
Concussion/Mild Traumatic Brain Injury (TBI) Induces Brain Insulin Resistance: A Positron Emission Tomography (PET) Scanning Study

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
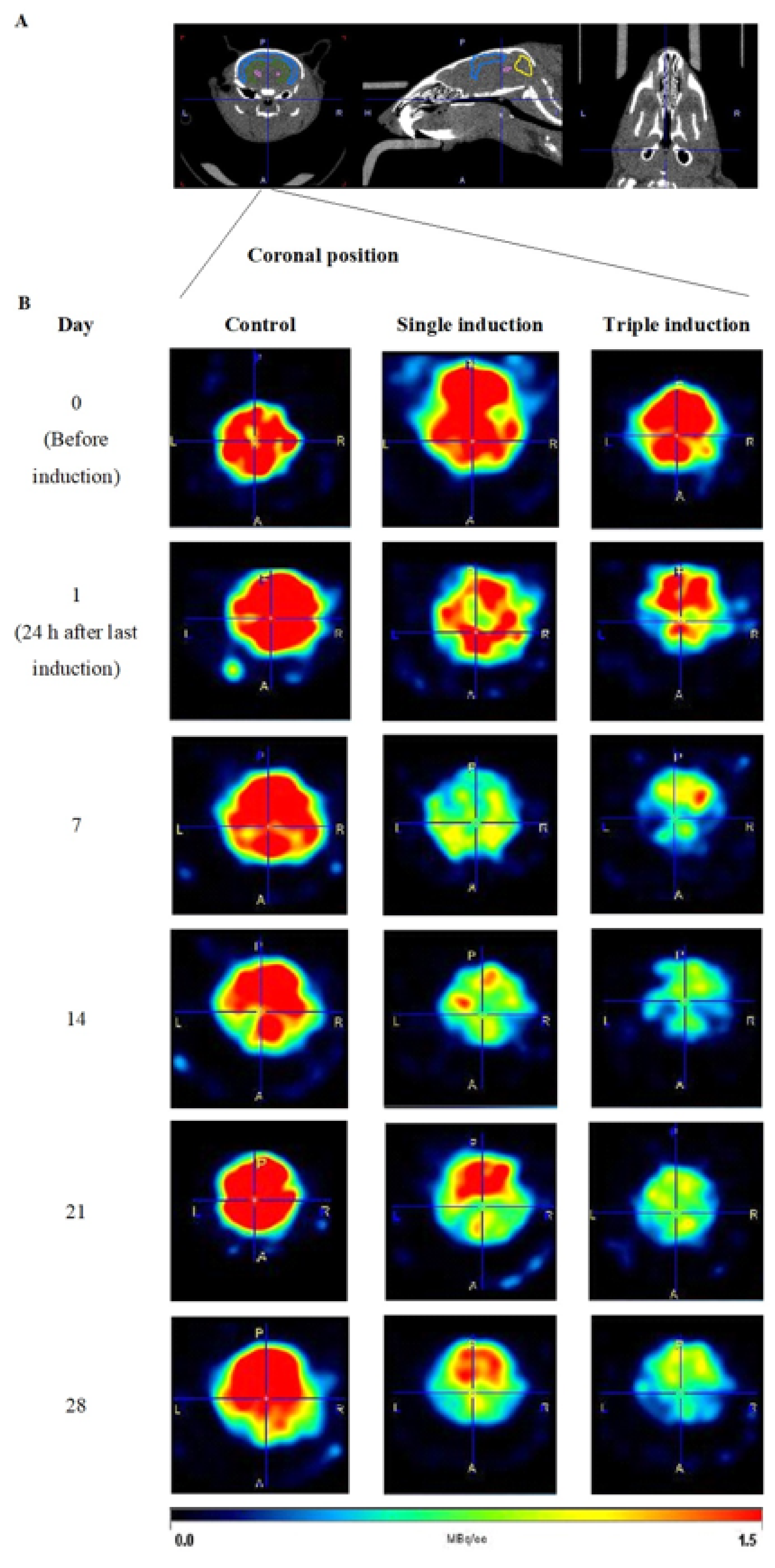
2.1. Determination of Glucose Uptake in TBI Mice Brains Following Single and Repeated Induction
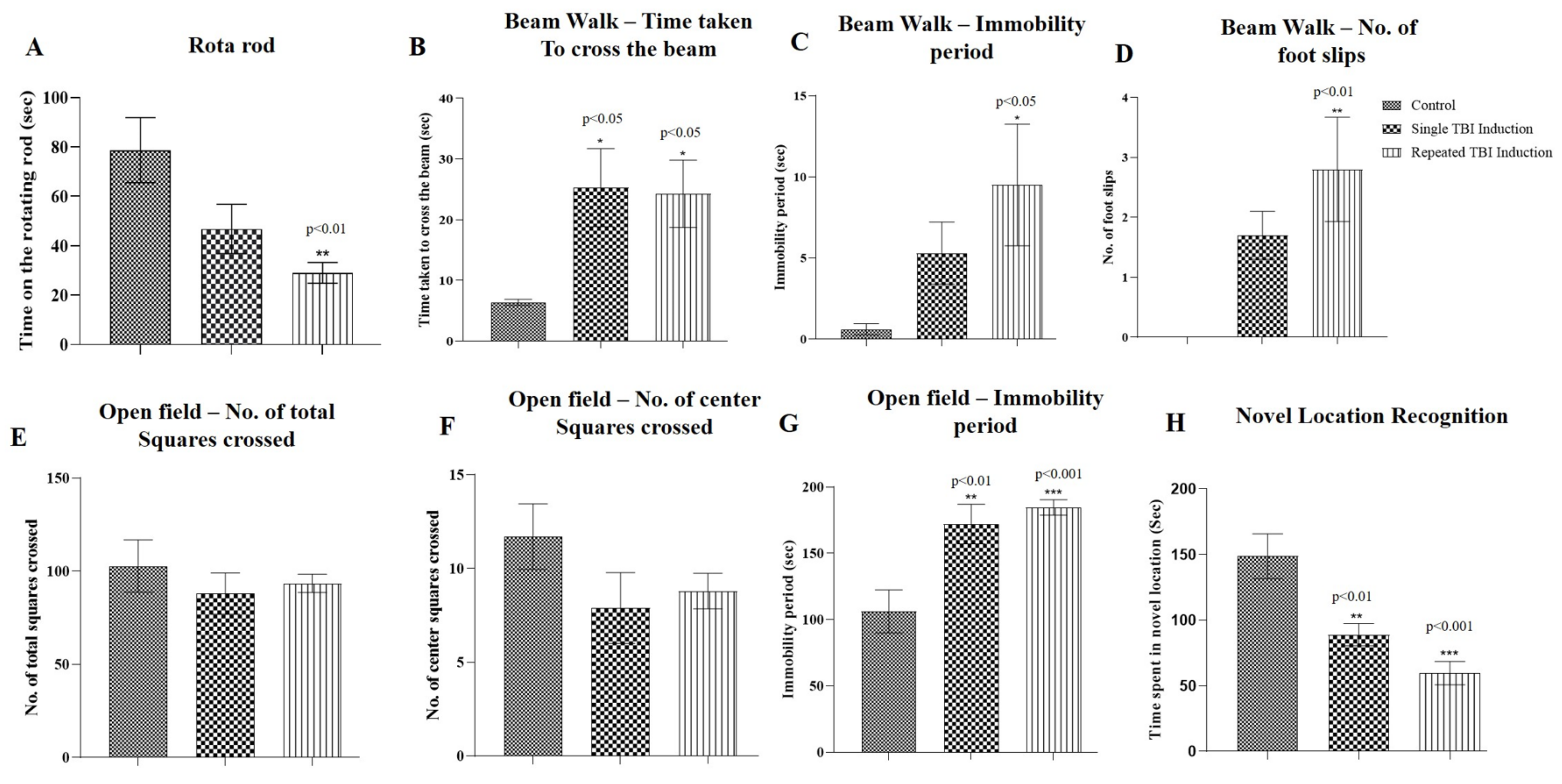
2.2. Determination of Functional Outcomes in TBI Mice following Single and Repeated Induction
2.2.1. Rota Rod Test
2.2.2. Beam Walk Test
2.2.3. Open Field Exploratory Test
2.2.4. Novel Location Recognition Test
2.3. Measurement of Plasma PrPc Level in TBI Mice following Single and Repeated Induction
2.4. Validation of PrPc, Flotillin, CAP, APS and TC10 Proteins in Lipid Raft Region of TBI Mice Brains Following Single and Repeated Induction
2.5. Interaction between the PrPc, Flotillin and CAP in TBI Mice Brains Following Single and Repeated Induction
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Animal Husbandry
4.3. Experimental Design
4.4. Evaluation of Glucose Uptake in Mouse Brains Using MicroPET/CT Imaging
4.5. Motor Function Tests
4.5.1. Rota Rod Test
4.5.2. Beam Walk Test
4.5.3. Open Field Test
4.6. Cognitive Function-Novel Location Recognition Test
4.7. Measurement of Plasma PrPc Level
4.8. Isolation of Lipid Raft
4.9. Co-Immunoprecipitation and Immunoblotting
4.10. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biessels, G.J.; De Leeuw, F.E.; Lindeboom, J.; Barkhof, F.; Scheltens, P. Increased cortical atrophy in patients with Alzheimer′s disease and type 2 diabetes mellitus. J. Neurol. Neurosurg. Psychiatry 2006, 77, 304–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2019, 130, 1039–1408. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Sen, N. Traumatic brain injury: A risk factor for neurodegenerative diseases. Rev. Neurosci. 2016, 27, 93–100. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Ikonomovic, M.D.; Gandy, S. Traumatic brain injury—Football, warfare, and longterm effects. N. Engl. J. Med. 2010, 363, 1293–1296. [Google Scholar] [CrossRef]
- Maroon, J.C.; Winkelman, R.; Bost, J.; Amos, A.; Mathyssek, C.; Miele, V. Chronic traumatic encephalopathy in contact sports: A systematic review of all reported pathological cases. PLoS ONE 2015, 10, e0117338. [Google Scholar]
- McKee, A.C.; Robinson, M.E. Military-related traumatic brain injury and neurodegeneration. Alzheimer′s Dement. 2014, 10, S242–S253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKee, A.C.; Daneshvar, D.H.; Alvarez, V.E.; Stein, T.D. The neuropathology of sport. Acta Neuropathol. 2014, 127, 29–51. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.; Kubilus, C.A.; Stern, R.A. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef]
- Omalu, B.; Bailes, J.; Hamilton, R.L.; Kamboh, M.I.; Hammers, J.; Case, M.; Fitzsimmons, R. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery 2011, 69, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Vincente Miranda, H.; El-Agnaf, O.M.; Outeiro, T.F. Glycation in Parkinson′s disease and Alzheimer′s disease. Mov. Disord. 2016, 31, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Verdile, G.; Fuller, S.J.; Martins, R.N. The role of type 2 diabetes in neurodegeneration. Neurobiol. Dis. 2015, 84, 22–38. [Google Scholar] [CrossRef]
- Sato, N.; Morishita, R. The roles of lipid and glucose metabolism in modulation of betaamyloid, tau, and neurodegeneration in the pathogenesis of Alzheimer disease. Front. Aging Neurosci. 2015, 7, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Monte, S.M. Type 3 diabetes is sporadic Alzheimers disease: Mini-review. Eur. Neuropsychopharmacol. 2014, 24, 1954–1960. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Jousilahti, P.; Bidel, S.; Antikainen, R.; Tuomilehto, J. Type 2 diabetes and the risk of Parkinson′s disease. Diabetes Care 2007, 30, 842–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Park, Y.; Huang, X.; Hollenbeck, A.; Blair, A.; Schatzkin, A.; Chen, H. Diabetes and risk of Parkinson’s disease. Diabetes Care 2011, 34, 910–915. [Google Scholar] [CrossRef] [Green Version]
- Vagelatos, N.T.; Eslick, G.D. Type 2 diabetes as a risk factor for Alzheimer′s disease: The confounders, interactions, and neuropathology associated with this relationship. Epidemiol. Rev. 2013, 35, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Trudeau, F.; Gagnon, S.; Massicotte, G. Hippocampal synaptic plasticity and glutamate receptor regulation: Influences of diabetes mellitus. Eur. J. Pharmacol. 2004, 490, 177–186. [Google Scholar] [CrossRef]
- Karelina, K.; Sarac, B.; Freeman, L.M.; Gaier, K.R.; Weil, Z.M. Traumatic brain injury and obesity induce persistent central insulin resistance. Eur. J. Neurosci. 2016, 43, 1034–1043. [Google Scholar] [CrossRef]
- Greenwood, C.E.; Winocur, G. High-fat diets, insulin resistance and declining cognitive function. Neurobiol. Aging 2005, 26, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Eakin, K.; Li, Y.; Chiang, Y.H.; Hoffer, B.J.; Rosenheim, H.; Greig, N.H.; Miller, J.P. Exendin-4 ameliorates traumatic brain injury-induced cognitive impairment in rats. PLoS ONE 2013, 8, e82016. [Google Scholar]
- Carlson, S.W.; Madathil, S.K.; Sama, D.M.; Gao, X.; Chen, J.; Saatman, K.E. Conditional overexpression of insulin-like growth factor-1 enhances hippocampal neurogenesis and restores immature neuron dendritic processes after traumatic brain injury. J. Neuropathol. Exp. Neurol. 2014, 73, 734–746. [Google Scholar] [CrossRef] [Green Version]
- Alf, M.F.; Wyss, M.T.; Buck, A.; Weber, B.; Schibli, R.; Kramer, S.D. Quantification of brain glucose metabolism by 18F-FDG PET with real-time arterial and image-derived input function in mice. J. Nucl. Med. 2013, 54, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Bergsneider, M.; Hovda, D.A.; McArthur, D.L.; Etchepare, M.; Huang, S.C.; Sehati, N.; Satz, P.; Phelps, M.E.; Becker, D.P. Metabolic recovery following human traumatic brain injury based on FDG-PET: Time course and relationship to neurological disability. J. Head Trauma. Rehabil. 2001, 16, 135–148. [Google Scholar] [CrossRef]
- Selwyn, R.; Hockenbury, N.; Jaiswal, S.; Mathur, S.; Armstrong, R.C.; Byrnes, K.R. Mild traumatic brain injury results in depressed cerebral glucose uptake: An FDG PET study. J. Neurotrauma 2013, 30, 1943–1953. [Google Scholar] [CrossRef]
- Yu, A.S.; Lin, H.D.; Huang, S.C.; Phelps, M.E.; Wu, H.M. Quantification of cerebral glucose metabolic rate in mice using 18F-FDG and small-animal PET. J. Nucl. Med. 2009, 50, 966–973. [Google Scholar] [CrossRef] [Green Version]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O′Connor, R.; Ravid, R.; O′Neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer′s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging 2010, 31, 224–243. [Google Scholar] [CrossRef] [PubMed]
- Meabon, J.S.; Huber, B.R.; Cross, D.J.; Richards, T.L.; Minoshima, S.; Pagulayan, K.F.; Li, G.; Meeker, K.D.; Kraemer, B.C.; Petrie, E.C.; et al. Repetitive blast exposure in mice and combat veterans causes persistent cerebellar dysfunction. Sci. Transl. Med. 2016, 8, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peskind, E.R.; Petrie, E.C.; Cross, D.J.; Pagulayan, K.; McCraw, K.; Hoff, D.; Hart, K.; Yu, C.; Raskind, M.A.; Cook, D.G.; et al. Cerebrocerebellar hypometabolism associated with repetitive blast exposure mild traumatic brain injury in 12 Iraq war Veterans with persistent postconcussive symptoms. Neuroimage 2011, 54, S76–S82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocker, R.P.J.; Cieply, M.A.; Paul, B.; Khan, H.; Henry, L.; Kontos, A.P.; Germain, A. Combat-related blast exposure and traumatic brain injury influence brain glucose metabolism during REM sleep in military veterans. Neuroimage 2014, 99, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, N.; Sawyer, T.W.; Wang, Y.; Jazii, F.R.; Vair, C.; Taghibiglou, C. Primary blast-induced traumatic brain injury in rats leads to increased prion protein in plasma: A potential biomarker for blast-induced traumatic brain injury. J. Neurotrauma 2015, 32, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Pham, N.; Akonasu, H.; Shishkin, R.; Taghibiglou, C. Plasma soluble prion protein, a potential biomarker for sport-related concussions: A pilot study. PLoS ONE 2015, 10, e0117286. [Google Scholar] [CrossRef]
- Sekar, S.; Zhang, Y.; Mahabadi, H.M.; Parvizi, A.; Taghibiglou, C. Low-field magnetic stimulation restores cognitive and motor functions in the mouse model of repeated traumatic brain injury: Role of cellular prion protein. J. Neurotrauma 2019, 36, 3103–3114. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, M.; Pessin, J.E. Insulin signaling: GLUT4 vesicles exit via the exocyst. Cur. Biol. 2003, 13, R574–R576. [Google Scholar] [CrossRef] [Green Version]
- Czech, M.P. Lipid rafts and insulin action. Nature 2000, 407, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Bodrikov, V.; Solis, G.P.; Stuermer, C.A. Prion protein promotes growth cone development through Reggie/Flotillin-Dependent N-Cadherin trafficking. J. Neurosci. 2011, 31, 18013–18025. [Google Scholar] [CrossRef] [PubMed]
- Mahabadi, H.M.; Taghibiglou, C. Cellular prion protein (PrPc): Putative interacting partners and consequences of the interaction. Int. J. Mol. Sci. 2020, 21, 7058. [Google Scholar] [CrossRef] [PubMed]
- Rovlias, A.; Kotsou, S. The influence of hyperglycemia on neurological outcome in patients with severe head injury. Neurosurgery 2000, 46, 335–342. [Google Scholar] [CrossRef]
- Cochran, A.; Scaife, E.R.; Hansen, K.W.; Downey, E.C. Hyperglycemia and outcomes from pediatric traumatic brain injury. J. Trauma 2003, 55, 1035–1038. [Google Scholar] [CrossRef]
- Xu, X.; Yang, M.; Zhangb, B.; Niu, F.; Dong, J.; Liua, B. Glucose metabolism: A link between traumatic brain injury and Alzheimer’s disease. Chin. J. Traumatol. 2021, 24, 5–10. [Google Scholar] [CrossRef]
- Jalloh, I.; Carpenter, K.L.H.; Helmy, A.; Carpenter, T.A.; Menon, D.K.; Hutchinson, P.J. Glucose metabolism following human traumatic brain injury: Methods of assessment and pathophysiological findings. Metab. Brain Dis. 2015, 30, 615–632. [Google Scholar] [CrossRef] [Green Version]
- O′Connell, M.T.; Seal, A.; Nortje, J.; Al-Rawi, P.G.; Coles, J.P.; Fryer, T.D.; Menon, D.K.; Pickard, J.D.; Hutchinson, P.J. Glucose metabolism in traumatic brain injury: A combined microdialysis and [18F]-2-fluoro-2-deoxy-D-glucose-positron emission tomography (FDG-PET) study. Acta Neurochir. 2005, 95, 165–168. [Google Scholar]
- Gimbel, D.A.; Nygaard, H.B.; Coffey, E.E.; Gunther, E.C.; Laurén, J.; Gimbel, Z.A.; Strittmatter, S.M. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 2010, 30, 6367–6374. [Google Scholar] [CrossRef]
- Persad, A.; Pham, N.; Moien-Afshari, F.; Gormley, W.; Yan, S.; Mannix, R.; Taghibiglou, C. Plasma PrPc and ADAM-10 as novel biomarkers for traumatic brain injury and concussion: A pilot study. Brain Inj. 2021, 35, 734–741. [Google Scholar] [CrossRef]
- Ley, E.J.; Srour, M.K.; Clond, M.A.; Barnajian, M.; Tillou, A.; Mirocha, J.; Salim, A. Diabetic patients with traumatic brain injury: Insulin deficiency is associated with increased mortality. J. Trauma Inj. Infect. Crit. Care 2011, 70, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Franklin, W.; Krishnan, B.; Taglialatela, G. Chronic synaptic insulin resistance after traumatic brain injury abolishes insulin protection from amyloid beta and tau oligomer-induced synaptic dysfunction. Sci. Rep. 2019, 9, 8228. [Google Scholar] [CrossRef] [PubMed]
- Rubin, T.G.; Lipton, M.L. Sex differences in animal models of traumatic brain injury. J. Exp. Neurosci. 2019, 13, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathiya, S.; Ranju, V.; Kalaivani, P.; Priya, R.J.; Sumathy, H.; Sunil, A.G.; Babu, C.S. Telmisartan attenuates MPTP induced dopaminergic degeneration and motor dysfunction through regulation of α-synuclein and neurotrophic factors (BDNF and GDNF) expression in C57BL/6J mice. Neuropharmacol 2013, 73, 98–110. [Google Scholar] [CrossRef]
- Salas, R.; Pieri, F.; Fung, B.; Dani, J.A.; De Biasi, M. Altered anxiety-related responses in mutant mice lacking the beta4 subunit of the nicotinic receptor. J. Neurosci. 2003, 23, 6255–6263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel-Ciernia, A.; Wood, M.A. Examining object location and object recognition memory in mice. Curr. Protoc. Neurosci. 2014, 69, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Taghibiglou, C.; Bradley, C.A.; Gaertner, T.; Li, Y.; Wang, Y.; Wang, Y.T. Mechanisms involved in cholesterol-induced neuronal insulin resistance. Neuropharmacol 2009, 57, 268–276. [Google Scholar] [CrossRef]
- Taghibiglou, C.; Ruddy, D.; Van Idersteine, S.C.; Aiton, A.; Cavallo, D.; Cheung, R.; Adeli, K. Intracellular mechanisms regulating apoB-containing lipoprotein assembly and secretion in primary hamster hepatocytes. J. Lipid Res. 2000, 41, 499–513. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sekar, S.; Viswas, R.S.; Miranzadeh Mahabadi, H.; Alizadeh, E.; Fonge, H.; Taghibiglou, C. Concussion/Mild Traumatic Brain Injury (TBI) Induces Brain Insulin Resistance: A Positron Emission Tomography (PET) Scanning Study. Int. J. Mol. Sci. 2021, 22, 9005. https://doi.org/10.3390/ijms22169005
Sekar S, Viswas RS, Miranzadeh Mahabadi H, Alizadeh E, Fonge H, Taghibiglou C. Concussion/Mild Traumatic Brain Injury (TBI) Induces Brain Insulin Resistance: A Positron Emission Tomography (PET) Scanning Study. International Journal of Molecular Sciences. 2021; 22(16):9005. https://doi.org/10.3390/ijms22169005
Chicago/Turabian StyleSekar, Sathiya, Raja Solomon Viswas, Hajar Miranzadeh Mahabadi, Elahe Alizadeh, Humphrey Fonge, and Changiz Taghibiglou. 2021. "Concussion/Mild Traumatic Brain Injury (TBI) Induces Brain Insulin Resistance: A Positron Emission Tomography (PET) Scanning Study" International Journal of Molecular Sciences 22, no. 16: 9005. https://doi.org/10.3390/ijms22169005