
CAR T-Cell Therapy in Hematological Malignancies


{kind=link}
Abstract
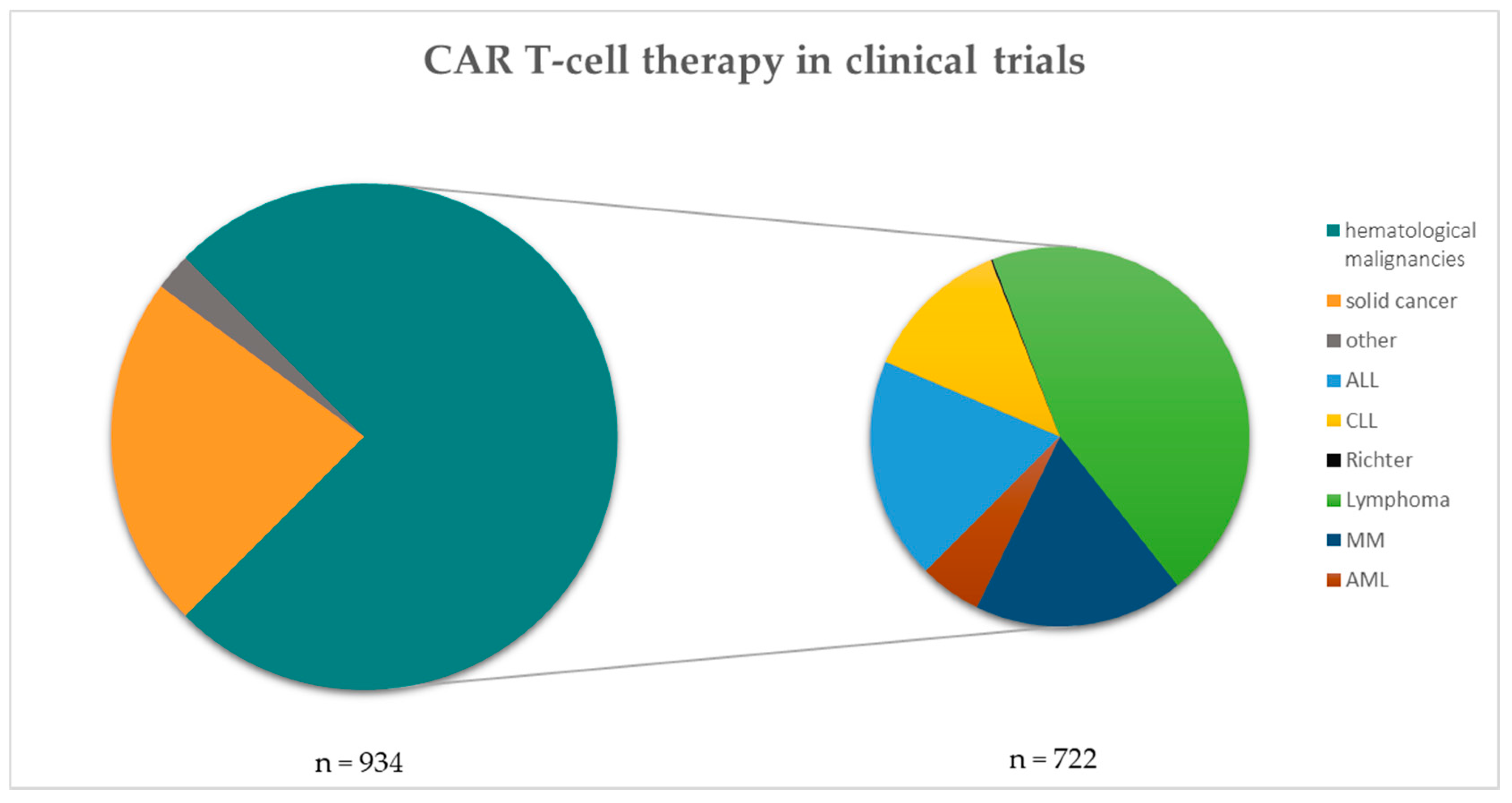
:1. Introduction
2. CAR T-Cell Therapy in Hematologic Malignant Neoplasms
2.1. CAR T-Cell Therapy in Acute Lymphoblastic Leukemia
2.2. CAR T-Cell Therapy in Chronic Lymphocytic Leukemia
CAR T-Cell Therapy in RICHTER’S Syndrome
2.3. CAR T-Cell Therapy in Lymphoma
2.4. CAR T-Cell Therapy in Multiple Myeloma
2.5. CAR T-Cell Therapy in Acute Myeloid Leukemia
3. Overcoming CAR T-Cell Dysfunction
4. CAR T-Cell Therapy and Combination Therapies
4.1. Monoclonal Antibodies
4.2. Small Molecule Inhibitors
4.3. Oncolytic Viruses
4.4. Proinflammatory Cytokines
5. Adverse Events of CAR T-Cell Therapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALL | acute lymphocytic leukemia |
| AML | acute myeloid leukemia |
| BCMA | B-cell maturation antigen |
| CAR | chimeric antigen receptor |
| CLL | chronic lymphocytic leukemia |
| CRS | cytokine release storm |
| CSR | chimeric switch receptors |
| DLBCL | diffuse large B-cell lymphoma |
| DNR | dominant negative receptor |
| FDA | food and drug administration |
| HL | Hodgkin lymphoma |
| HSCT | hematopoietic stem cell transplant |
| HSPCs | hematopoietic stem and progenitor cells |
| MAPK | mitogen-activated protein kinase |
| MCL | mantle cell lymphoma |
| MHC | major histocompatibility complex |
| MM | multiple myeloma |
| NHL | Non-Hodgkin lymphomas |
| scFv | single-chain variable fragment |
| TAA | tumor-associated antigens |
| TanCAR | tandem CAR |
| T-cells | T-lymphocytes |
| TCR | T-cell receptor |
| TILs | tumor infiltrating lymphocytes |
| TME | tumor microenvironment |
| TRUCKS | T-cells redirected for universal cytokine killing |
| VEGF-A | vascular endothelial growth factor A |
References
- Quezada, S.; Peggs, K.S.; Simpson, T.R.; Allison, J.P. Shifting the equilibrium in cancer immunoediting: From tumor tolerance to eradication. Immunol. Rev. 2011, 241, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Houot, R.; Schultz, L.; Marabelle, A.; Kohrt, H. T-cell–based Immunotherapy: Adoptive Cell Transfer and Checkpoint Inhibition. Cancer Immunol. Res. 2015, 3, 1115–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miliotou, A.; Papadopoulou, L. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tariq, S.M.; Haider, S.A.; Hasan, M.; Tahir, A.; Khan, M.; Rehan, A.; Kamal, A. Chimeric Antigen Receptor T-Cell Therapy: A Beacon of Hope in the Fight Against Cancer. Cureus 2018, 10, e3486. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Riddell, S.R. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015, 36, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.L. Performance-enhancing drugs: Design and production of redirected chimeric antigen receptor (CAR) T cells. Cancer Gene Ther. 2015, 22, 79–84. [Google Scholar] [CrossRef]
- Mueller, K.T.; Maude, S.L.; Porter, D.L.; Frey, N.; Wood, P.; Han, X.; Waldron, E.; Chakraborty, A.; Awasthi, R.; Levine, B.L.; et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 2017, 130, 2317–2325. [Google Scholar] [CrossRef]
- Landoni, E.; Savoldo, B. Treating hematological malignancies with cell therapy: Where are we now? Expert Opin. Biol. Ther. 2017, 18, 65–75. [Google Scholar] [CrossRef]
- FDA. Anna Kwilas, PhD, Chair of the Review Committee, OTAT/DCGT. Summary Basis for Regulatory Action—Abecma, 26 March 2021. Available online: https://www.fda.gov/vaccines-blood-biologics/abecma-idecabtagene-vicleucel (accessed on 10 June 2021).
- Fiorenza, S.; Turtle, C.J. CAR-T Cell Therapy for Acute Myeloid Leukemia: Preclinical Rationale, Current Clinical Progress, and Barriers to Success. BioDrugs 2021, 35, 281–302. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [Green Version]
- NIH U.S. National library of Medicine. National library of Medicine. “ClinicalTrials.gov,” NIH U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/results?term=CAR&cond=Multiple+Myeloma&Search=Apply&recrs=b&recrs=a&recrs=f&recrs=d&recrs=e&age_v=&gndr=&type=&rslt= (accessed on 27 May 2021).
- Onciu, M. Acute lymphoblastic leukemia. Hematol. Oncol. Clin. N. Am. 2009, 23, 655–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Beldjord, K.; Chevret, S.; Asnafi, V.; Huguet, F.; Boulland, M.L.; Leguay, T.; Thomas, X.; Cayuela, J.M.; Grardel, N.; Cha-lan-don, Y.; et al. Oncogenetics and minimal residual disease are independent outcome predictors in adult pa-tients with acute lymphoblastic leukemia. Blood J. Am. Soc. Hematol. 2014, 123, 3739–3749. [Google Scholar]
- Paul, S.; Rausch, C.R.; Nasnas, P.E.; Kantarjian, H.; Jabbour, E.J. Treatment of relapsed/refractory acute lym-pho-blastic leukemia. Clin. Adv. Hematol. Oncol. 2019, 17, 166–175. [Google Scholar]
- Hunger, S.P.; Raetz, E.A. How I treat relapsed acute lymphoblastic leukemia in the pediatric population. Blood 2020, 136, 1803–1812. [Google Scholar] [CrossRef] [PubMed]
- FDA. Xiaobin Victor Lu, Ph.D., Chair of the Review Committee, Summary Basis for Regulatory Action—Kymriah, 30 August 2017. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/kymriah-tisagenlecleucel (accessed on 10 June 2021).
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.-A.N.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhao, L.; Zhang, Y.; Qin, Y.; Guan, Y.; Zhang, T.; Liu, C.; Zhou, J. Understanding the Mechanisms of Resistance to CAR T-Cell Therapy in Malignancies. Front. Oncol. 2019, 9, 1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NIH U.S. National library of Medicine. “ClinicalTrials.gov,” NIH U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/results?term=CAR+T+cell+therapy&cond=Acute+Lymphoid+Leukemia&Search=Clear&age_v=&gndr=&type=&rslt= (accessed on 4 June 2021).
- Rodríguez-Vicente, A.E.; Díaz, M.G.; Hernández-Rivas, J.M. Chronic lymphocytic leukemia: A clinical and molecular heterogenous disease. Cancer Genet. 2013, 206, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef] [Green Version]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood J. Am. Soc. Hematol. 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, M.B.; Rivière, I.; Sénéchal, B.; Wang, X.; Wang, Y.; Purdon, T.J.; Hsu, M.; Devlin, S.M.; Palomba, M.L.; Halton, E.; et al. Safety and tolerability of conditioning chemotherapy followed by CD19-targeted CAR T cells for re-lapsed/refractory CLL. JCI Insight 2019, 4, e122627. [Google Scholar] [CrossRef]
- Frey, N.V.; Gill, S.; Hexner, E.O.; Schuster, S.; Nasta, S.; Loren, A.; Svoboda, J.; Stadtmauer, E.; Landsburg, D.J.; Mato, A.; et al. Long-term outcomes from a randomized dose optimization study of chimeric antigen receptor modi-fied T cells in relapsed chronic lymphocytic leukemia. J. Clin. Oncol. 2020, 38, 2862–2871. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.B.; Rivière, I.; Sénéchal, B.; Wang, X.; Wang, Y.; Purdon, T.J.; Hsu, M.; Devlin, S.M.; Halton, E.; Lamanna, N.; et al. Autologous CD19-targeted CAR T cells in patients with residual CLL following initial purine ana-log-based therapy. Mol. Ther. 2018, 26, 1896–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, P.; O’Brien, S. Richter’s transformation in chronic lymphocytic leukemia. Oncology 2012, 26, 1146. [Google Scholar]
- Parikh, S.A.; Kay, N.E.; Shanafelt, T.D. How we treat Richter syndrome. Blood J. Am. Soc. Hematol. 2014, 123, 1647–1657. [Google Scholar] [CrossRef]
- Kittai, A.S.; Bond, D.A.; William, B.; Saad, A.; Penza, S.; Efebera, Y.; Larkin, K.; Wall, S.A.; Choe, H.K.; Bhatnagar, B.; et al. Clinical activity of axicabtagene ciloleucel in adult patients with Richter syndrome. Blood Adv. 2020, 4, 4648–4652. [Google Scholar] [CrossRef]
- Li, S.; Young, K.H.; Medeiros, L.J. Diffuse large B-cell lymphoma. Pathology 2018, 50, 74–87. [Google Scholar] [CrossRef] [Green Version]
- FDA. Michael Havert, PhD, Chair of the Review Committee, Summary Basis for Regulatory Action—Yescarta, 18 October 2017. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/yescarta-axicabtagene-ciloleucel (accessed on 10 June 2021).
- Viardot, A.; Wais, V.; Sala, E.; Koerper, S. Chimeric antigen receptor (CAR) T-cell therapy as a treatment option for patients with B-cell lymphomas: Perspectives on the therapeutic potential of Axicabtagene ciloleucel. Cancer Manag. Res. 2019, 11, 2393–2404. [Google Scholar] [CrossRef] [Green Version]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Tim-merman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lym-pho-ma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- FDA. Kimberly LW Schultz, PhD, Chair of the Review Committee, Summary Basis for Regulatory Action Template—Breyanzi, 5 Feburary 2021. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/breyanzi-lisocabtagene-maraleucel (accessed on 10 June 2021).
- FDA. Graeme Price PhD, Chair of the Review Committee, Summary Basis for Regulatory Action—Tecartus, 23 July 2020. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/tecartus-brexucabtagene-autoleucel (accessed on 10 June 2021).
- Wang, Y.; Zhang, W.-Y.; Han, Q.-W.; Liu, Y.; Dai, H.-R.; Guo, Y.-L.; Bo, J.; Fan, H.; Zhang, Y.; Zhang, Y.-J.; et al. Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T cells. Clin. Immunol. 2014, 155, 160–175. [Google Scholar] [CrossRef]
- Wang, C.-M.; Wu, Z.-Q.; Wang, Y.; Guo, Y.-L.; Dai, H.-R.; Wang, X.-H.; Li, X.; Zhang, Y.-J.; Zhang, W.-Y.; Chen, M.-X.; et al. Autologous T Cells Expressing CD30 Chimeric Antigen Receptors for Relapsed or Refractory Hodgkin Lymphoma: An Open-Label Phase I Trial. Clin. Cancer Res. 2016, 23, 1156–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, J.C.; Bachmeier, C.; Kharfan-Dabaja, M.A. CAR T-cell therapy for B-cell lymphomas: Clinical trial results of available products. Ther. Adv. Hematol. 2019, 10, 2040620719841581. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell Maturation Antigen Is a Promising Target for Adoptive T-cell Therapy of Multiple Myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.K.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Zeldenrust, S.R.; Dingli, D.; Russell, S.J.; Lust, J.A.; et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008, 111, 2516–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanackovic, D.; Radhakrishnan, S.V.; Bhardwaj, N.; Luetkens, T. Chimeric Antigen Receptor (CAR) therapy for multiple myeloma. Br. J. Haematol. 2016, 172, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Chen, M.; Han, Q.; Hui, F.; Dai, H.; Zhang, W.; Zhang, Y.; Wang, Y.; Zhu, H.; Han, W. CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma. J. Cell. Immunother. 2016, 2, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Murati, A.; Brecqueville, M.; Devillier, R.; Mozziconacci, M.J.; Gelsi-Boyer, V.; Birnbaum, D. Myeloid malignancies: Mutations, models and management. BMC Cancer 2012, 12, 304. [Google Scholar] [CrossRef] [Green Version]
- Dinardo, C.D.; Wei, A.H. How I treat acute myeloid leukemia in the era of new drugs. Blood 2020, 135, 85–96. [Google Scholar] [CrossRef]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Hönemann, D.; et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leuke-mia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehninger, A.; Kramer, M.; Röllig, C.; Thiede, C.; Bornhäuser, M.; Von Bonin, M.; Wermke, M.; Feldmann, A.; Bachmann, M.; Ehninger, G.; et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute mye-loid leukemia. Blood Cancer J. 2014, 4, e218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haubner, S.; Perna, F.; Köhnke, T.; Schmidt, C.; Berman, S.; Augsberger, C.; Schnorfeil, F.M.; Krupka, C.; Lichtenegger, F.S.; Liu, X.; et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia 2018, 33, 64–74. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2018, 7, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Martyniszyn, A.; Krahl, A.-C.; André, M.C.; Hombach, A.A.; Abken, H. CD20-CD19 Bispecific CAR T Cells for the Treatment of B-Cell Malignancies. Hum. Gene Ther. 2017, 28, 1147–1157. [Google Scholar] [CrossRef]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef]
- Condomines, M.; Arnason, J.; Benjamin, R.; Gunset, G.; Plotkin, J.; Sadelain, M. Tumor-Targeted Human T Cells Expressing CD28-Based Chimeric Antigen Receptors Circumvent CTLA-4 Inhibition. PLoS ONE 2015, 10, e0130518. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pule, M.; Straathof, K.C.; Dotti, G.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. 2005, 12, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Song, D.G.; Ye, Q.; Poussin, M.; Harms, G.M.; Figini, M.; Powell, D.J., Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood J. Am. Soc. Hematol. 2012, 119, 696–706. [Google Scholar] [CrossRef]
- Frigault, M.J.; Lee, J.; Basil, M.C.; Carpenito, C.; Motohashi, S.; Scholler, J.; Kawalekar, O.U.; Guedan, S.; McGettigan, S.; Posey, J.A.; et al. Identification of Chimeric Antigen Receptors That Mediate Constitutive or Inducible Proliferation of T Cells. Cancer Immunol. Res. 2015, 3, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv. Cell Gene Ther. 2020, 3, e84. [Google Scholar] [CrossRef]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef] [Green Version]
- Gholami, M.D.; Saeedi, Y.; Heydari, S.; Garssen, J.; Falak, R. Exhaustion of T lymphocytes in the tumor microenvironment: Significance and effective mechanisms. Cell. Immunol. 2017, 322, 1–14. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mognol, G.P.; Spreafico, R.; Wong, V.; Scott-Browne, J.P.; Togher, S.; Hoffmann, A.; Hogan, P.G.; Rao, A.; Trifari, S. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2776–E2785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Morello, A.; Tano, Z.; Adusumilli, P.S. CAR T-cell intrinsic PD-1 checkpoint blockade: A two-in-one approach for solid tumor immunotherapy. OncoImmunology 2017, 6, e1273302. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Lei, W.; Zhang, C.; Yang, C.; Wei, J.; Guo, Q.; Guo, X.; Chen, Z.; Lu, Y.; Young, K.H.; et al. CD19-specific CAR T Cells that Express a PD-1/CD28 Chimeric Switch-Receptor are Effective in Patients with PD-L1–positive B-Cell Lymphoma. Clin. Cancer Res. 2020, 27, 473–484. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, H.; Lu, Z.; Lei, W.; Zhang, C.; Li, P.; Liang, A.; Young, K.H.; Qian, W. CD19 CAR-T expressing PD-1/CD28 chimeric switch receptor as a salvage therapy for DLBCL patients treated with different CD19-directed CAR T-cell therapies. J. Hematol. Oncol. 2021, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)–modified T cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2015, 103, 26–48. [Google Scholar] [CrossRef]
- Ott, P.A.; Adams, S. Small-molecule protein kinase inhibitors and their effects on the immune system: Implications for cancer treatment. Immunotherapy 2011, 3, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deniger, D.C.; Kwong, M.L.M.; Pasetto, A.; Dudley, M.E.; Wunderlich, J.R.; Langhan, M.M.; Lee, C.-C.; Rosenberg, S.A. A Pilot Trial of the Combination of Vemurafenib with Adoptive Cell Therapy in Patients with Metastatic Melanoma. Clin. Cancer Res. 2016, 23, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Urak, R.; Walter, M.; Lim, L.; Wong, C.W.; Budde, L.E.; Thomas, S.; Forman, S.J.; Wang, X. Ex vivo Akt inhibition promotes the generation of potent CD19CAR T cells for adoptive immunotherapy. J. Immunother. Cancer 2017, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Huye, L.E.; Nakazawa, Y.; Patel, M.P.; Yvon, E.; Sun, J.; Savoldo, B.; Wilson, M.H.; Dotti, G.; Rooney, C.M. Com-bining mTor inhibitors with rapamycin-resistant T cells: A two-pronged approach to tumor elimination. Mol. Ther. 2011, 19, 2239–2248. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, M.S.; Lemoine, N.; Wang, Y. Systemic Delivery of Oncolytic Viruses: Hopes and Hurdles. Adv. Virol. 2012, 2012, 805629. [Google Scholar] [CrossRef] [PubMed]
- Senac, J.S.; Doronin, K.; Russell, S.J.; Jelinek, D.F.; Greipp, P.R.; Barry, M.A. Infection and killing of multiple myeloma by adenoviruses. Hum. Gene Ther. 2010, 21, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed Oncolytic Virus Enhances Immune Functions of Chimeric Antigen Receptor–Modified T Cells in Solid Tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic mela-noma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Kaartinen, T.; Luostarinen, A.; Maliniemi, P.; Keto, J.; Arvas, M.; Belt, H.; Koponen, J.; Loskog, A.; Mustjoki, S.; Porkka, K.; et al. Low interleukin-2 concentration favors generation of early memory T cells over effector pheno-types during chimeric antigen receptor T-cell expansion. Cytotherapy 2017, 19, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood J. Am. Soc. Hematol. 2014, 124, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.; Frey, N.; Wood, P.A.; Weng, Y.; Grupp, S.A. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J. Hematol. Oncol. 2018, 11, 35. [Google Scholar] [CrossRef]
- Adkins, S. CAR T-cell therapy: Adverse events and management. J. Adv. Pract. Oncol. 2019, 10 (Suppl. 3), 21. [Google Scholar]
- Howard, S.C.; Jones, D.P.; Pui, C.H. The tumor lysis syndrome. N. Engl. J. Med. 2011, 364, 1844–1845. [Google Scholar] [CrossRef]
- Mei, H.; Jiang, H.; Wu, Y.; Guo, T.; Xia, L.; Jin, R.; Hu, Y. Neurological toxicities and coagulation disorders in the cytokine release syndrome during CAR-T therapy. Br. J. Haematol. 2017, 181, 689–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhao, Y. Increasing the safety and efficacy of chimeric antigen receptor T cell therapy. Protein Cell 2017, 8, 573–589. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haslauer, T.; Greil, R.; Zaborsky, N.; Geisberger, R. CAR T-Cell Therapy in Hematological Malignancies. Int. J. Mol. Sci. 2021, 22, 8996. https://doi.org/10.3390/ijms22168996
Haslauer T, Greil R, Zaborsky N, Geisberger R. CAR T-Cell Therapy in Hematological Malignancies. International Journal of Molecular Sciences. 2021; 22(16):8996. https://doi.org/10.3390/ijms22168996
Chicago/Turabian StyleHaslauer, Theresa, Richard Greil, Nadja Zaborsky, and Roland Geisberger. 2021. "CAR T-Cell Therapy in Hematological Malignancies" International Journal of Molecular Sciences 22, no. 16: 8996. https://doi.org/10.3390/ijms22168996
APA StyleHaslauer, T., Greil, R., Zaborsky, N., & Geisberger, R. (2021). CAR T-Cell Therapy in Hematological Malignancies. International Journal of Molecular Sciences, 22(16), 8996. https://doi.org/10.3390/ijms22168996