machine learning |
| DL | deep learning |
| ALS | amyotrophic lateral sclerosis |
| R&D | research and development |
| α-MNs | alpha motor neurons |
| FL-SMN | full length, functional SMN |
| OMIM | Online Mendelian Inheritance in Man |
| PDB | Protein Data Bank |
| Ge2BD | Gemin2 binding domain |
| snRNP | spliceosomal small nuclear ribonucleoprotein |
| sDMA | symmetric dimethylarginine |
| ASO | antisense oligonucleotide |
| CNS | central nervous system |
| AAV9 | adeno-associated virus 9 |
| cDNA | complementary DNA |
| hnRNP | heterogenous nuclear ribonucleoprotein |
| FSTA | fast skeletal muscle troponin activator |
| BBB | blood–brain barrier |
| 6MWT | six-minute walk test |
| MEP | maximal expiratory pressure |
| CADD | computer-aided drug design |
| ADME | absorption, distribution, metabolism and extraction |
| HTS | high throughput screening |
| NDD | network-driven drug discovery |
| AID | artificial intelligence (AI)-assisted drug discovery |
| HDAC | Histone deacetylase inhibitors |
| VPA | valproic acid |
| SMILES | Simplified Molecular Input Line Entry System |
| InChl | International Chemical Identifier |
| Tc | Tanimoto coefficient |
| DTIs | drug-target interactions |
| NCBI | National Center for Biotechnology Information |
| GEO | Gene Expression Omnibus |
| dbSNP | Single Nucleotide Polymorphism database |
| SRA | Sequence Read Archive |
| ADEs | adverse drug events |
| SIDER | side effect resource |
| CMap | connectivity map |
| NIH | National Institutes of Health |
| LINCS | library of integrated network based cellular signatures |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| ROCK | RhoA/Rho kinase |
| cAMP | cyclic adenosine monophosphate |
| ERK | extracellular regulated kinase |
| JNK | c-Jun N-terminal Kinase |
| GPUs | graphical processing units |
| TPUs | tensor processing units |
| QSAR | quantitative structure-activity relationship |
| OCD | obsessive compulsive disorder |
| RF | random forests |
| SVM | support vector machines |
| NN | neural networks |
| GAN | generative adversarial neural network |
| BERT | Bidirectional Encoder Representations from Transformers |
| CNN | convolutional neural networks |
| CI | concordance index |
References
- Cherry, J.J.; Kobayashi, D.T.; Lynes, M.M.; Naryshkin, N.N.; Tiziano, F.D.; Zaworski, P.G.; Rubin, L.L.; Jarecki, J. Assays for the Identification and Prioritization of Drug Candidates for Spinal Muscular Atrophy. Assay Drug Dev. Technol. 2014, 12, 315–341. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Neurol. Clin. 2015, 33, 831–846. [Google Scholar] [CrossRef]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy: A Timely Review. Arch. Neurol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between Severity and SMN Protein Level in Spinal Muscular Atrophy. Nat. Genet. 1997, 16, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Wurster, C.D.; Ludolph, A.C. Nusinersen for Spinal Muscular Atrophy. Ther. Adv. Neurol. Disord. 2018, 11, 175628561875445. [Google Scholar] [CrossRef]
- Mahajan, R. Onasemnogene Abeparvovec for Spinal Muscular Atrophy: The Costlier Drug Ever. Int. J. Appl. Basic Med. Res. 2019, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Tabet, R.; El Bitar, S.; Zaidan, J.; Dabaghian, G. Spinal Muscular Atrophy: The Treatment Approved. Cureus 2017. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.-K.; Pichika, M.R. Artificial Intelligence in Drug Development: Present Status and Future Prospects. Drug Discov. Today 2019, 24, 773–780. [Google Scholar] [CrossRef]
- Otsuki, N.; Arakawa, R.; Kaneko, K.; Aoki, R.; Arakawa, M.; Saito, K. A New Biomarker Candidate for Spinal Muscular Atrophy: Identification of a Peripheral Blood Cell Population Capable of Monitoring the Level of Survival Motor Neuron Protein. PLoS ONE 2018, 13, e0201764. [Google Scholar] [CrossRef]
- Kolb, S.J. Spinal Muscular Atrophy. Arch. Neurol. 2011, 68, 979. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.O.; Pardo, C.A. The Neurobiology of Childhood Spinal Muscular Atrophy. Neurobiol. Dis. 1996, 3, 97–110. [Google Scholar] [CrossRef]
- Markowitz, J.A.; Tinkle, M.B.; Fischbeck, K.H. Spinal Muscular Atrophy in the Neonate. J. Obstet. Gynecol. Neonatal Nurs. 2004, 33, 12–20. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, C.J.; Parks, R.J.; Kothary, R. Development of a Gene Therapy Strategy for the Restoration of Survival Motor Neuron Protein Expression: Implications for Spinal Muscular Atrophy Therapy. Hum. Gene Ther. 2003, 14, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, S.; Pellizzoni, L. Disease Mechanisms and Therapeutic Approaches in Spinal Muscular Atrophy. J. Neurosci. 2015, 35, 8691–8700. [Google Scholar] [CrossRef]
- Sumner, C.J.; Crawford, T.O. Two Breakthrough Gene-Targeted Treatments for Spinal Muscular Atrophy: Challenges Remain. J. Clin. Investig. 2018, 128, 3219–3227. [Google Scholar] [CrossRef]
- Lunn, M.R.; Wang, C.H. Spinal Muscular Atrophy. Lancet 2008, 371, 2120–2133. [Google Scholar] [CrossRef]
- Roberts, D.F.; Chavez, J.; Court, S.D.M. The Genetic Component in Child Mortality. Arch. Dis. Child. 1970, 45, 33–38. [Google Scholar] [CrossRef]
- Wirth, B. An Update of the Mutation Spectrum of the Survival Motor Neuron Gene (SMN1) in Autosomal Recessive Spinal Muscular Atrophy (SMA). Hum. Mutat. 2000, 15, 228–237. [Google Scholar] [CrossRef]
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal Muscular Atrophy. Orphanet J. Rare Dis. 2011. [Google Scholar] [CrossRef]
- Ogino, S.; Leonard, D.G.B.; Rennert, H.; Ewens, W.J.; Wilson, R.B. Genetic Risk Assessment in Carrier Testing for Spinal Muscular Atrophy. Am. J. Med Genet. 2002, 110, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Prior, T.W.; Snyder, P.J.; Rink, B.D.; Pearl, D.K.; Pyatt, R.E.; Mihal, D.C.; Conlan, T.; Schmalz, B.; Montgomery, L.; Ziegler, K.; et al. Newborn and Carrier Screening for Spinal Muscular Atrophy. Am. J. Med Genet. Part A 2010, 152A, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Schorling, D.C.; Pechmann, A.; Kirschner, J. Advances in Treatment of Spinal Muscular Atrophy—New Phenotypes, New Challenges, New Implications for Care. J. Neuromuscul. Dis. 2020, 7, 1–13. [Google Scholar] [CrossRef]
- Feng, Y.; Ge, X.; Meng, L.; Scull, J.; Li, J.; Tian, X.; Zhang, T.; Jin, W.; Cheng, H.; Wang, X.; et al. The next Generation of Population-Based Spinal Muscular Atrophy Carrier Screening: Comprehensive Pan-Ethnic SMN1 Copy-Number and Sequence Variant Analysis by Massively Parallel Sequencing. Genet. Med. 2017, 19, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Robertson, A.; Wilson, I.J.; Aartsma-Rus, A.; Cameron, S.; Jones, C.C.; Cook, S.F.; Lochmüller, H. Prevalence, Incidence and Carrier Frequency of 5q–Linked Spinal Muscular Atrophy—A Literature Review. Orphanet J. Rare Dis. 2017, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Sugarman, E.A.; Nagan, N.; Zhu, H.; Akmaev, V.R.; Zhou, Z.; Rohlfs, E.M.; Flynn, K.; Hendrickson, B.C.; Scholl, T.; Sirko-Osadsa, D.A.; et al. Pan-Ethnic Carrier Screening and Prenatal Diagnosis for Spinal Muscular Atrophy: Clinical Laboratory Analysis of >72,400 Specimens. Eur. J. Hum. Genet. 2012, 20, 27–32. [Google Scholar] [CrossRef]
- Kvasnicová, M.; Styková, J.; Hudec, P. Incidence of Spinal Muscular Atrophy and Duchenne’s Muscular Dystrophy in the Juvenile Population of Central Slovakia. Bratislavské Lekárske Listy 1994, 95, 78–82. [Google Scholar] [PubMed]
- Ludvigsson, P.; Olafsson, E.; Hauser, W.A. Spinal Muscular Atrophy: Incidence in Iceland. Neuroepidemiology 1999, 18, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Zaldivar, T.; Montejo, Y.; Acevedo, A.M.; Guerra, R.; Vargas, J.; Garofalo, N.; Alvarez, R.; Alvarez, M.A.; Hardiman, O. Evidence of Reduced Frequency of Spinal Muscular Atrophy Type I in the Cuban Population. Neurology 2005, 65, 636–638. [Google Scholar] [CrossRef]
- Hendrickson, B.C.; Donohoe, C.; Akmaev, V.R.; Sugarman, E.A.; Labrousse, P.; Boguslavskiy, L.; Flynn, K.; Rohlfs, E.M.; Walker, A.; Allitto, B.; et al. Differences in SMN1 Allele Frequencies among Ethnic Groups within North America. J. Med Genet. 2009, 46, 641–644. [Google Scholar] [CrossRef]
- MacDonald, W.K.; Hamilton, D.; Kuhle, S. SMA Carrier Testing: A Meta-Analysis of Differences in Test Performance by Ethnic Group. Prenat. Diagn. 2014, 34, 1219–1226. [Google Scholar] [CrossRef]
- Russman, B.S. Spinal Muscular Atrophy: Clinical Classification and Disease Heterogeneity. J. Child Neurol. 2007, 22, 946–951. [Google Scholar] [CrossRef]
- Arnold, W.D.; Kassar, D.; Kissel, J.T. Spinal Muscular Atrophy: Diagnosis and Management in a New Therapeutic Era. Muscle Nerve 2015, 51, 157–167. [Google Scholar] [CrossRef]
- Campbell, L.; Potter, A.; Ignatius, J.; Dubowitz, V.; Davies, K. Genomic Variation and Gene Conversion in Spinal Muscular Atrophy: Implications for Disease Process and Clinical Phenotype. Am. J. Hum. Genet. 1997, 61, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, M.; Becker, C.G.; Yáñez-Muñoz, R.J.; Ning, K.; Wood, M.J.A.; Gillingwater, T.H.; Talbot, K. Therapeutic Strategies for Spinal Muscular Atrophy: SMN and Beyond. Dis. Models Mech. 2017, 10, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Paushkin, S.; Charroux, B.; Abel, L.; Perkinson, R.A.; Pellizzoni, L.; Dreyfuss, G. The Survival Motor Neuron Protein of Schizosacharomyces Pombe. J. Biol. Chem. 2000, 275, 23841–23846. [Google Scholar] [CrossRef] [PubMed]
- Tariq, F.; Holcik, M.; MacKenzie, A. Spinal Muscular Atrophy: Classification, Diagnosis, Background, Molecular Mechanism and Development of Therapeutics. In Neurodegenerative Diseases; InTech: London, UK, 2013. [Google Scholar] [CrossRef]
- Cartegni, L.; Krainer, A.R. Disruption of an SF2/ASF-Dependent Exonic Splicing Enhancer in SMN2 Causes Spinal Muscular Atrophy in the Absence of SMN1. Nat. Genet. 2002, 30, 377–384. [Google Scholar] [CrossRef]
- Kashima, T.; Manley, J.L. A Negative Element in SMN2 Exon 7 Inhibits Splicing in Spinal Muscular Atrophy. Nat. Genet. 2003, 34, 460–463. [Google Scholar] [CrossRef]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A Single Nucleotide Difference That Alters Splicing Patterns Distinguishes the SMA Gene SMN1 from the Copy Gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Gennarelli, M.; Lucarelli, M.; Capon, F.; Pizzuti, A.; Merlini, L.; Angelini, C.; Novelli, G.; Dallapiccola, B. Survival Motor-Neuron Gene Transcript Analysis in Muscles from Spinal Muscular-Atrophy Patients. Biochem. Biophys. Res. Commun. 1995, 213, 342–348. [Google Scholar] [CrossRef]
- Rad, I.A. Mutation Spectrum of Survival Motor Neuron Gene in Spinal Muscular Atrophy. J. Down Syndr. Chromosome Abnorm. 2017, 3, 1–2. [Google Scholar] [CrossRef]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA Type and SMN2 Copy Number Revisited: An Analysis of 625 Unrelated Spanish Patients and a Compilation of 2834 Reported Cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef]
- Al Dakhoul, S. Very Severe Spinal Muscular Atrophy (Type 0). Avicenna J. Med. 2017, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Feldkötter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative Analyses of SMN1 and SMN2 Based on Real-Time LightCycler PCR: Fast and Highly Reliable Carrier Testing and Prediction of Severity of Spinal Muscular Atrophy. Am. J. Hum. Genet. 2002, 70, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Wirth, B.; Brichta, L.; Schrank, B.; Lochmüller, H.; Blick, S.; Baasner, A.; Heller, R. Mildly Affected Patients with Spinal Muscular Atrophy Are Partially Protected by an Increased SMN2 Copy Number. Hum. Genet. 2006, 119, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Cuscó, I.; Barceló, M.J.; Rojas–García, R.; Illa, I.; Gámez, J.; Cervera, C.; Pou, A.; Izquierdo, G.; Baiget, M.; Tizzano, E.F. SMN2 Copy Number Predicts Acute or Chronic Spinal Muscular Atrophy but Does Not Account for Intrafamilial Variability in Siblings. J. Neurol. 2006, 253, 21–25. [Google Scholar] [CrossRef]
- Burghes, A.H.M.; Beattie, C.E. Spinal Muscular Atrophy: Why Do Low Levels of Survival Motor Neuron Protein Make Motor Neurons Sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Monani, U.R.; Sendtner, M.; Coovert, D.D.; Parsons, D.W.; Andreassi, C.; Le, T.T.; Jablonka, S.; Schrank, B.; Rossol, W.; Prior, T.W.; et al. The Human Centromeric Survival Motor Neuron Gene (SMN2) Rescues Embryonic Lethality in Smn-/-Mice and Results in a Mouse with Spinal Muscular Atrophy. Hum. Mol. Genet. 2000, 9, 333–339. [Google Scholar] [CrossRef]
- Munsat, T.L. International SMA Collaboration. Neuromuscul. Disord. 1991. [Google Scholar] [CrossRef]
- Dubowitz, V. Very Severe Spinal Muscular Atrophy (SMA Type 0): An Expanding Clinical Phenotype. Eur. J. Paediatr. Neurol. 1999, 3, 49–51. [Google Scholar] [CrossRef]
- Rudnik-Schoneborn, S.; Heller, R.; Berg, C.; Betzler, C.; Grimm, T.; Eggermann, T.; Eggermann, K.; Wirth, R.; Wirth, B.; Zerres, K. Congenital Heart Disease Is a Feature of Severe Infantile Spinal Muscular Atrophy. J. Med Genet. 2008, 45, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Grotto, S.; Cuisset, J.-M.; Marret, S.; Drunat, S.; Faure, P.; Audebert-Bellanger, S.; Desguerre, I.; Flurin, V.; Grebille, A.-G.; Guerrot, A.-M.; et al. Type 0 Spinal Muscular Atrophy: Further Delineation of Prenatal and Postnatal Features in 16 Patients. J. Neuromuscul. Dis. 2016, 3, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Menke, L.A.; Poll-The, B.T.; Clur, S.-A.; Bilardo, C.M.; van der Wal, A.C.; Lemmink, H.H.; Cobben, J.M. Congenital Heart Defects in Spinal Muscular Atrophy Type I: A Clinical Report of Two Siblings and a Review of the Literature. Am. J. Med Genet. Part A 2008, 146A, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Zerres, K. Natural History in Proximal Spinal Muscular Atrophy. Arch. Neurol. 1995, 52, 518. [Google Scholar] [CrossRef] [PubMed]
- Butchbach, M.E.R. Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Front. Mol. Biosci. 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Zerres, K.; Davies, K.E. 59th ENMC International Workshop: Spinal Muscular Atrophies: Recent Progress and Revised Diagnostic Criteria 17–19 April 1998, Soestduinen, The Netherlands. Neuromuscul. Disord. 1999, 9, 272–278. [Google Scholar] [CrossRef]
- O’Hagen, J.M.; Glanzman, A.M.; McDermott, M.P.; Ryan, P.A.; Flickinger, J.; Quigley, J.; Riley, S.; Sanborn, E.; Irvine, C.; Martens, W.B.; et al. An Expanded Version of the Hammersmith Functional Motor Scale for SMA II and III Patients. Neuromuscul. Disord. 2007, 17, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.H.; Dubowitz, V. The Natural History of Type I (Severe) Spinal Muscular Atrophy. Neuromuscul. Disord. 1994, 4, 497–502. [Google Scholar] [CrossRef]
- Dawood, A.A.; Moosa, A. Hand and ECG Tremor in Spinal Muscular Atrophy. Arch. Dis. Child. 1983, 58, 376–378. [Google Scholar] [CrossRef]
- Sharawat, I.K.; Kohli, T.S.; Saini, L. Trembling Hands and Trembling ECG. BMJ Case Rep. 2019, 12, 230618. [Google Scholar] [CrossRef] [PubMed]
- Shorrock, H.K.; Gillingwater, T.H.; Groen, E.J.N. Overview of Current Drugs and Molecules in Development for Spinal Muscular Atrophy Therapy. Drugs 2018, 78, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Moosa, A.; Dubowitz, V. Spinal Muscular Atrophy in Childhood: Two Clues to Clinical Diagnosis. Arch. Dis. Child. 1973, 48, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Piepers, S.; Berg, L.H.; Brugman, F.; Scheffer, H.; Ruiterkamp-Versteeg, M.; Engelen, B.G.; Faber, C.G.; Visser, M.; Pol, W.-L.; Wokke, J.H.J. A Natural History Study of Late Onset Spinal Muscular Atrophy Types 3b and 4. J. Neurol. 2008, 255, 1400–1404. [Google Scholar] [CrossRef] [PubMed]
- Melki, J.; Lefebvre, S.; Burglen, L.; Burlet, P.; Clermont, O.; Millasseau, P.; Reboullet, S.; Benichou, B.; Zeviani, M.; Le Paslier, D.; et al. De Novo and Inherited Deletions of the 5q13 Region in Spinal Muscular Atrophies. Science 1994, 264, 1474–1477. [Google Scholar] [CrossRef] [PubMed]
- Oskoui, M.; Darras, B.T.; De Vivo, D.C. Spinal Muscular Atrophy. In Spinal Muscular Atrophy; Elsevier: Amsterdam, The Netherlands, 2017; pp. 3–19. [Google Scholar] [CrossRef]
- Battaglia, G.; Princivalle, A.; Forti, F.; Lizier, C.; Zeviani, M. Expression of the SMN Gene, the Spinal Muscular Atrophy Determining Gene, in the Mammalian Central Nervous System. Hum. Mol. Genet. 1997, 6, 1961–1971. [Google Scholar] [CrossRef]
- Renvoise, B. Distinct Domains of the Spinal Muscular Atrophy Protein SMN Are Required for Targeting to Cajal Bodies in Mammalian Cells. J. Cell Sci. 2006, 119, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Chaytow, H.; Huang, Y.-T.; Gillingwater, T.H.; Faller, K.M.E. The Role of Survival Motor Neuron Protein (SMN) in Protein Homeostasis. Cell. Mol. Life Sci. 2018, 75, 3877–3894. [Google Scholar] [CrossRef]
- So, B.R.; Zhang, Z.; Dreyfuss, G. The Function of Survival Motor Neuron Complex and Its Role in Spinal Muscular Atrophy Pathogenesis. In Spinal Muscular Atrophy; Elsevier: Amsterdam, The Netherlands, 2017; pp. 99–111. [Google Scholar] [CrossRef]
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse Role of Survival Motor Neuron Protein. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2017, 1860, 299–315. [Google Scholar] [CrossRef]
- Ogawa, C.; Usui, K.; Aoki, M.; Ito, F.; Itoh, M.; Kai, C.; Kanamori-Katayama, M.; Hayashizaki, Y.; Suzuki, H. Gemin2 Plays an Important Role in Stabilizing the Survival of Motor Neuron Complex. J. Biol. Chem. 2007, 282, 11122–11134. [Google Scholar] [CrossRef]
- Young, P.J.; Day, P.M.; Zhou, J.; Androphy, E.J.; Morris, G.E.; Lorson, C.L. A Direct Interaction between the Survival Motor Neuron Protein and P53 and Its Relationship to Spinal Muscular Atrophy. J. Biol. Chem. 2002, 277, 2852–2859. [Google Scholar] [CrossRef]
- Ponting, C.P. Tudor Domains in Proteins That Interact with RNA. Trends Biochem. Sci. 1997, 22, 51–52. [Google Scholar] [CrossRef]
- Brahms, H.; Meheus, L.; De Brabandere, V.; Fischer, U.; Luhrmann, R. Symmetrical Dimethylation of Arginine Residues in Spliceosomal Sm Protein B/B′ and the Sm-like Protein LSm4, and Their Interaction with the SMN Protein. RNA 2001, 7. [Google Scholar] [CrossRef]
- Martin, R.; Gupta, K.; Ninan, N.S.; Perry, K.; Van Duyne, G.D. The Survival Motor Neuron Protein Forms Soluble Glycine Zipper Oligomers. Structure 2012, 20, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Selenko, P.; Sprangers, R.; Stier, G.; Bühler, D.; Fischer, U.; Sattler, M. SMN Tudor Domain Structure and Its Interaction with the Sm Proteins. Nat. Struct. Biol. 2001. [Google Scholar] [CrossRef]
- Giesemann, T.; Rathke-Hartlieb, S.; Rothkegel, M.; Bartsch, J.W.; Buchmeier, S.; Jockusch, B.M.; Jockusch, H. A Role for Polyproline Motifs in the Spinal Muscular Atrophy Protein SMN. J. Biol. Chem. 1999, 274, 37908–37914. [Google Scholar] [CrossRef] [PubMed]
- Nölle, A.; Zeug, A.; van Bergeijk, J.; Tönges, L.; Gerhard, R.; Brinkmann, H.; Al Rayes, S.; Hensel, N.; Schill, Y.; Apkhazava, D.; et al. The Spinal Muscular Atrophy Disease Protein SMN Is Linked to the Rho-Kinase Pathway via Profilin. Hum. Mol. Genet. 2011, 20, 4865–4878. [Google Scholar] [CrossRef]
- Charroux, B.; Pellizzoni, L.; Perkinson, R.A.; Shevchenko, A.; Mann, M.; Dreyfuss, G. Gemin3. J. Cell Biol. 1999, 147, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Gangwani, L.; Mikrut, M.; Theroux, S.; Sharma, M.; Davis, R.J. Spinal Muscular Atrophy Disrupts the Interaction of ZPR1 with the SMN Protein. Nat. Cell Biol. 2001, 3, 376–383. [Google Scholar] [CrossRef]
- Zou, J.; Barahmand-pour, F.; Blackburn, M.L.; Matsui, Y.; Chansky, H.A.; Yang, L. Survival Motor Neuron (SMN) Protein Interacts with Transcription Corepressor MSin3A. J. Biol. Chem. 2004, 279, 14922–14928. [Google Scholar] [CrossRef]
- Cho, S.; Dreyfuss, G. A Degron Created by SMN2 Exon 7 Skipping Is a Principal Contributor to Spinal Muscular Atrophy Severity. Genes Dev. 2010, 24, 438–442. [Google Scholar] [CrossRef]
- Lorson, M.A.; Lorson, C.L. SMN-Inducing Compounds for the Treatment of Spinal Muscular Atrophy. Future Med. Chem. 2012, 4, 2067–2084. [Google Scholar] [CrossRef] [PubMed]
- Fan, L. Survival Motor Neuron (SMN) Protein: Role in Neurite Outgrowth and Neuromuscular Maturation during Neuronal Differentiation and Development. Hum. Mol. Genet. 2002, 11, 1605–1614. [Google Scholar] [CrossRef]
- Martínez-Hernández, R.; Soler-Botija, C.; Also, E.; Alias, L.; Caselles, L.; Gich, I.; Bernal, S.; Tizzano, E.F. The Developmental Pattern of Myotubes in Spinal Muscular Atrophy Indicates Prenatal Delay of Muscle Maturation. J. Neuropathol. Exp. Neurol. 2009, 68, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.G.; Bowerman, M.; Kothary, R. The Many Faces of SMN: Deciphering the Function Critical to Spinal Muscular Atrophy Pathogenesis. Future Neurol. 2010, 5, 873–890. [Google Scholar] [CrossRef]
- Pruss, R.M.; Giraudon-Paoli, M.; Morozova, S.; Berna, P.; Abitbol, J.-L.; Bordet, T. Drug Discovery and Development for Spinal Muscular Atrophy: Lessons from Screening Approaches and Future Challenges for Clinical Development. Future Med. Chem. 2010, 2, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. FDA Approves First Drug for Spinal Muscular Atrophy. Mol. Cell. Pharmacol. 2017, 15. [Google Scholar]
- Zanetta, C.; Nizzardo, M.; Simone, C.; Monguzzi, E.; Bresolin, N.; Comi, G.P.; Corti, S. Molecular Therapeutic Strategies for Spinal Muscular Atrophies: Current and Future Clinical Trials. Clin. Ther. 2014, 36, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense Correction of SMN2 Splicing in the CNS Rescues Necrosis in a Type III SMA Mouse Model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense Masking of an HnRNP A1/A2 Intronic Splicing Silencer Corrects SMN2 Splicing in Transgenic Mice. Am. J. Hum. Genet. 2008, 82, 834–848. [Google Scholar] [CrossRef]
- Porensky, P.N.; Burghes, A.H.M. Antisense Oligonucleotides for the Treatment of Spinal Muscular Atrophy. Hum. Gene Ther. 2013, 24, 489–498. [Google Scholar] [CrossRef]
- Passini, M.A.; Bu, J.; Richards, A.M.; Kinnecom, C.; Sardi, S.P.; Stanek, L.M.; Hua, Y.; Rigo, F.; Matson, J.; Hung, G.; et al. Antisense Oligonucleotides Delivered to the Mouse CNS Ameliorate Symptoms of Severe Spinal Muscular Atrophy. Sci. Transl. Med. 2011, 3, 72ra18. [Google Scholar] [CrossRef]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, Biodistribution and Cell Uptake of Antisense Oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef]
- Dodou, K. Intrathecal Route of Drug Delivery Can Save Lives or Improve Quality of Life. Pharm. J. 2012, 289, 501. [Google Scholar]
- Chiriboga, C.A. Nusinersen for the Treatment of Spinal Muscular Atrophy. Expert Rev. Neurother. 2017, 17, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Nusinersen: First Global Approval. Drugs 2017, 77, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. FDA Approves Innovative Gene Therapy to Treat Pediatric Patients with Spinal Muscular Atrophy, a Rare Disease and Leading Genetic Cause of Infant Mortality. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease (accessed on 29 March 2020). [CrossRef]
- Hensel, N.; Kubinski, S.; Claus, P. The Need for SMN-Independent Treatments of Spinal Muscular Atrophy (SMA) to Complement SMN-Enhancing Drugs. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- (Hoy, S.M. Onasemnogene Abeparvovec: First Global Approval. Drugs 2019, 79, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Food and Drug Administration. FDA Approves Oral Treatment for Spinal Muscular Atrophy. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy (accessed on 28 August 2020).
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef]
- Song, J.Y.; Kim, H.S.; Park, S.-J.; Lee, J.; Lee, J. Nusinersen Administration in Spinal Muscular Atrophy Patients with Severe Scoliosis: Interlaminar Approaches at the Lumbar Level. Ann. Child Neurol. 2020, 28, 49–56. [Google Scholar] [CrossRef]
- Zuluaga-Sanchez, S.; Teynor, M.; Knight, C.; Thompson, R.; Lundqvist, T.; Ekelund, M.; Forsmark, A.; Vickers, A.D.; Lloyd, A. Cost Effectiveness of Nusinersen in the Treatment of Patients with Infantile-Onset and Later-Onset Spinal Muscular Atrophy in Sweden. PharmacoEconomics 2019, 37, 845–865. [Google Scholar] [CrossRef]
- SMA News Today. Zolgensma. Available online: https://smanewstoday.com/avxs-101-avexis (accessed on 1 April 2020).
- Roche. FDA Approves Roche’s Evrysdi (Risdiplam) for Treatment of Spinal Muscular Atrophy (SMA) in Adults and Children 2 Months and Older. Available online: https://www.roche.com/investors/updates/inv-update-2020-08-10b.htm (accessed on 28 August 2020).
- Palacino, J.; Swalley, S.E.; Song, C.; Cheung, A.K.; Shu, L.; Zhang, X.; Van Hoosear, M.; Shin, Y.; Chin, D.N.; Keller, C.G.; et al. SMN2 Splice Modulators Enhance U1–Pre-MRNA Association and Rescue SMA Mice. Nat. Chem. Biol. 2015, 11, 511–517. [Google Scholar] [CrossRef]
- Hensel, N.; Claus, P. The Actin Cytoskeleton in SMA and ALS: How Does It Contribute to Motoneuron Degeneration? Neuroscientist 2018, 24, 54–72. [Google Scholar] [CrossRef] [PubMed]
- Parente, V.; Corti, S. Advances in Spinal Muscular Atrophy Therapeutics. Ther. Adv. Neurol. Disord. 2018, 11, 175628561875450. [Google Scholar] [CrossRef]
- Calder, A.N.; Androphy, E.J.; Hodgetts, K.J. Small Molecules in Development for the Treatment of Spinal Muscular Atrophy. J. Med. Chem. 2016, 59, 10067–10083. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.A.; Miller, T.M.; Vijayakumar, V.; Stoltz, R.; James, J.K.; Meng, L.; Wolff, A.A.; Malik, F.I. CK-2127107 Amplifies Skeletal Muscle Response to Nerve Activation in Humans. Muscle Nerve 2018, 57, 729–734. [Google Scholar] [CrossRef]
- Long, K.K.; O’Shea, K.M.; Khairallah, R.J.; Howell, K.; Paushkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific Inhibition of Myostatin Activation Is Beneficial in Mouse Models of SMA Therapy. Hum. Mol. Genet. 2019, 28, 1076–1089. [Google Scholar] [CrossRef] [PubMed]
- Duelen, R.; Corvelyn, M.; Tortorella, I.; Leonardi, L.; Chai, Y.C.; Sampaolesi, M. Medicinal Biotechnology for Disease Modeling, Clinical Therapy, and Drug Discovery and Development. In Introduction to Biotech Entrepreneurship: From Idea to Business; Springer International Publishing: Cham, Switzerland, 2019; pp. 89–128. [Google Scholar] [CrossRef]
- Prieto-Martínez, F.D.; López-López, E.; Eurídice Juárez-Mercado, K.; Medina-Franco, J.L. Computational Drug Design Methods—Current and Future Perspectives. In Silico Drug Design; Elsevier: Amsterdam, The Netherlands, 2019; pp. 19–44. [Google Scholar] [CrossRef]
- Danon, J.J.; Reekie, T.A.; Kassiou, M. Challenges and Opportunities in Central Nervous System Drug Discovery. Trends Chem. 2019, 1, 612–624. [Google Scholar] [CrossRef]
- Bagchi, S.; Chhibber, T.; Lahooti, B.; Verma, A.; Borse, V.; Jayant, R.D. In-Vitro Blood-Brain Barrier Models for Drug Screening and Permeation Studies: An Overview. Drug Des. Dev. Ther. 2019, 13, 3591–3605. [Google Scholar] [CrossRef]
- Sun, W.; Zheng, W.; Simeonov, A. Drug Discovery and Development for Rare Genetic Disorders. Am. J. Med Genet. Part A 2017, 173, 2307–2322. [Google Scholar] [CrossRef]
- Hoolachan, J.M.; Sutton, E.R.; Bowerman, M. Teaching an Old Drug New Tricks: Repositioning Strategies for Spinal Muscular Atrophy. Future Neurol. 2019, 14, FNL25. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug Repositioning: Identifying and Developing New Uses for Existing Drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-G.; Hsieh-Li, H.-M.; Jong, Y.-J.; Wang, N.M.; Tsai, C.-H.; Li, H. Treatment of Spinal Muscular Atrophy by Sodium Butyrate. Proc. Natl. Acad. Sci. USA 2001, 98, 9808–9813. [Google Scholar] [CrossRef]
- Andreassi, C.; Angelozzi, C.; Tiziano, F.D.; Vitali, T.; De Vincenzi, E.; Boninsegna, A.; Villanova, M.; Bertini, E.; Pini, A.; Neri, G.; et al. Phenylbutyrate Increases SMN Expression in Vitro: Relevance for Treatment of Spinal Muscular Atrophy. Eur. J. Hum. Genet. 2004, 12, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Brahe, C.; Vitali, T.; Tiziano, F.D.; Angelozzi, C.; Pinto, A.M.; Borgo, F.; Moscato, U.; Bertini, E.; Mercuri, E.; Neri, G. Phenylbutyrate Increases SMN Gene Expression in Spinal Muscular Atrophy Patients. Eur. J. Hum. Genet. 2005, 13, 256–259. [Google Scholar] [CrossRef]
- Brichta, L. Valproic Acid Increases the SMN2 Protein Level: A Well-Known Drug as a Potential Therapy for Spinal Muscular Atrophy. Hum. Mol. Genet. 2003, 12, 2481–2489. [Google Scholar] [CrossRef]
- Minucci, S.; Pelicci, P.G. Histone Deacetylase Inhibitors and the Promise of Epigenetic (and More) Treatments for Cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef]
- Sumner, C.J.; Huynh, T.N.; Markowitz, J.A.; Perhac, J.S.; Hill, B.; Coovert, D.D.; Schussler, K.; Chen, X.; Jarecki, J.; Burghes, A.H.M.; et al. Valproic Acid Increases SMN Levels in Spinal Muscular Atrophy Patient Cells. Ann. Neurol. 2003, 54, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, V.; Guerra, M.; Di Rosa, V.; Compagnucci, C.; Sette, C. Combined Treatment with the Histone Deacetylase Inhibitor LBH589 and a Splice-switch Antisense Oligonucleotide Enhances SMN2 Splicing and SMN Expression in Spinal Muscular Atrophy Cells. J. Neurochem. 2020, 153, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Milane, A.; Fernandez, C.; Vautier, S.; Bensimon, G.; Meininger, V.; Farinotti, R. Minocycline and Riluzole Brain Disposition: Interactions with p-Glycoprotein at the Blood-Brain Barrier. J. Neurochem. 2007, 103, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Sohraby, F.; Bagheri, M.; Aryapour, H. Performing an In Silico Repurposing of Existing Drugs by Combining Virtual Screening and Molecular Dynamics Simulation. Methods Mol. Biol. 2019, 1903, 23–43. [Google Scholar] [CrossRef]
- Jin, G.; Wong, S.T.C. Toward Better Drug Repositioning: Prioritizing and Integrating Existing Methods into Efficient Pipelines. Drug Discov. Today 2014, 19, 637–644. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A Web-Accessible Database of Experimentally Determined Protein-Ligand Binding Affinities. Nucleic Acids Res. 2007. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A Public Database for Medicinal Chemistry, Computational Chemistry and Systems Pharmacology. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Stark, C. BioGRID: A General Repository for Interaction Datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID Database: A Comprehensive Biomedical Resource of Curated Protein, Genetic, and Chemical Interactions. Protein Sci. 2020, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL Web Services: Streamlining Access to Drug Discovery Data and Utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards Direct Deposition of Bioassay Data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Chen, J.H.; Linstead, E.; Swamidass, S.J.; Wang, D.; Baldi, P. ChemDB Update—Full-Text Search and Virtual Chemical Space. Bioinformatics 2007, 23, 2348–2351. [Google Scholar] [CrossRef]
- Pence, H.E.; Williams, A. ChemSpider: An Online Chemical Information Resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Lamb, J. The Connectivity Map: A New Tool for Biomedical Research. Nat. Rev. Cancer 2007, 7, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- Salwinski, L. The Database of Interacting Proteins: 2004 Update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef]
- Xenarios, I. DIP: The Database of Interacting Proteins. Nucleic Acids Res. 2000, 28, 289–291. [Google Scholar] [CrossRef]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-Generation Drug Library and Information Resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.; Bologa, C.G.; Holmes, J.; Bocci, G.; Wilson, T.B.; Nguyen, D.-T.; Curpan, R.; Halip, L.; Bora, A.; Yang, J.J.; et al. DrugCentral 2021 Supports Drug Discovery and Repositioning. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Sun, J.; Jeliazkova, N.; Chupakhin, V.; Golib-Dzib, J.-F.; Engkvist, O.; Carlsson, L.; Wegner, J.; Ceulemans, H.; Georgiev, I.; Jeliazkov, V.; et al. ExCAPE-DB: An Integrated Large Scale Dataset Facilitating Big Data Analysis in Chemogenomics. J. Cheminformatics 2017, 9, 17. [Google Scholar] [CrossRef]
- Fahey, M.E.; Bennett, M.J.; Mahon, C.; Jäger, S.; Pache, L.; Kumar, D.; Shapiro, A.; Rao, K.; Chanda, S.K.; Craik, C.S.; et al. GPS-Prot: A Web-Based Visualization Platform for Integrating Host-Pathogen Interaction Data. BMC Bioinform. 2011. [Google Scholar] [CrossRef]
- Peri, S. Development of Human Protein Reference Database as an Initial Platform for Approaching Systems Biology in Humans. Genome Res. 2003, 13, 2363–2371. [Google Scholar] [CrossRef]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database—2009 Update. Nucleic Acids Res. 2009. [Google Scholar] [CrossRef]
- Stathias, V.; Turner, J.; Koleti, A.; Vidovic, D.; Cooper, D.; Fazel-Najafabadi, M.; Pilarczyk, M.; Terryn, R.; Chung, C.; Umeano, A.; et al. LINCS Data Portal 2.0: Next Generation Access Point for Perturbation-Response Signatures. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zanzoni, A.; Montecchi-Palazzi, L.; Quondam, M.; Ausiello, G.; Helmer-Citterich, M.; Cesareni, G. MINT: A Molecular INTeraction Database. FEBS Lett. 2002, 513, 135–140. [Google Scholar] [CrossRef]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT, the Molecular Interaction Database: 2012 Update. Nucleic Acids Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Prilusky, J.; Sussman, J.L. Proteopedia: A Collaborative, Virtual 3D Web-Resource for Protein and Biomolecule Structure and Function. Biochem. Mol. Biol. Educ. 2010. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 Update: Improved Access to Chemical Data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Novick, P.A.; Ortiz, O.F.; Poelman, J.; Abdulhay, A.Y.; Pande, V.S. SWEETLEAD: An in Silico Database of Approved Drugs, Regulated Chemicals, and Herbal Isolates for Computer-Aided Drug Discovery. PLoS ONE 2013, 8, e79568. [Google Scholar] [CrossRef]
- Siramshetty, V.B.; Eckert, O.A.; Gohlke, B.O.; Goede, A.; Chen, Q.; Devarakonda, P.; Preissner, S.; Preissner, R. SuperDRUG2: A One Stop Resource for Approved/Marketed Drugs. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Huang, R.; Southall, N.; Wang, Y.; Yasgar, A.; Shinn, P.; Jadhav, A.; Nguyen, D.-T.; Austin, C.P. The NCGC Pharmaceutical Collection: A Comprehensive Resource of Clinically Approved Drugs Enabling Repurposing and Chemical Genomics. Sci. Transl. Med. 2011, 3, 80ps16. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: A Worldwide Hub of Protein Knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Modeling 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Iorio, F.; Bosotti, R.; Scacheri, E.; Belcastro, V.; Mithbaokar, P.; Ferriero, R.; Murino, L.; Tagliaferri, R.; Brunetti-Pierri, N.; Isacchi, A.; et al. Discovery of Drug Mode of Action and Drug Repositioning from Transcriptional Responses. Proc. Natl. Acad. Sci. USA 2010, 107, 14621–14626. [Google Scholar] [CrossRef]
- Carrella, D.; Napolitano, F.; Rispoli, R.; Miglietta, M.; Carissimo, A.; Cutillo, L.; Sirci, F.; Gregoretti, F.; Di Bernardo, D. Mantra 2.0: An Online Collaborative Resource for Drug Mode of Action and Repurposing by Network Analysis. Bioinformatics 2014, 30, 1787–1788. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.G.; Gross, B.E.; Demir, E.; Rodchenkov, I.; Babur, O.; Anwar, N.; Schultz, N.; Bader, G.D.; Sander, C. Pathway Commons, a Web Resource for Biological Pathway Data. Nucleic Acids Res. 2011, 39, D685–D690. [Google Scholar] [CrossRef] [PubMed]
- Rodchenkov, I.; Babur, O.; Luna, A.; Aksoy, B.A.; Wong, J.V.; Fong, D.; Franz, M.; Siper, M.C.; Cheung, M.; Wrana, M.; et al. Pathway Commons 2019 Update: Integration, Analysis and Exploration of Pathway Data. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome Pathway Analysis: A High-Performance in-Memory Approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef] [PubMed]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress Update—From Bulk to Single-Cell Expression Data. Nucleic Acids Res. 2018, 47, D711–D715. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2012, 41, D36–D42. [Google Scholar] [CrossRef]
- Barrett, T.; Edgar, R. Gene Expression Omnibus: Microarray Data Storage, Submission, Retrieval, and Analysis. Methods Enzymol. 2006, 411, 352–369. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk, A.; Ingelman-Sundberg, M.; Miller, N.A.; Leeder, J.S.; Whirl-Carrillo, M.; Klein, T.E. The Pharmacogene Variation (PharmVar) Consortium: Incorporation of the Human Cytochrome P450 (CYP) Allele Nomenclature Database. Clin. Pharmacol. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef]
- Smigielski, E.M. DbSNP: A Database of Single Nucleotide Polymorphisms. Nucleic Acids Res. 2000, 28, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Mattingly, C.J.; Colby, G.T.; Forrest, J.N.; Boyer, J.L. The Comparative Toxicogenomics Database (CTD). Environ. Health Perspect. 2003. [Google Scholar] [CrossRef]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A Comprehensive Platform Integrating Information on Human Disease-Associated Genes and Variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef]
- Fang, H.; Su, Z.; Wang, Y.; Miller, A.; Liu, Z.; Howard, P.C.; Tong, W.; Lin, S.M. Exploring the FDA Adverse Event Reporting System to Generate Hypotheses for Monitoring of Disease Characteristics. Clin. Pharmacol. Ther. 2014, 95, 496–498. [Google Scholar] [CrossRef]
- Hamosh, A. Online Mendelian Inheritance in Man (OMIM), a Knowledgebase of Human Genes and Genetic Disorders. Nucleic Acids Res. 2004, 33, D514–D517. [Google Scholar] [CrossRef]
- Carvalho-Silva, D.; Pierleoni, A.; Pignatelli, M.; Ong, C.; Fumis, L.; Karamanis, N.; Carmona, M.; Faulconbridge, A.; Hercules, A.; McAuley, E.; et al. Open Targets Platform: New Developments and Updates Two Years On. Nucleic Acids Res. 2019, 47, D1056–D1065. [Google Scholar] [CrossRef]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics Knowledge for Personalized Medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER Database of Drugs and Side Effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Li, F.; Zhou, Y.; Zhang, Y.; Wang, Z.; Zhang, R.; Zhu, J.; Ren, Y.; Tan, Y.; et al. Therapeutic Target Database 2020: Enriched Resource for Facilitating Research and Early Development of Targeted Therapeutics. Nucleic Acids Res. 2020, 48, D1031–D1041. [Google Scholar] [CrossRef]
- Jia, J.; An, Z.; Ming, Y.; Guo, Y.; Li, W.; Liang, Y.; Guo, D.; Li, X.; Tai, J.; Chen, G.; et al. ERAM: Encyclopedia of Rare Disease Annotations for Precision Medicine. Nucleic Acids Res. 2018, 46, D937–D943. [Google Scholar] [CrossRef] [PubMed]
- Weininger, D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Modeling 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Cheminformatics 2015, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular Similarity in Medicinal Chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef]
- Hodos, R.A.; Kidd, B.A.; Shameer, K.; Readhead, B.P.; Dudley, J.T. In Silico Methods for Drug Repurposing and Pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 186–210. [Google Scholar] [CrossRef]
- Rognan, D. Chemogenomic Approaches to Rational Drug Design. Br. J. Pharmacol. 2007, 152, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Nidhi; Glick, M.; Davies, J.W.; Jenkins, J.L. Prediction of Biological Targets for Compounds Using Multiple-Category Bayesian Models Trained on Chemogenomics Databases. J. Chem. Inf. Modeling 2006, 46, 1124–1133. [Google Scholar] [CrossRef]
- Cao, D.-S.; Liang, Y.-Z.; Deng, Z.; Hu, Q.-N.; He, M.; Xu, Q.-S.; Zhou, G.-H.; Zhang, L.-X.; Deng, Z.; Liu, S. Genome-Scale Screening of Drug-Target Associations Relevant to Ki Using a Chemogenomics Approach. PLoS ONE 2013, 8, e57680. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, Y.; Liu, F.; Luo, F.; Tian, G.; Li, X. Predicting Potential Drug-Drug Interactions by Integrating Chemical, Biological, Phenotypic and Network Data. BMC Bioinform. 2017, 18, 18. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Agarwal, P. Systematic Drug Repositioning Based on Clinical Side-Effects. PLoS ONE 2011, 6, e28025. [Google Scholar] [CrossRef]
- Bisgin, H.; Liu, Z.; Fang, H.; Kelly, R.; Xu, X.; Tong, W. A Phenome-Guided Drug Repositioning through a Latent Variable Model. BMC Bioinform. 2014, 15, 267. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Liu, Q.; Wei, J. Construction of Drug Network Based on Side Effects and Its Application for Drug Repositioning. PLoS ONE 2014, 9, e87864. [Google Scholar] [CrossRef]
- Sridhar, D.; Fakhraei, S.; Getoor, L. A Probabilistic Approach for Collective Similarity-Based Drug-Drug Interaction Prediction. Bioinformatics 2016. [Google Scholar] [CrossRef]
- GNS, H.S.; GR, S.; Murahari, M.; Krishnamurthy, M. An Update on Drug Repurposing: Re-Written Saga of the Drug’s Fate. Biomed. Pharmacother. 2019, 110, 700–716. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER Server for Protein 3D Structure Prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved Protein Structure Prediction Using Predicted Interresidue Orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational Protein–Ligand Docking and Virtual Drug Screening with the AutoDock Suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Polak, V.; Shatsky, M.; Halperin, I.; Benyamini, H.; Barzilai, A.; Dror, O.; Haspel, N.; Nussinov, R.; et al. Taking Geometry to Its Edge: Fast Unbound Rigid (and Hinge-Bent) Docking. Proteins Struct. Funct. Genet. 2003, 52, 107–112. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Zhi, D.G. Ligand-Protein Inverse Docking and Its Potential Use in the Computer Search of Protein Targets of a Small Molecule. Proteins Struct. Funct. Genet. 2001, 43, 217–226. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Modeling 2005. [Google Scholar] [CrossRef]
- Liu, X.; Ouyang, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Li, H.; Jiang, H. PharmMapper Server: A Web Server for Potential Drug Target Identification Using Pharmacophore Mapping Approach. Nucleic Acids Res. 2010, 38 (Suppl. S2), W609–W614. [Google Scholar] [CrossRef]
- Wang, X.; Shen, Y.; Wang, S.; Li, S.; Zhang, W.; Liu, X.; Lai, L.; Pei, J.; Li, H. PharmMapper 2017 Update: A Web Server for Potential Drug Target Identification with a Comprehensive Target Pharmacophore Database. Nucleic Acids Res. 2017, 45, W356–W360. [Google Scholar] [CrossRef]
- Koes, D.R.; Camacho, C.J. ZINCPharmer: Pharmacophore Search of the ZINC Database. Nucleic Acids Res. 2012, 40, W409–W414. [Google Scholar] [CrossRef] [PubMed]
- Backman, T.W.H.; Cao, Y.; Girke, T. ChemMine Tools: An Online Service for Analyzing and Clustering Small Molecules. Nucleic Acids Res. 2011, 39, W486–W491. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Carlson, H.A. ChemTreeMap: An Interactive Map of Biochemical Similarity in Molecular Datasets. Bioinformatics 2016, 32, 3584–3592. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Alam, Z.; Peddinti, G.; Aittokallio, T. C-SPADE: A Web-Tool for Interactive Analysis and Visualization of Drug Screening Experiments through Compound-Specific Bioactivity Dendrograms. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nickel, J.; Gohlke, B.-O.; Erehman, J.; Banerjee, P.; Rong, W.W.; Goede, A.; Dunkel, M.; Preissner, R. SuperPred: Update on Drug Classification and Target Prediction. Nucleic Acids Res. 2014, 42, W26–W31. [Google Scholar] [CrossRef]
- Jia, Z.; Liu, Y.; Guan, N.; Bo, X.; Luo, Z.; Barnes, M.R. Cogena, a Novel Tool for Co-Expressed Gene-Set Enrichment Analysis, Applied to Drug Repositioning and Drug Mode of Action Discovery. BMC Genom. 2016, 17, 414. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Kong, S.W.; Kohane, I.S.; Patel, C.J. KsRepo: A Generalized Platform for Computational Drug Repositioning. BMC Bioinform. 2016, 17, 78. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Huang, J.; Zhong, Y.; Huang, Q. DrugSig: A Resource for Computational Drug Repositioning Utilizing Gene Expression Signatures. PLoS ONE 2017, 12, e0177743. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Li, M.; Wang, S.; Liu, Q.; Li, Y.; Wang, J. Computational Drug Repositioning Using Low-Rank Matrix Approximation and Randomized Algorithms. Bioinformatics 2018, 34, 1904–1912. [Google Scholar] [CrossRef]
- Martínez, V.; Navarro, C.; Cano, C.; Fajardo, W.; Blanco, A. DrugNet: Network-Based Drug–Disease Prioritization by Integrating Heterogeneous Data. Artif. Intell. Med. 2015, 63, 41–49. [Google Scholar] [CrossRef]
- Mullen, J.; Cockell, S.J.; Woollard, P.; Wipat, A. An Integrated Data Driven Approach to Drug Repositioning Using Gene-Disease Associations. PLoS ONE 2016, 11, e0155811. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Choo, S.; Park, J.; Jung, J.; Kang, Y.; Lee, D. Prediction of Drugs Having Opposite Effects on Disease Genes in a Directed Network. BMC Syst. Biol. 2016, 10, S2. [Google Scholar] [CrossRef]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA Prediction Server: Biological Network Integration for Gene Prioritization and Predicting Gene Function. Nucleic Acids Res. 2010, 38 (Suppl. S2), W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, A.; Egorov, S.; Daraselia, N.; Mazo, I. Pathway Studio--the Analysis and Navigation of Molecular Networks. Bioinformatics 2003, 19, 2155–2157. [Google Scholar] [CrossRef] [PubMed]
- Demir, E.; Babur, O.; Dogrusoz, U.; Gursoy, A.; Nisanci, G.; Cetin-Atalay, R.; Ozturk, M. PATIKA: An Integrated Visual Environment for Collaborative Construction and Analysis of Cellular Pathways. Bioinformatics 2002, 18, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Mellor, J.; Wu, J.; DeLisi, C. VisANT: An Online Visualization and Analysis Tool for Biological Interaction Data. BMC Bioinform. 2004, 5, 17. [Google Scholar] [CrossRef]
- Engin, H.; Gursoy, A.; Nussinov, R.; Keskin, O. Network-Based Strategies Can Help Mono- and Poly-Pharmacology Drug Discovery: A Systems Biology View. Curr. Pharm. Des. 2014, 20, 1201–1207. [Google Scholar] [CrossRef]
- Fotis, C.; Antoranz, A.; Hatziavramidis, D.; Sakellaropoulos, T.; Alexopoulos, L.G. Network-Based Technologies for Early Drug Discovery. Drug Discov. Today 2018, 23, 626–635. [Google Scholar] [CrossRef]
- Sidders, B.; Karlsson, A.; Kitching, L.; Torella, R.; Karila, P.; Phelan, A. Network-Based Drug Discovery: Coupling Network Pharmacology with Phenotypic Screening for Neuronal Excitability. J. Mol. Biol. 2018, 430, 3005–3015. [Google Scholar] [CrossRef]
- Borg, R.; Cauchi, R.J. GEMINs: Potential Therapeutic Targets for Spinal Muscular Atrophy? Front. Neurosci. 2014, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.W.; Morrison, K.D.; Shirran, S.L.; Groen, E.J.N.; Gillingwater, T.H.; Botting, C.H.; Sleeman, J.E. Neurochondrin Interacts with the SMN Protein Suggesting a Novel Mechanism for Spinal Muscular Atrophy Pathology. J. Cell Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Curmi, F.; Cauchi, R.J. The Multiple Lives of DEAD-Box RNA Helicase DP103/DDX20/Gemin3. Biochem. Soc. Trans. 2018, 46, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Wadman, R.I.; Jansen, M.D.; Curial, C.A.D.; Groen, E.J.N.; Stam, M.; Wijngaarde, C.A.; Medic, J.; Sodaar, P.; van Eijk, K.R.; Huibers, M.M.H.; et al. Analysis of FUS, PFN2, TDP-43, and PLS3 as Potential Disease Severity Modifiers in Spinal Muscular Atrophy. Neurol. Genet. 2020, 6, e386. [Google Scholar] [CrossRef]
- Tabei, Y.; Kotera, M.; Sawada, R.; Yamanishi, Y. Network-Based Characterization of Drug-Protein Interaction Signatures with a Space-Efficient Approach. BMC Syst. Biol. 2019, 13, 39. [Google Scholar] [CrossRef]
- Hensel, N.; Stockbrügger, I.; Rademacher, S.; Broughton, N.; Brinkmann, H.; Grothe, C.; Claus, P. Bilateral Crosstalk of Rho- and Extracellular-Signal-Regulated-Kinase (ERK) Pathways Is Confined to an Unidirectional Mode in Spinal Muscular Atrophy (SMA). Cell. Signal. 2014, 26, 540–548. [Google Scholar] [CrossRef]
- Fu, P.-C.; Tang, R.-H.; Yu, Z.-Y.; Xie, M.-J.; Wang, W.; Luo, X. The Rho-Associated Kinase Inhibitors Y27632 and Fasudil Promote Microglial Migration in the Spinal Cord via the ERK Signaling Pathway. Neural Regen. Res. 2018, 13, 677. [Google Scholar] [CrossRef]
- Mack, S.G.; Cook, D.J.; Dhurjati, P.; Butchbach, M.E.R. Systems Biology Investigation of CAMP Modulation to Increase SMN Levels for the Treatment of Spinal Muscular Atrophy. PLoS ONE 2014, 9, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Hensel, N.; Ratzka, A.; Brinkmann, H.; Klimaschewski, L.; Grothe, C.; Claus, P. Analysis of the Fibroblast Growth Factor System Reveals Alterations in a Mouse Model of Spinal Muscular Atrophy. PLoS ONE 2012, 7, e31202. [Google Scholar] [CrossRef]
- Schellino, R.; Boido, M.; Borsello, T.; Vercelli, A. Pharmacological C-Jun NH2-Terminal Kinase (JNK) Pathway Inhibition Reduces Severity of Spinal Muscular Atrophy Disease in Mice. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef]
- Simon, C.M.; Dai, Y.; Van Alstyne, M.; Koutsioumpa, C.; Pagiazitis, J.G.; Chalif, J.I.; Wang, X.; Rabinowitz, J.E.; Henderson, C.E.; Pellizzoni, L.; et al. Converging Mechanisms of P53 Activation Drive Motor Neuron Degeneration in Spinal Muscular Atrophy. Cell Rep. 2017, 21, 3767–3780. [Google Scholar] [CrossRef]
- Bowerman, M.; Beauvais, A.; Anderson, C.L.; Kothary, R. Rho-Kinase Inactivation Prolongs Survival of an Intermediate SMA Mouse Model. Hum. Mol. Genet. 2010, 19, 1468–1478. [Google Scholar] [CrossRef]
- Bowerman, M.; Murray, L.M.; Boyer, J.G.; Anderson, C.L.; Kothary, R. Fasudil Improves Survival and Promotes Skeletal Muscle Development in a Mouse Model of Spinal Muscular Atrophy. BMC Med. 2012, 10, 24. [Google Scholar] [CrossRef]
- Demeler, B.; Zhou, G. Neural Network Optimization for E.Coli Promoter Prediction. Nucleic Acids Res. 1991, 19, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Holley, L.H.; Karplus, M. Protein Secondary Structure Prediction with a Neural Network. Proc. Natl. Acad. Sci. USA 1989, 86, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep Learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Gawehn, E.; Hiss, J.A.; Brown, J.B.; Schneider, G. Advancing Drug Discovery via GPU-Based Deep Learning. Expert Opin. Drug Discov. 2018, 13, 579–582. [Google Scholar] [CrossRef]
- Li, G.-B.; Yang, L.-L.; Wang, W.-J.; Li, L.-L.; Yang, S.-Y. ID-Score: A New Empirical Scoring Function Based on a Comprehensive Set of Descriptors Related to Protein–Ligand Interactions. J. Chem. Inf. Modeling 2013, 53, 592–600. [Google Scholar] [CrossRef]
- Rensi, S.E.; Altman, R.B. Shallow Representation Learning via Kernel PCA Improves QSAR Modelability. J. Chem. Inf. Modeling 2017, 57, 1859–1867. [Google Scholar] [CrossRef]
- Hartenfeller, M.; Zettl, H.; Walter, M.; Rupp, M.; Reisen, F.; Proschak, E.; Weggen, S.; Stark, H.; Schneider, G. DOGS: Reaction-Driven de Novo Design of Bioactive Compounds. PLoS Comput. Biol. 2012, 8, e1002380. [Google Scholar] [CrossRef]
- Merk, D.; Friedrich, L.; Grisoni, F.; Schneider, G. De Novo Design of Bioactive Small Molecules by Artificial Intelligence. Mol. Inform. 2018, 37, 1700153. [Google Scholar] [CrossRef]
- Napolitano, F.; Zhao, Y.; Moreira, V.M.; Tagliaferri, R.; Kere, J.; D’Amato, M.; Greco, D. Drug Repositioning: A Machine-Learning Approach through Data Integration. J. Cheminformatics 2013, 5, 30. [Google Scholar] [CrossRef]
- Karlov, D.S.; Sosnin, S.; Tetko, I.V.; Fedorov, M.V. Chemical Space Exploration Guided by Deep Neural Networks. RSC Adv. 2019, 9, 5151–5157. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of Machine Learning in Drug Discovery and Development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Burki, T. A New Paradigm for Drug Development. Lancet Digit. Health 2020, 2, e226–e227. [Google Scholar] [CrossRef]
- The World Federation of Neurology. Platform Communications: Abstract Book—30th International Symposium on ALS/MND. Amyotroph. Lateral Scler. Front. Degener. 2019, 20 (Suppl. S1), 1–99. [Google Scholar] [CrossRef] [PubMed]
- Stopford, M.; Myszczynska, M.; Markus, N.; Sheppard, D.; Richardson, P.; Phelan, A.; Mead, R.; Ferraiuolo, L. C29 Harnessing Machine Learning and Artificial Intelligence to Identify Novel ALS Therapeutics. Amyotroph. Lateral Scler. Front. Degener. 2017, 18 (Suppl. S2), 20–21. [Google Scholar] [CrossRef]
- Querin, G.; El Mendili, M.-M.; Bede, P.; Delphine, S.; Lenglet, T.; Marchand-Pauvert, V.; Pradat, P.-F. Multimodal Spinal Cord MRI Offers Accurate Diagnostic Classification in ALS. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1220–1221. [Google Scholar] [CrossRef]
- D’hulst, L.; Van Weehaeghe, D.; Chiò, A.; Calvo, A.; Moglia, C.; Canosa, A.; Cistaro, A.; Willekens, S.M.; De Vocht, J.; Van Damme, P.; et al. Multicenter Validation of (18 F)-FDG PET and Support-Vector Machine Discriminant Analysis in Automatically Classifying Patients with Amyotrophic Lateral Sclerosis versus Controls. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Van der Burgh, H.K.; Schmidt, R.; Westeneng, H.-J.; de Reus, M.A.; van den Berg, L.H.; van den Heuvel, M.P. Deep Learning Predictions of Survival Based on MRI in Amyotrophic Lateral Sclerosis. NeuroImage Clin. 2017, 13, 361–369. [Google Scholar] [CrossRef]
- Prykhodko, O.; Johansson, S.V.; Kotsias, P.-C.; Arús-Pous, J.; Bjerrum, E.J.; Engkvist, O.; Chen, H. A de Novo Molecular Generation Method Using Latent Vector Based Generative Adversarial Network. J. Cheminformatics 2019, 11, 74. [Google Scholar] [CrossRef]
- Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. DruGAN: An Advanced Generative Adversarial Autoencoder Model for de Novo Generation of New Molecules with Desired Molecular Properties in Silico. Mol. Pharm. 2017, 14, 3098–3104. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep Reinforcement Learning for de Novo Drug Design. Sci. Adv. 2018. [Google Scholar] [CrossRef] [PubMed]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep Drug–Target Binding Affinity Prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef] [PubMed]
- Zitnik, M.; Agrawal, M.; Leskovec, J. Modeling Polypharmacy Side Effects with Graph Convolutional Networks. Bioinformatics 2018, 34, i457–i466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xiao, C.; Glass, L.M.; Sun, J. DeepEnroll: Patient-Trial Matching with Deep Embedding and Entailment Prediction. arXiv 2020, arXiv:2001.08179. [Google Scholar]
- Ismail, H.M.; Dorchies, O.M.; Scapozza, L. The Potential and Benefits of Repurposing Existing Drugs to Treat Rare Muscular Dystrophies. Expert Opin. Orphan Drugs 2018, 6, 259–271. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name|Structure|DrugBank ID | Mechanism of Action | Route of Administration | Clinical Trial Stages | Effect of Drugs | Type of SMA | Sponsor |
|---|---|---|---|---|---|---|
Risdiplam (RG7916) * DrugBank ID: DB15305 | SMN2 splicing modifier | Oral (daily; through a g-tube) | II (JEWELFISH, RAINBOWFISH) | Two-fold increment in SMN protein concentration after 12 weeks of therapy | All types of SMA | Hoffmann-La Roche, PTC Therapeutics, SMA Foundation |
Branaplam (LMI070, NVS-SM1) DrugBank ID: DB14918 | SMN2 splicing modifier | Oral | II | N/A | Type I | Novartis |
Reldesemtiv (CK-2127107; 2-aminoalkyl-5-N-heteroarylpyrimidine) DrugBank ID: DB15256 | Fast skeletal muscle troponin activator (FSTA) | Oral | II | Mild improvement in the six-minute walk test (6MWT) after 4 and 8 weeks of treatment | Type II/III/IV | Cytokinetics, Astellas |
| SRK-015# | Myostatin inhibitor | Intravenous (IV) injection | II (TOPAZ) | Positive results in animal model | Type II/III | Scholar Rock |
| Category | Database | URL | Reference |
|---|---|---|---|
| Drug and target database | Binding Database (BindingDB) | https://www.bindingdb.org/bind/index.jsp (accessed on 29 December 2020) | [132,133] |
| Biological General Repository for Interaction Datasets (BioGRID) | https://thebiogrid.org/ (accessed on 29 December 2020) | [134,135] | |
| ChEMBL | https://www.ebi.ac.uk/chembl/ (accessed on 29 December 2020) | [136,137] | |
| ChemDB | http://chemdb.ics.uci.edu/ (accessed on 29 December 2020) | [138] | |
| ChemSpider | http://www.chemspider.com/ (accessed on 29 December 2020) | [139] | |
| Connectivity Map (CMap) | https://portals.broadinstitute.org/cmap/ (accessed on 29 December 2020) | [140,141] | |
| Database of Interacting Proteins (DIP) | https://dip.doe-mbi.ucla.edu/dip/Main.cgi (accessed on 29 December 2020) | [142,143] | |
| Drug Repurposing Hub | https://clue.io/repurposing (accessed on 29 December 2020) (accessed on 29 December 2020) | [144] | |
| DrugBank | http://www.drugbank.ca/ (accessed on 29 December 2020) | [145] | |
| DrugCentral | http://drugcentral.org (accessed on 29 December 2020) | [146] | |
| Exascale Compound Activity Prediction Engine (ExCAPE-DB) | https://solr.ideaconsult.net/search/excape/ (accessed on 29 December 2020) | [147] | |
| GPS-Prot | http://gpsprot.org/ (accessed on 29 December 2020) | [148] | |
| Human Protein Reference Database (HPRD) | http://www.hprd.org/ (accessed on 29 December 2020) | [149,150] | |
| Library of Integrated Network-based Cellular Signatures (LINCS) | https://lincs.hms.harvard.edu/db/ (accessed on 29 December 2020) | [151] | |
| Molecular INTeraction database (MINT) | https://mint.bio.uniroma2.it/ (accessed on 29 December 2020) | [152,153] | |
| Proteopedia | http://proteopedia.org (accessed on 29 December 2020) | [154] | |
| PubChem | http://pubchem.ncbi.nlm.nih.gov (accessed on 29 December 2020) | [155] | |
| RCSB Protein Data Bank (PDB) | https://www.rcsb.org/ (accessed on 29 January 2021) | [156] | |
| Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) | https://string-db.org/ (accessed on 29 December 2020) | [157] | |
| Structures of Well-curated Extracts, Existing Therapies, and Legally regulated Entities for Accelerated Discovery (SWEETLEAD) | https://simtk.org/projects/sweetlead (accessed on 29 December 2020) | [158] | |
| SuperDRUG2 | http://cheminfo.charite.de/superdrug2/ (accessed on 29 December 2020) | [159] | |
| The NCGC Pharmaceutical Collection (NPC) | https://tripod.nih.gov/npc/ (accessed on 29 December 2020) | [160] | |
| The Universal Protein Resource (UniProt) | https://www.uniprot.org/ (accessed on 29 December 2020) | [161] | |
| ZINC | https://zinc.docking.org/ (accessed on 29 December 2020) | [162] | |
| Pathway omics data | Kyoto Encyclopedia of Genes and Genomes (KEGG) | https://www.genome.jp/kegg/ (accessed on 29 December 2020) | [163,164] |
| Mode of Action by NeTwoRk Analysis (MANTRA) | https://mantra.tigem.it/ (accessed on 29 December 2020) | [165,166] | |
| PathwayCommons | https://www.pathwaycommons.org/ (accessed on 29 December 2020) | [167,168] | |
| Reactome | https://reactome.org/ (accessed on 29 December 2020) | [169] | |
| Genomics data | ArrayExpress | https://www.ebi.ac.uk/arrayexpress/ (accessed on 29 December 2020) | [170] |
| GenBank | http://www.ncbi.nlm.nih.gov (accessed on 29 December 2020) | [171] | |
| Gene Expression Omnibus (NCBI-GEO) | http://www.ncbi.nlm.nih.gov/geo/ (accessed on 29 December 2020) | [172] | |
| Pharmacogene Variation (PharmVar) | https://www.pharmvar.org/ (accessed on 29 December 2020) | [173] | |
| Sequence Read Archive (SRA) | https://trace.ncbi.nlm.nih.gov/Traces/sra/ (accessed on 29 December 2020) | [174] | |
| Single Nucleotide Polymorphism database (dbSNP) | https://www.ncbi.nlm.nih.gov/snp/ (accessed on 29 December 2020) | [175] | |
| Clinical and disease information | ClinicalTrials | https://clinicaltrials.gov/ (accessed on 29 December 2020) | - |
| Comparative Toxicogenomics Database (CTD) | http://ctdbase.org/ (accessed on 29 December 2020) | [176] | |
| DisGeNET | https://www.disgenet.org/ (accessed on 29 December 2020) | [177] | |
| Drugs@FDA | https://www.accessdata.fda.gov/scripts/cder/daf/ (accessed on 29 December 2020) | - | |
| Genome-wide Association Studies (GWAS Catalog) | https://www.ebi.ac.uk/gwas/ (accessed on 29 December 2020) | [178] | |
| FDA Adverse Event Reporting System (FAERS) | https://open.fda.gov/data/faers/ (accessed on 29 December 2020) | [179] | |
| Online Mendelian in Man (OMIM) | https://www.ncbi.nlm.nih.gov/omim (accessed on 28 August 2020) | [180] | |
| OpenTargets | https://www.opentargets.org/ (accessed on 29 December 2020) | [181] | |
| Pharmacogenomics Knowledgebase (PharmGKB) | https://www.pharmgkb.org/ (accessed on 29 December 2020) | [182] | |
| Side Effect Resource (SIDER) | http://sideeffects.embl.de/ (accessed on 29 December 2020) | [183] | |
| Therapeutic Target Database (TTD) | http://db.idrblab.net/ttd/ (accessed on 29 December 2020) | [184] | |
| Rare disease and orphan drugs | eRAM | http://www.unimd.org/eram/ (accessed on 29 December 2020) | [185] |
| Orphanet (Oprhadata and Oprhanet Rare Disease Ontology (ORDO)) | http://www.orpha.net (accessed on 29 December 2020) | - |
| Method | Approach | Required Data | Software Tools (Tool Name|Tool URL) | |
|---|---|---|---|---|
| Drug-oriented | In silico screening | Protein 3D structure, chemical structure, chemical information (targets and ligands) | Protein structure prediction tools | |
| I-TASSER [199] | https://zhanglab.ccmb.med.umich.edu/I-TASSER/ (accessed on 29 December 2020) | |||
| Modeller [200] | https://salilab.org/modeller/ (accessed on 29 December 2020) | |||
| transform-restrained Rosetta [201] | http://robetta.bakerlab.org/ (accessed on 29 December 2020) | |||
| Docking | ||||
| Ligand based screening and molecular docking | AutoDock [202] | http://autodock.scripps.edu/ (accessed on 29 December 2020) | ||
| AutoDock Vina [203] | http://vina.scripps.edu/ (accessed on 29 December 2020) | |||
| High Ambiguity Driven protein-protein DOCKing (HADDOCK [204]) | https://wenmr.science.uu.nl/haddock2.4/ (accessed on 29 December 2020) | |||
| PatchDock [205] | https://bioinfo3d.cs.tau.ac.il/PatchDock/ (accessed on 29 December 2020) | |||
| Pharmacophore mapping and inverse virtual docking (IVD) programs | ||||
| Fragment-based screening | BIOVIA Discovery Studio | https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 29 December 2020) | ||
| INVDOCK [206] | http://bidd.group/group/softwares/invdock.htm (accessed on 29 December 2020) | |||
| LigandScout [207] | http://www.inteligand.com/ligandscout/ (accessed on 29 December 2020) | |||
| PharmMap | http://www.meilerlab.org/index.php/research/show?w_text_id=32 (accessed on 29 December 2020) | |||
| PharmMapper [208,209] | http://www.lilab-ecust.cn/pharmmapper/ (accessed on 29 December 2020) | |||
| ZINCPharmer [210] | http://zincpharmer.csb.pitt.edu/ (accessed on 29 December 2020) | |||
| Drug similarity studies | Chemical structure, chemical information of drugs, clinical trial information, side effects and adverse events, FDA approval labels | Drug-drug similarities prediction and visualization | ||
| ChemMine Tools [211] | http://chemmine.ucr.edu/ (accessed on 29 December 2020) | |||
| ChemTreeMap [212] | https://chemtreemap.readthedocs.io/en/latest/ (accessed on 29 December 2020) | |||
| Compound Specific bioActivity DENdrogram (C-SPACE [213]) | http://cspade.fimm.fi/ (accessed on 29 December 2020) | |||
| Drug-drug similarities and drug-target interaction prediction | ||||
| SuperPred [214] | https://prediction.charite.de/ (accessed on 29 December 2020) | |||
| Disease-/therapy-oriented | Signature-based drug repurposing | Gene signatures information, disease/genetics data, drug omics data | Signature-based drug repurposing tool | |
| Cogena [215] | http://bioconductor.org/packages/release/bioc/html/cogena.html (accessed on 29 December 2020) | |||
| ksRepo [216] | https://github.com/adam-sam-brown/ksRepo (accessed on 29 December 2020) | |||
| DrugSig [217] | http://biotechlab.fudan.edu.cn/database/drugsig/ (accessed on 29 December 2020) | |||
| Pathway-/network-based drug repurposing | General drug information, pathway information | Network-based drug repurposing tool | ||
| Drug Repurposing Recommendation System (DRRS [218]) | http://bioinformatics.csu.edu.cn/resources/softs/DrugRepositioning/DRRS/index.html (accessed on 29 December 2020) | |||
| DrugNet [219] | http://genome.ugr.es:9000/drugnet (accessed on 29 December 2020) | |||
| GeneDiseaseRepositioning [220] | https://bitbucket.org/ncl-intbio/genediseaserepositioning/src/master/ (accessed on 29 December 2020) | |||
| Predicting Drugs having Opposite effects on Disease genes (PDOD [221]) | http://gto.kaist.ac.kr/pdod/index.php/main (accessed on 29 December 2020) | |||
| Targeted mechanism-based drug repurposing | Network visualization | |||
| Cytoscape [222] | https://cytoscape.org/ (accessed on 29 December 2020) | |||
| GeneMANIA [223] | https://genemania.org/ (accessed on 29 December 2020) | |||
| Pathway Studio [224] | https://www.pathwaystudio.com/ (accessed on 29 December 2020) | |||
| PATIKAweb [225] | http://www.cs.bilkent.edu.tr/~patikaweb/ (accessed on 29 December 2020) | |||
| VisANT [226] | http://www.visantnet.org/visantnet.html (accessed on 29 December 2020) | |||
| Machine Learning Methods | Area of Drug Development | Reference |
|---|---|---|
| Design and Discovery and Preclinical Research
| [261,262,263] |
| Clinical Research and Safety Monitoring
| [264,265,266] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chong, L.C.; Gandhi, G.; Lee, J.M.; Yeo, W.W.Y.; Choi, S.-B. Drug Discovery of Spinal Muscular Atrophy (SMA) from the Computational Perspective: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 8962. https://doi.org/10.3390/ijms22168962
Chong LC, Gandhi G, Lee JM, Yeo WWY, Choi S-B. Drug Discovery of Spinal Muscular Atrophy (SMA) from the Computational Perspective: A Comprehensive Review. International Journal of Molecular Sciences. 2021; 22(16):8962. https://doi.org/10.3390/ijms22168962
Chicago/Turabian StyleChong, Li Chuin, Gayatri Gandhi, Jian Ming Lee, Wendy Wai Yeng Yeo, and Sy-Bing Choi. 2021. "Drug Discovery of Spinal Muscular Atrophy (SMA) from the Computational Perspective: A Comprehensive Review" International Journal of Molecular Sciences 22, no. 16: 8962. https://doi.org/10.3390/ijms22168962
APA StyleChong, L. C., Gandhi, G., Lee, J. M., Yeo, W. W. Y., & Choi, S.-B. (2021). Drug Discovery of Spinal Muscular Atrophy (SMA) from the Computational Perspective: A Comprehensive Review. International Journal of Molecular Sciences, 22(16), 8962. https://doi.org/10.3390/ijms22168962