
(Dys)function Follows Form: Nucleic Acid Structure, Repeat Expansion, and Disease Pathology in FMR1 Disorders


{kind=link}
{kind=link}
Abstract
:1. Introduction
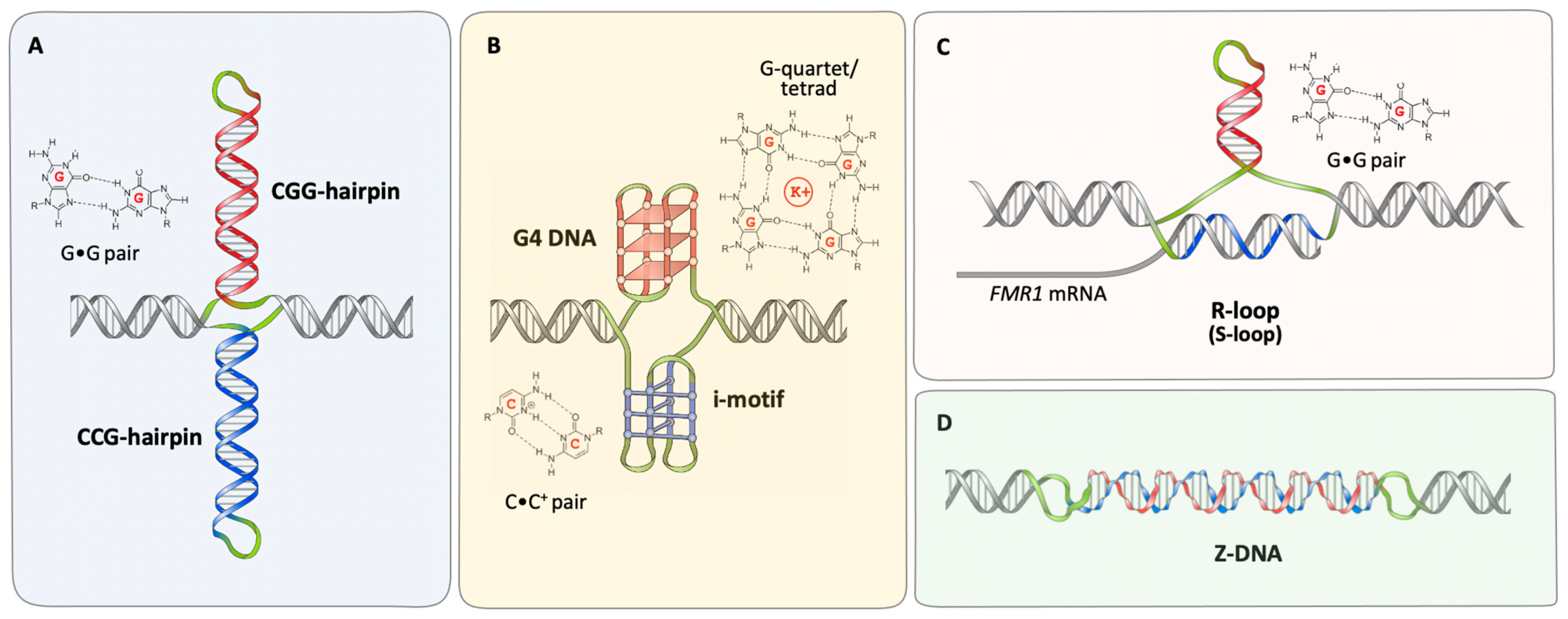
2. Secondary Structures Formed by FX Repeats
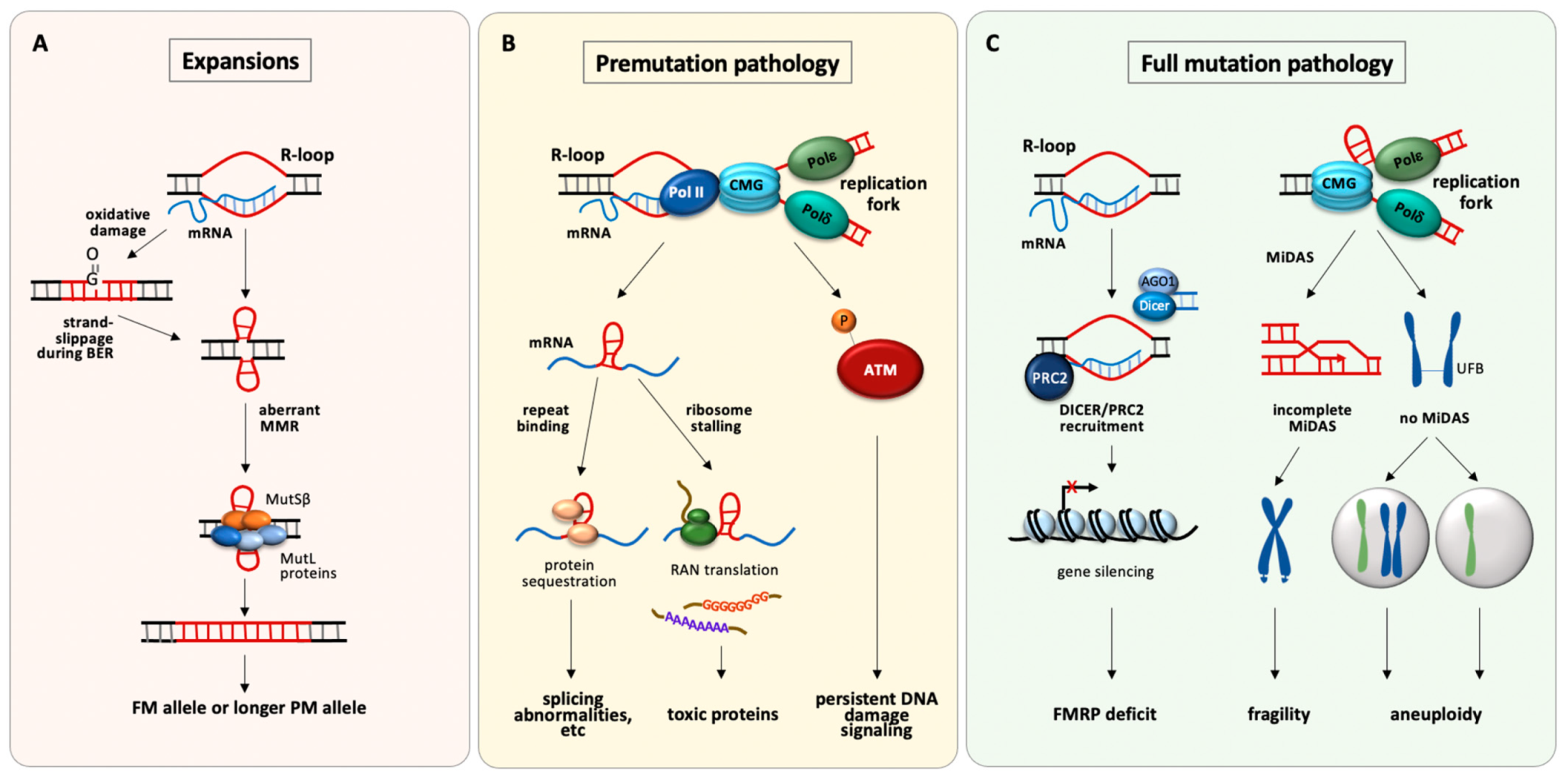
3. Repeat Expansion
4. Consequences of Repeat Expansion
4.1. Pathology in PM Carriers
4.2. Pathology in FM Carriers
4.2.1. FMR1 Gene Silencing
4.2.2. Chromosome Fragility
5. Concluding Remarks
Funding
Conflicts of Interest
References
- Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Loesch, D.; Hagerman, R. Unstable mutations in the FMR1 gene and the phenotypes. Adv. Exp. Med. Biol. 2012, 769, 78–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, C.M.; Wright, S.E.; Kearse, M.G.; Haenfler, J.M.; Flores, B.N.; Liu, Y.; Ifrim, M.F.; Glineburg, M.R.; Krans, A.; Jafar-Nejad, P.; et al. A native function for RAN translation and CGG repeats in regulating fragile X protein synthesis. Nat. Neurosci. 2020, 23, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Tortora, N.; Allen, E.; Macpherson, J.; Mila, M.; Vianna-Morgante, A.M.; Sherman, S.L.; Dobkin, C.; Latham, G.J.; et al. Expansions and contractions of the FMR1 CGG repeat in 5508 transmissions of normal, intermediate, and premutation alleles. Am. J. Med. Genet. A 2019, 179, 1148–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokanga, R.A.; Entezam, A.; Kumari, D.; Yudkin, D.; Qin, M.; Smith, C.B.; Usdin, K. Somatic expansion in mouse and human carriers of fragile X premutation alleles. Hum. Mutat. 2013, 34, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Ciosi, M.; Maxwell, A.; Cumming, S.A.; Hensman Moss, D.J.; Alshammari, A.M.; Flower, M.D.; Durr, A.; Leavitt, B.R.; Roos, R.A.C.; Team, T.-H.; et al. A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine 2019, 48, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Genetic Modifiers of Huntington’s Disease Consortium. CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset. Cell 2019, 178, 887–900.e814. [Google Scholar] [CrossRef] [Green Version]
- Tassone, F.; Hagerman, R.J.; Taylor, A.K.; Gane, L.W.; Godfrey, T.E.; Hagerman, P.J. Elevated levels of FMR1 mRNA in carrier males: A new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 2000, 66, 6–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkerk, A.J.; Holden, J.J.; Fenwick, R.G., Jr.; Warren, S.T.; et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Hagerman, R.; Hagerman, P. Fragile X-associated tremor/ataxia syndrome: Pathophysiology and management. Curr. Opin. Neurol. 2021, 34, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.G.; Charen, K.; Hipp, H.S.; Shubeck, L.; Amin, A.; He, W.; Hunter, J.E.; Sherman, S.L. Clustering of comorbid conditions among women who carry an FMR1 premutation. Genet. Med. 2020, 22, 758–766. [Google Scholar] [CrossRef] [Green Version]
- Ennis, S.; Ward, D.; Murray, A. Nonlinear association between CGG repeat number and age of menopause in FMR1 premutation carriers. Eur. J. Hum. Genet. 2006, 14, 253–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, A.K.; Marcus, M.; Epstein, M.P.; Allen, E.G.; Anido, A.E.; Paquin, J.J.; Yadav-Shah, M.; Sherman, S.L. Association of FMR1 repeat size with ovarian dysfunction. Hum. Reprod. 2005, 20, 402–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, E.; Tang, H.T.; AlOlaby, R.R.; Hickey, L.; Tassone, F. Altered expression of the FMR1 splicing variants landscape in premutation carriers. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Arocena, D.G.; Iwahashi, C.K.; Won, N.; Beilina, A.; Ludwig, A.L.; Tassone, F.; Schwartz, P.H.; Hagerman, P.J. Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum. Mol. Genet. 2005, 14, 3661–3671. [Google Scholar] [CrossRef] [Green Version]
- Iwahashi, C.K.; Yasui, D.H.; An, H.J.; Greco, C.M.; Tassone, F.; Nannen, K.; Babineau, B.; Lebrilla, C.B.; Hagerman, R.J.; Hagerman, P.J. Protein composition of the intranuclear inclusions of FXTAS. Brain 2006, 129, 256–271. [Google Scholar] [CrossRef]
- Ma, L.; Herren, A.W.; Espinal, G.; Randol, J.; McLaughlin, B.; Martinez-Cerdeno, V.; Pessah, I.N.; Hagerman, R.J.; Hagerman, P.J. Composition of the Intranuclear Inclusions of Fragile X-associated Tremor/Ataxia Syndrome. Acta Neuropathol. Commun. 2019, 7, 143. [Google Scholar] [CrossRef] [Green Version]
- Yrigollen, C.M.; Martorell, L.; Durbin-Johnson, B.; Naudo, M.; Genoves, J.; Murgia, A.; Polli, R.; Zhou, L.; Barbouth, D.; Rupchock, A.; et al. AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J. Neurodev. Disord. 2014, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Reyniers, E.; Vits, L.; De Boulle, K.; Van Roy, B.; Van Velzen, D.; de Graaff, E.; Verkerk, A.J.; Jorens, H.Z.; Darby, J.K.; Oostra, B.; et al. The full mutation in the FMR-1 gene of male fragile X patients is absent in their sperm. Nat. Genet. 1993, 4, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Azarang, A.; Wilaisakditipakorn, T.; Hagerman, R.J. Fragile X syndrome: A review of clinical management. Intractable Rare Dis. Res. 2016, 5, 145–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef]
- Richter, J.D.; Zhao, X. The molecular biology of FMRP: New insights into fragile X syndrome. Nat. Rev. Neurosci. 2021, 22, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Dobkin, C.; Radu, G.; Ding, X.H.; Brown, W.T.; Nolin, S.L. Fragile X prenatal analyses show full mutation females at high risk for mosaic Turner syndrome: Fragile X leads to chromosome loss. Am. J. Med. Genet. A 2009, 149A, 2152–2157. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Jensen, K.B.; Jin, P.; Brown, V.; Warren, S.T.; Darnell, R.B. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 2001, 107, 489–499. [Google Scholar] [CrossRef]
- Chakraborty, A.; Jenjaroenpun, P.; Li, J.; El Hilali, S.; McCulley, A.; Haarer, B.; Hoffman, E.A.; Belak, A.; Thorland, A.; Hehnly, H.; et al. Replication stress induces global chromosome breakage in the fragile X genome. Cell Rep. 2021, 34, 108838. [Google Scholar] [CrossRef] [PubMed]
- Usdin, K.; Woodford, K.J. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 1995, 23, 4202–4209. [Google Scholar] [CrossRef] [Green Version]
- Mitas, M.; Yu, A.; Dill, J.; Haworth, I.S. The trinucleotide repeat sequence d(CGG)15 forms a heat-stable hairpin containing Gsyn. Ganti base pairs. Biochemistry 1995, 34, 12803–12811. [Google Scholar] [CrossRef]
- Yu, A.; Barron, M.D.; Romero, R.M.; Christy, M.; Gold, B.; Dai, J.; Gray, D.M.; Haworth, I.S.; Mitas, M. At physiological pH, d(CCG)15 forms a hairpin containing protonated cytosines and a distorted helix. Biochemistry 1997, 36, 3687–3699. [Google Scholar] [CrossRef] [PubMed]
- Nadel, Y.; Weisman-Shomer, P.; Fry, M. The fragile X syndrome single strand d(CGG)n nucleotide repeats readily fold back to form unimolecular hairpin structures. J. Biol. Chem. 1995, 270, 28970–28977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Mariappan, S.V.; Catasti, P.; Ratliff, R.; Moyzis, R.K.; Laayoun, A.; Smith, S.S.; Bradbury, E.M.; Gupta, G. Hairpins are formed by the single DNA strands of the fragile X triplet repeats: Structure and biological implications. Proc. Natl. Acad. Sci. USA 1995, 92, 5199–5203. [Google Scholar] [CrossRef] [Green Version]
- Gacy, A.M.; Goellner, G.; Juranic, N.; Macura, S.; McMurray, C.T. Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 1995, 81, 533–540. [Google Scholar] [CrossRef] [Green Version]
- Mitas, M. Trinucleotide repeats associated with human disease. Nucleic Acids Res. 1997, 25, 2245–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Chen, X.; Bissler, J.J.; Sinden, R.R.; Leffak, M. Replication-dependent instability at (CTG) x (CAG) repeat hairpins in human cells. Nat. Chem. Biol. 2010, 6, 652–659. [Google Scholar] [CrossRef] [Green Version]
- Zumwalt, M.; Ludwig, A.; Hagerman, P.J.; Dieckmann, T. Secondary structure and dynamics of the r(CGG) repeat in the mRNA of the fragile X mental retardation 1 (FMR1) gene. RNA Biol. 2007, 4, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handa, V.; Saha, T.; Usdin, K. The fragile X syndrome repeats form RNA hairpins that do not activate the interferon-inducible protein kinase, PKR, but are cut by Dicer. Nucleic Acids Res. 2003, 31, 6243–6248. [Google Scholar] [CrossRef]
- Sobczak, K.; de Mezer, M.; Michlewski, G.; Krol, J.; Krzyzosiak, W.J. RNA structure of trinucleotide repeats associated with human neurological diseases. Nucleic Acids Res. 2003, 31, 5469–5482. [Google Scholar] [CrossRef] [PubMed]
- Khateb, S.; Weisman-Shomer, P.; Hershco, I.; Loeb, L.A.; Fry, M. Destabilization of tetraplex structures of the fragile X repeat sequence (CGG)n is mediated by homolog-conserved domains in three members of the hnRNP family. Nucleic Acids Res. 2004, 32, 4145–4154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khateb, S.; Weisman-Shomer, P.; Hershco-Shani, I.; Ludwig, A.L.; Fry, M. The tetraplex (CGG)n destabilizing proteins hnRNP A2 and CBF-A enhance the in vivo translation of fragile X premutation mRNA. Nucleic Acids Res. 2007, 35, 5775–5788. [Google Scholar] [CrossRef]
- Weisman-Shomer, P.; Cohen, E.; Fry, M. Distinct domains in the CArG-box binding factor A destabilize tetraplex forms of the fragile X expanded sequence d(CGG)n. Nucleic Acids Res. 2002, 30, 3672–3681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisman-Shomer, P.; Cohen, E.; Hershco, I.; Khateb, S.; Wolfovitz-Barchad, O.; Hurley, L.H.; Fry, M. The cationic porphyrin TMPyP4 destabilizes the tetraplex form of the fragile X syndrome expanded sequence d(CGG)n. Nucleic Acids Res. 2003, 31, 3963–3970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettani, A.; Kumar, R.A.; Patel, D.J. Solution structure of a DNA quadruplex containing the fragile X syndrome triplet repeat. J. Mol. Biol. 1995, 254, 638–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malgowska, M.; Gudanis, D.; Kierzek, R.; Wyszko, E.; Gabelica, V.; Gdaniec, Z. Distinctive structural motifs of RNA G-quadruplexes composed of AGG, CGG and UGG trinucleotide repeats. Nucleic Acids Res. 2014, 42, 10196–10207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binas, O.; Bessi, I.; Schwalbe, H. Structure Validation of G-Rich RNAs in Noncoding Regions of the Human Genome. ChemBioChem 2020, 21, 1656–1663. [Google Scholar] [CrossRef]
- Asamitsu, S.; Yabuki, Y.; Ikenoshita, S.; Kawakubo, K.; Kawasaki, M.; Usuki, S.; Nakayama, Y.; Adachi, K.; Kugoh, H.; Ishii, K.; et al. CGG repeat RNA G-quadruplexes interact with FMRpolyG to cause neuronal dysfunction in fragile X-related tremor/ataxia syndrome. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Chen, Y.W.; Satange, R.; Wu, P.C.; Jhan, C.R.; Chang, C.K.; Chung, K.R.; Waring, M.J.; Lin, S.W.; Hsieh, L.C.; Hou, M.H. Co(II)(Chromomycin)(2) Complex Induces a Conformational Change of CCG Repeats from i-Motif to Base-Extruded DNA Duplex. Int. J. Mol. Sci. 2018, 19, 2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Rodgers, M.T. Base-pairing energies of proton-bound heterodimers of cytosine and modified cytosines: Implications for the stability of DNA i-motif conformations. J. Am. Chem. Soc. 2014, 136, 282–290. [Google Scholar] [CrossRef]
- Fojtik, P.; Vorlickova, M. The fragile X chromosome (GCC) repeat folds into a DNA tetraplex at neutral pH. Nucleic Acids Res. 2001, 29, 4684–4690. [Google Scholar] [CrossRef] [Green Version]
- Loomis, E.W.; Sanz, L.A.; Chedin, F.; Hagerman, P.J. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 2014, 10, e1004294. [Google Scholar] [CrossRef]
- Kumari, D.; Usdin, K. Sustained expression of FMR1 mRNA from reactivated fragile X syndrome alleles after treatment with small molecules that prevent trimethylation of H3K27. Hum. Mol. Genet. 2016, 25, 3689–3698. [Google Scholar] [CrossRef] [Green Version]
- Abu Diab, M.; Mor-Shaked, H.; Cohen, E.; Cohen-Hadad, Y.; Ram, O.; Epsztejn-Litman, S.; Eiges, R. The G-rich Repeats in FMR1 and C9orf72 Loci Are Hotspots for Local Unpairing of DNA. Genetics 2018, 210, 1239–1252. [Google Scholar] [CrossRef] [Green Version]
- Groh, M.; Lufino, M.M.; Wade-Martins, R.; Gromak, N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef] [Green Version]
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.; Chedin, F. GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013, 23, 1590–1600. [Google Scholar] [CrossRef] [Green Version]
- De Magis, A.; Manzo, S.G.; Russo, M.; Marinello, J.; Morigi, R.; Sordet, O.; Capranico, G. DNA damage and genome instability by G-quadruplex ligands are mediated by R loops in human cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 816–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, K.; Tam, M.; Bowater, R.P.; Barber, M.; Tomlinson, M.; Nichol Edamura, K.; Wang, Y.H.; Pearson, C.E. Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res. 2011, 39, 1749–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renciuk, D.; Kypr, J.; Vorlickova, M. CGG repeats associated with fragile X chromosome form left-handed Z-DNA structure. Biopolymers 2011, 95, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Genetic Modifiers of Huntington’s Disease Consortium. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 2015, 162, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Moss, D.J.H.; Pardinas, A.F.; Langbehn, D.; Lo, K.; Leavitt, B.R.; Roos, R.; Durr, A.; Mead, S.; TRACK-HD Investigators; REGISTRY Investigators; et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef]
- Lee, J.M.; Chao, M.J.; Harold, D.; Abu Elneel, K.; Gillis, T.; Holmans, P.; Jones, L.; Orth, M.; Myers, R.H.; Kwak, S.; et al. A modifier of Huntington’s disease onset at the MLH1 locus. Hum. Mol. Genet. 2017, 26, 3859–3867. [Google Scholar] [CrossRef]
- Bettencourt, C.; Hensman-Moss, D.; Flower, M.; Wiethoff, S.; Brice, A.; Goizet, C.; Stevanin, G.; Koutsis, G.; Karadima, G.; Panas, M.; et al. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann. Neurol. 2016, 79, 983–990. [Google Scholar] [CrossRef]
- Goold, R.; Flower, M.; Moss, D.H.; Medway, C.; Wood-Kaczmar, A.; Andre, R.; Farshim, P.; Bates, G.P.; Holmans, P.; Jones, L.; et al. FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum. Mol. Genet. 2019, 28, 650–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, F.; Vasquez, M.; Santamaria, C.; Cuenca, P.; Corrales, E.; Monckton, D.G. A polymorphism in the MSH3 mismatch repair gene is associated with the levels of somatic instability of the expanded CTG repeat in the blood DNA of myotonic dystrophy type 1 patients. DNA Repair (Amst) 2016, 40, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Holmans, P.A.; Massey, T.H.; Jones, L. Genetic modifiers of Mendelian disease: Huntington’s disease and the trinucleotide repeat disorders. Hum. Mol. Genet. 2017, 26, R83–R90. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.N.; Lokanga, R.; Allette, K.; Gazy, I.; Wu, D.; Usdin, K. A MutSbeta-Dependent Contribution of MutSalpha to Repeat Expansions in Fragile X Premutation Mice? PLoS Genet. 2016, 12, e1006190. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kumari, D.; Miller, C.J.; Kim, G.Y.; Hayward, B.; Vitalo, A.G.; Pinto, R.M.; Usdin, K. Modifiers of Somatic Repeat Instability in Mouse Models of Friedreich Ataxia and the Fragile X-Related Disorders: Implications for the Mechanism of Somatic Expansion in Huntington’s Disease. J. Huntingt. Dis. 2021, 10, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, V.C.; Dion, V. Modifiers of CAG/CTG Repeat Instability: Insights from Mammalian Models. J. Huntingt. Dis. 2021, 10, 123–148. [Google Scholar] [CrossRef] [PubMed]
- Owen, B.A.; Yang, Z.; Lai, M.; Gajec, M.; Badger, J.D., 2nd; Hayes, J.J.; Edelmann, W.; Kucherlapati, R.; Wilson, T.M.; McMurray, C.T. (CAG)(n)-hairpin DNA binds to Msh2-Msh3 and changes properties of mismatch recognition. Nat. Struct. Mol. Biol. 2005, 12, 663–670. [Google Scholar] [CrossRef]
- Shelbourne, P.F.; Keller-McGandy, C.; Bi, W.L.; Yoon, S.R.; Dubeau, L.; Veitch, N.J.; Vonsattel, J.P.; Wexler, N.S.; Group, U.S.-V.C.R.; Arnheim, N.; et al. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum. Mol. Genet. 2007, 16, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Kovtun, I.V.; Johnson, K.O.; McMurray, C.T. Cockayne syndrome B protein antagonizes OGG1 in modulating CAG repeat length in vivo. Aging 2011, 3, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Jarem, D.A.; Wilson, N.R.; Delaney, S. Structure-dependent DNA damage and repair in a trinucleotide repeat sequence. Biochemistry 2009, 48, 6655–6663. [Google Scholar] [CrossRef]
- Mollersen, L.; Rowe, A.D.; Illuzzi, J.L.; Hildrestrand, G.A.; Gerhold, K.J.; Tveteras, L.; Bjolgerud, A.; Wilson, D.M., 3rd; Bjoras, M.; Klungland, A. Neil1 is a genetic modifier of somatic and germline CAG trinucleotide repeat instability in R6/1 mice. Hum. Mol. Genet. 2012, 21, 4939–4947. [Google Scholar] [CrossRef]
- Entezam, A.; Biacsi, R.; Orrison, B.; Saha, T.; Hoffman, G.E.; Grabczyk, E.; Nussbaum, R.L.; Usdin, K. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene 2007, 395, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Jonson, I.; Ougland, R.; Klungland, A.; Larsen, E. Oxidative stress causes DNA triplet expansion in Huntington’s disease mouse embryonic stem cells. Stem Cell Res. 2013, 11, 1264–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budworth, H.; Harris, F.R.; Williams, P.; Lee, D.Y.; Holt, A.; Pahnke, J.; Szczesny, B.; Acevedo-Torres, K.; Ayala-Pena, S.; McMurray, C.T. Suppression of Somatic Expansion Delays the Onset of Pathophysiology in a Mouse Model of Huntington’s Disease. PLoS Genet. 2015, 11, e1005267. [Google Scholar] [CrossRef] [PubMed]
- Mollersen, L.; Moldestad, O.; Rowe, A.D.; Bjolgerud, A.; Holm, I.; Tveteras, L.; Klungland, A.; Retterstol, L. Effects of Anthocyanins on CAG Repeat Instability and Behaviour in Huntington’s Disease R6/1 Mice. PLoS Curr. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Lokanga, R.A.; Zhao, X.N.; Entezam, A.; Usdin, K. X inactivation plays a major role in the gender bias in somatic expansion in a mouse model of the fragile X-related disorders: Implications for the mechanism of repeat expansion. Hum. Mol. Genet. 2014, 23, 4985–4994. [Google Scholar] [CrossRef] [Green Version]
- Kovtun, I.V.; Liu, Y.; Bjoras, M.; Klungland, A.; Wilson, S.H.; McMurray, C.T. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 2007, 447, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Lokanga, R.A.; Senejani, A.G.; Sweasy, J.B.; Usdin, K. Heterozygosity for a hypomorphic Polbeta mutation reduces the expansion frequency in a mouse model of the Fragile X-related disorders. PLoS Genet. 2015, 11, e1005181. [Google Scholar] [CrossRef] [Green Version]
- Lokanga, R.A.; Zhao, X.N.; Usdin, K. The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model. Hum. Mutat. 2014, 35, 129–136. [Google Scholar] [CrossRef]
- Miller, C.J.; Kim, G.Y.; Zhao, X.; Usdin, K. All three mammalian MutL complexes are required for repeat expansion in a mouse cell model of the Fragile X-related disorders. PLoS Genet 2020, 16, e1008902. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Wilkins, K.; Edelmann, W.; Usdin, K. MutLgamma promotes repeat expansion in a Fragile X mouse model while EXO1 is protective. PLoS Genet. 2018, 14, e1007719. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.N.; Kumari, D.; Gupta, S.; Wu, D.; Evanitsky, M.; Yang, W.; Usdin, K. Mutsbeta generates both expansions and contractions in a mouse model of the Fragile X-associated disorders. Hum. Mol. Genet. 2015, 24, 7087–7096. [Google Scholar] [CrossRef] [Green Version]
- Gazy, I.; Hayward, B.; Potapova, S.; Zhao, X.; Usdin, K. Double-strand break repair plays a role in repeat instability in a fragile X mouse model. DNA Repair (Amst) 2019, 74, 63–69. [Google Scholar] [CrossRef]
- Sellier, C.; Freyermuth, F.; Tabet, R.; Tran, T.; He, F.; Ruffenach, F.; Alunni, V.; Moine, H.; Thibault, C.; Page, A.; et al. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2013, 3, 869–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, P.K.; Oh, S.Y.; Krans, A.; He, F.; Sellier, C.; Frazer, M.; Renoux, A.J.; Chen, K.C.; Scaglione, K.M.; Basrur, V.; et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 2013, 78, 440–455. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.Y.; He, F.; Krans, A.; Frazer, M.; Taylor, J.P.; Paulson, H.L.; Todd, P.K. RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X-associated tremor ataxia syndrome. Hum. Mol. Genet. 2015, 24, 4317–4326. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.; Cleary, J.D.; Ranum, L.P.W. Repeat-Associated Non-ATG Translation: Molecular Mechanisms and Contribution to Neurological Disease. Annu. Rev. Neurosci. 2019, 42, 227–247. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Torlai Triglia, E.; Warburton, M.; Voigt, P.; Bird, A.; Pombo, A. R-Loops Enhance Polycomb Repression at a Subset of Developmental Regulator Genes. Mol. Cell 2019, 73, 930–945.e934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skourti-Stathaki, K.; Kamieniarz-Gdula, K.; Proudfoot, N.J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 2014, 516, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Garribba, L.; Bjerregaard, V.A.; Goncalves Dinis, M.M.; Ozer, O.; Wu, W.; Sakellariou, D.; Pena-Diaz, J.; Hickson, I.D.; Liu, Y. Folate stress induces SLX1- and RAD51-dependent mitotic DNA synthesis at the fragile X locus in human cells. Proc. Natl. Acad. Sci. USA 2020, 117, 16527–16536. [Google Scholar] [CrossRef]
- Hayward, B.E.; Steinbach, P.J.; Usdin, K. A point mutation in the nuclease domain of MLH3 eliminates repeat expansions in a mouse stem cell model of the Fragile X-related disorders. Nucleic Acids Res. 2020, 48, 7856–7863. [Google Scholar] [CrossRef]
- Kadyrova, L.Y.; Gujar, V.; Burdett, V.; Modrich, P.L.; Kadyrov, F.A. Human MutLgamma, the MLH1-MLH3 heterodimer, is an endonuclease that promotes DNA expansion. Proc. Natl. Acad. Sci. USA 2020, 117, 3535–3542. [Google Scholar] [CrossRef] [Green Version]
- Handa, V.; Goldwater, D.; Stiles, D.; Cam, M.; Poy, G.; Kumari, D.; Usdin, K. Long CGG-repeat tracts are toxic to human cells: Implications for carriers of Fragile X premutation alleles. FEBS Lett. 2005, 579, 2702–2708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, P.; Zarnescu, D.C.; Zhang, F.; Pearson, C.E.; Lucchesi, J.C.; Moses, K.; Warren, S.T. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron 2003, 39, 739–747. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, R.; Hoogeveen-Westerveld, M.; Reis, S.; Holstege, J.; Severijnen, L.A.; Nieuwenhuizen, I.M.; Schrier, M.; van Unen, L.; Tassone, F.; Hoogeveen, A.T.; et al. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum. Mol. Genet. 2003, 12, 949–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashem, V.; Galloway, J.N.; Mori, M.; Willemsen, R.; Oostra, B.A.; Paylor, R.; Nelson, D.L. Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum. Mol. Genet. 2009, 18, 2443–2451. [Google Scholar] [CrossRef] [Green Version]
- Hoem, G.; Raske, C.R.; Garcia-Arocena, D.; Tassone, F.; Sanchez, E.; Ludwig, A.L.; Iwahashi, C.K.; Kumar, M.; Yang, J.E.; Hagerman, P.J. CGG-repeat length threshold for FMR1 RNA pathogenesis in a cellular model for FXTAS. Hum. Mol. Genet. 2011, 20, 2161–2170. [Google Scholar] [CrossRef]
- Greco, C.M.; Hagerman, R.J.; Tassone, F.; Chudley, A.E.; Del Bigio, M.R.; Jacquemont, S.; Leehey, M.; Hagerman, P.J. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 2002, 125, 1760–1771. [Google Scholar] [CrossRef]
- Tassone, F.; Iwahashi, C.; Hagerman, P.J. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS). RNA Biol. 2004, 1, 103–105. [Google Scholar] [CrossRef] [Green Version]
- Powell, W.T.; Coulson, R.L.; Gonzales, M.L.; Crary, F.K.; Wong, S.S.; Adams, S.; Ach, R.A.; Tsang, P.; Yamada, N.A.; Yasui, D.H.; et al. R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proc. Natl. Acad. Sci. USA 2013, 110, 13938–13943. [Google Scholar] [CrossRef] [Green Version]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [Green Version]
- Arab, K.; Karaulanov, E.; Musheev, M.; Trnka, P.; Schafer, A.; Grummt, I.; Niehrs, C. GADD45A binds R-loops and recruits TET1 to CpG island promoters. Nat. Genet. 2019, 51, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Redstone, S.C.J.; Fleming, A.M.; Burrows, C.J. Oxidative Modification of the Potential G-Quadruplex Sequence in the PCNA Gene Promoter Can Turn on Transcription. Chem. Res. Toxicol. 2019, 32, 437–446. [Google Scholar] [CrossRef]
- Sellier, C.; Rau, F.; Liu, Y.; Tassone, F.; Hukema, R.K.; Gattoni, R.; Schneider, A.; Richard, S.; Willemsen, R.; Elliott, D.J.; et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010, 29, 1248–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.G.; Green, K.M.; Krans, A.; Rodriguez, C.M.; Linsalata, A.E.; Goldstrohm, A.C.; Todd, P.K. CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-Dependent Scanning Mechanism of Initiation to Produce Toxic Proteins. Mol. Cell 2016, 62, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Krans, A.; Kearse, M.G.; Todd, P.K. Repeat-associated non-AUG translation from antisense CCG repeats in fragile X tremor/ataxia syndrome. Ann. Neurol. 2016, 80, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Buijsen, R.A.M.; He, F.; Natla, S.; Jung, L.; Tropel, P.; Gaucherot, A.; Jacobs, H.; Meziane, H.; Vincent, A.; et al. Translation of Expanded CGG Repeats into FMRpolyG Is Pathogenic and May Contribute to Fragile X Tremor Ataxia Syndrome. Neuron 2017, 93, 331–347. [Google Scholar] [CrossRef] [Green Version]
- Kozak, M. Evaluation of the fidelity of initiation of translation in reticulocyte lysates from commercial sources. Nucleic Acids Res. 1990, 18, 2828. [Google Scholar] [CrossRef] [Green Version]
- Krans, A.; Skariah, G.; Zhang, Y.; Bayly, B.; Todd, P.K. Neuropathology of RAN translation proteins in fragile X-associated tremor/ataxia syndrome. Acta Neuropathol. Commun. 2019, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- Bonapace, G.; Gullace, R.; Concolino, D.; Iannello, G.; Procopio, R.; Gagliardi, M.; Arabia, G.; Barbagallo, G.; Lupo, A.; Manfredini, L.I.; et al. Intracellular FMRpolyG-Hsp70 complex in fibroblast cells from a patient affected by fragile X tremor ataxia syndrome. Heliyon 2019, 5, e01954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buijsen, R.A.; Sellier, C.; Severijnen, L.A.; Oulad-Abdelghani, M.; Verhagen, R.F.; Berman, R.F.; Charlet-Berguerand, N.; Willemsen, R.; Hukema, R.K. FMRpolyG-positive inclusions in CNS and non-CNS organs of a fragile X premutation carrier with fragile X-associated tremor/ataxia syndrome. Acta Neuropathol. Commun. 2014, 2, 162. [Google Scholar] [CrossRef] [Green Version]
- Friedman-Gohas, M.; Elizur, S.E.; Dratviman-Storobinsky, O.; Aizer, A.; Haas, J.; Raanani, H.; Orvieto, R.; Cohen, Y. FMRpolyG accumulates in FMR1 premutation granulosa cells. J. Ovarian Res. 2020, 13, 22. [Google Scholar] [CrossRef] [Green Version]
- Tian, B.; White, R.J.; Xia, T.; Welle, S.; Turner, D.H.; Mathews, M.B.; Thornton, C.A. Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA 2000, 6, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, T.; Guo, S.; Bardhi, O.; Ryskamp, D.A.; Li, J.; Khoramian Tusi, S.; Engelbrecht, A.; Klippel, K.; Chakrabarty, P.; Nguyen, L.; et al. Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc. Natl. Acad. Sci. USA 2020, 117, 18591–18599. [Google Scholar] [CrossRef] [PubMed]
- Green, K.M.; Glineburg, M.R.; Kearse, M.G.; Flores, B.N.; Linsalata, A.E.; Fedak, S.J.; Goldstrohm, A.C.; Barmada, S.J.; Todd, P.K. RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nat. Commun. 2017, 8, 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haify, S.N.; Mankoe, R.S.D.; Boumeester, V.; van der Toorn, E.C.; Verhagen, R.F.M.; Willemsen, R.; Hukema, R.K.; Bosman, L.W.J. Lack of a Clear Behavioral Phenotype in an Inducible FXTAS Mouse Model Despite the Presence of Neuronal FMRpolyG-Positive Aggregates. Front. Mol. Biosci. 2020, 7, 599101. [Google Scholar] [CrossRef]
- Holm, K.N.; Herren, A.W.; Taylor, S.L.; Randol, J.L.; Kim, K.; Espinal, G.; Martiinez-Cerdeno, V.; Pessah, I.N.; Hagerman, R.J.; Hagerman, P.J. Human Cerebral Cortex Proteome of Fragile X-Associated Tremor/Ataxia Syndrome. Front. Mol. Biosci. 2020, 7, 600840. [Google Scholar] [CrossRef]
- Rodriguez, S.; Sahin, A.; Schrank, B.R.; Al-Lawati, H.; Costantino, I.; Benz, E.; Fard, D.; Albers, A.D.; Cao, L.; Gomez, A.C.; et al. Genome-encoded cytoplasmic double-stranded RNAs, found in C9ORF72 ALS-FTD brain, propagate neuronal loss. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Cabal-Herrera, A.M.; Tassanakijpanich, N.; Salcedo-Arellano, M.J.; Hagerman, R.J. Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS): Pathophysiology and Clinical Implications. Int. J. Mol. Sci. 2020, 21, 4391. [Google Scholar] [CrossRef]
- Cristini, A.; Ricci, G.; Britton, S.; Salimbeni, S.; Huang, S.N.; Marinello, J.; Calsou, P.; Pommier, Y.; Favre, G.; Capranico, G.; et al. Dual Processing of R-Loops and Topoisomerase I Induces Transcription-Dependent DNA Double-Strand Breaks. Cell Rep. 2019, 28, 3167–3181.e3166. [Google Scholar] [CrossRef] [Green Version]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Primerano, B.; Tassone, F.; Hagerman, R.J.; Hagerman, P.; Amaldi, F.; Bagni, C. Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. RNA 2002, 8, 1482–1488. [Google Scholar] [PubMed]
- Schneider, A.; Winarni, T.I.; Cabal-Herrera, A.M.; Bacalman, S.; Gane, L.; Hagerman, P.; Tassone, F.; Hagerman, R. Elevated FMR1-mRNA and lowered FMRP-A double-hit mechanism for psychiatric features in men with FMR1 premutations. Transl. Psychiatry 2020, 10, 205. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhang, F.; Lokey, L.K.; Chastain, J.L.; Lakkis, L.; Eberhart, D.; Warren, S.T. Translational suppression by trinucleotide repeat expansion at FMR1. Science 1995, 268, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Usdin, K. Polycomb group complexes are recruited to reactivated FMR1 alleles in Fragile X syndrome in response to FMR1 transcription. Hum. Mol. Genet. 2014, 23, 6575–6583. [Google Scholar] [CrossRef] [Green Version]
- Alecki, C.; Chiwara, V.; Sanz, L.A.; Grau, D.; Arias Perez, O.; Boulier, E.L.; Armache, K.J.; Chedin, F.; Francis, N.J. RNA-DNA strand exchange by the Drosophila Polycomb complex PRC2. Nat. Commun. 2020, 11, 1781. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Goodrich, K.J.; Gooding, A.R.; Naeem, H.; Archer, S.; Paucek, R.D.; Youmans, D.T.; Cech, T.R.; Davidovich, C. Targeting of Polycomb Repressive Complex 2 to RNA by Short Repeats of Consecutive Guanines. Mol. Cell 2017, 65, 1056–1067.e1055. [Google Scholar] [CrossRef] [Green Version]
- Jani, K.S.; Jain, S.U.; Ge, E.J.; Diehl, K.L.; Lundgren, S.M.; Muller, M.M.; Lewis, P.W.; Muir, T.W. Histone H3 tail binds a unique sensing pocket in EZH2 to activate the PRC2 methyltransferase. Proc. Natl. Acad. Sci. USA 2019, 116, 8295–8300. [Google Scholar] [CrossRef] [Green Version]
- Yuan, W.; Xu, M.; Huang, C.; Liu, N.; Chen, S.; Zhu, B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 2011, 286, 7983–7989. [Google Scholar] [CrossRef] [Green Version]
- Schmitges, F.W.; Prusty, A.B.; Faty, M.; Stutzer, A.; Lingaraju, G.M.; Aiwazian, J.; Sack, R.; Hess, D.; Li, L.; Zhou, S.; et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 2011, 42, 330–341. [Google Scholar] [CrossRef] [Green Version]
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Pankotai, T.; Bonhomme, C.; Chen, D.; Soutoglou, E. DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat. Struct. Mol. Biol. 2012, 19, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Kumari, D.; Sciascia, N.; Usdin, K. CGG-repeat dynamics and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons. Mol. Autism 2016, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Niehrs, C.; Luke, B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Tabib, A.; Kahan, T.; Orlanski, S.; Gropp, M.; Tabach, Y.; Yanuka, O.; Benvenisty, N.; Keshet, I.; Cedar, H. Epigenetic mechanism of FMR1 inactivation in Fragile X syndrome. Int. J. Dev. Biol. 2017, 61, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Ladd, P.D.; Smith, L.E.; Rabaia, N.A.; Moore, J.M.; Georges, S.A.; Hansen, R.S.; Hagerman, R.J.; Tassone, F.; Tapscott, S.J.; Filippova, G.N. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 2007, 16, 3174–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, D.; Sciascia, N.; Usdin, K. Small Molecules Targeting H3K9 Methylation Prevent Silencing of Reactivated FMR1 Alleles in Fragile X Syndrome Patient Derived Cells. Genes 2020, 11, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukusa, T.; Fryns, J.P. Human chromosome fragility. Biochim. Biophys. Acta 2008, 1779, 3–16. [Google Scholar] [CrossRef]
- Sutherland, G.R. Heritable fragile sites on human chromosomes. III. Detection of fra(X)(q27) in males with X-linked mental retardation and in their female relatives. Hum. Genet. 1979, 53, 23–27. [Google Scholar] [CrossRef]
- Voineagu, I.; Surka, C.F.; Shishkin, A.A.; Krasilnikova, M.M.; Mirkin, S.M. Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat. Struct. Mol. Biol. 2009, 16, 226–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhardt, J.; Tomishima, M.J.; Zaninovic, N.; Colak, D.; Yan, Z.; Zhan, Q.; Rosenwaks, Z.; Jaffrey, S.R.; Schildkraut, C.L. The DNA replication program is altered at the FMR1 locus in fragile X embryonic stem cells. Mol. Cell 2014, 53, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Derbis, M.; Kul, E.; Niewiadomska, D.; Sekrecki, M.; Piasecka, A.; Taylor, K.; Hukema, R.K.; Stork, O.; Sobczak, K. Short antisense oligonucleotides alleviate the pleiotropic toxicity of RNA harboring expanded CGG repeats. Nat. Commun. 2021, 12, 1265. [Google Scholar] [CrossRef] [PubMed]
- Disney, M.D.; Liu, B.; Yang, W.Y.; Sellier, C.; Tran, T.; Charlet-Berguerand, N.; Childs-Disney, J.L. A small molecule that targets r(CGG)(exp) and improves defects in fragile X-associated tremor ataxia syndrome. ACS Chem. Biol. 2012, 7, 1711–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, A.K.; Khan, E.; Mishra, S.K.; Mishra, A.; Charlet-Berguerand, N.; Kumar, A. Curcumin Regulates the r(CGG)(exp) RNA Hairpin Structure and Ameliorate Defects in Fragile X-Associated Tremor Ataxia Syndrome. Front. Neurosci. 2020, 14, 295. [Google Scholar] [CrossRef]
- Haify, S.N.; Buijsen, R.A.M.; Verwegen, L.; Severijnen, L.; de Boer, H.; Boumeester, V.; Monshouwer, R.; Yang, W.Y.; Cameron, M.D.; Willemsen, R.; et al. Small molecule 1a reduces FMRpolyG-mediated toxicity in in vitro and in vivo models for FMR1 premutation. Hum. Mol. Genet. 2021. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Usdin, K. (Dys)function Follows Form: Nucleic Acid Structure, Repeat Expansion, and Disease Pathology in FMR1 Disorders. Int. J. Mol. Sci. 2021, 22, 9167. https://doi.org/10.3390/ijms22179167
Zhao X, Usdin K. (Dys)function Follows Form: Nucleic Acid Structure, Repeat Expansion, and Disease Pathology in FMR1 Disorders. International Journal of Molecular Sciences. 2021; 22(17):9167. https://doi.org/10.3390/ijms22179167
Chicago/Turabian StyleZhao, Xiaonan, and Karen Usdin. 2021. "(Dys)function Follows Form: Nucleic Acid Structure, Repeat Expansion, and Disease Pathology in FMR1 Disorders" International Journal of Molecular Sciences 22, no. 17: 9167. https://doi.org/10.3390/ijms22179167
APA StyleZhao, X., & Usdin, K. (2021). (Dys)function Follows Form: Nucleic Acid Structure, Repeat Expansion, and Disease Pathology in FMR1 Disorders. International Journal of Molecular Sciences, 22(17), 9167. https://doi.org/10.3390/ijms22179167