
Chemical Chaperones Modulate the Formation of Metabolite Assemblies

 , , ,
, , , 
Abstract
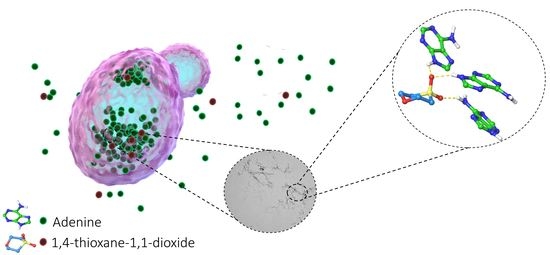
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
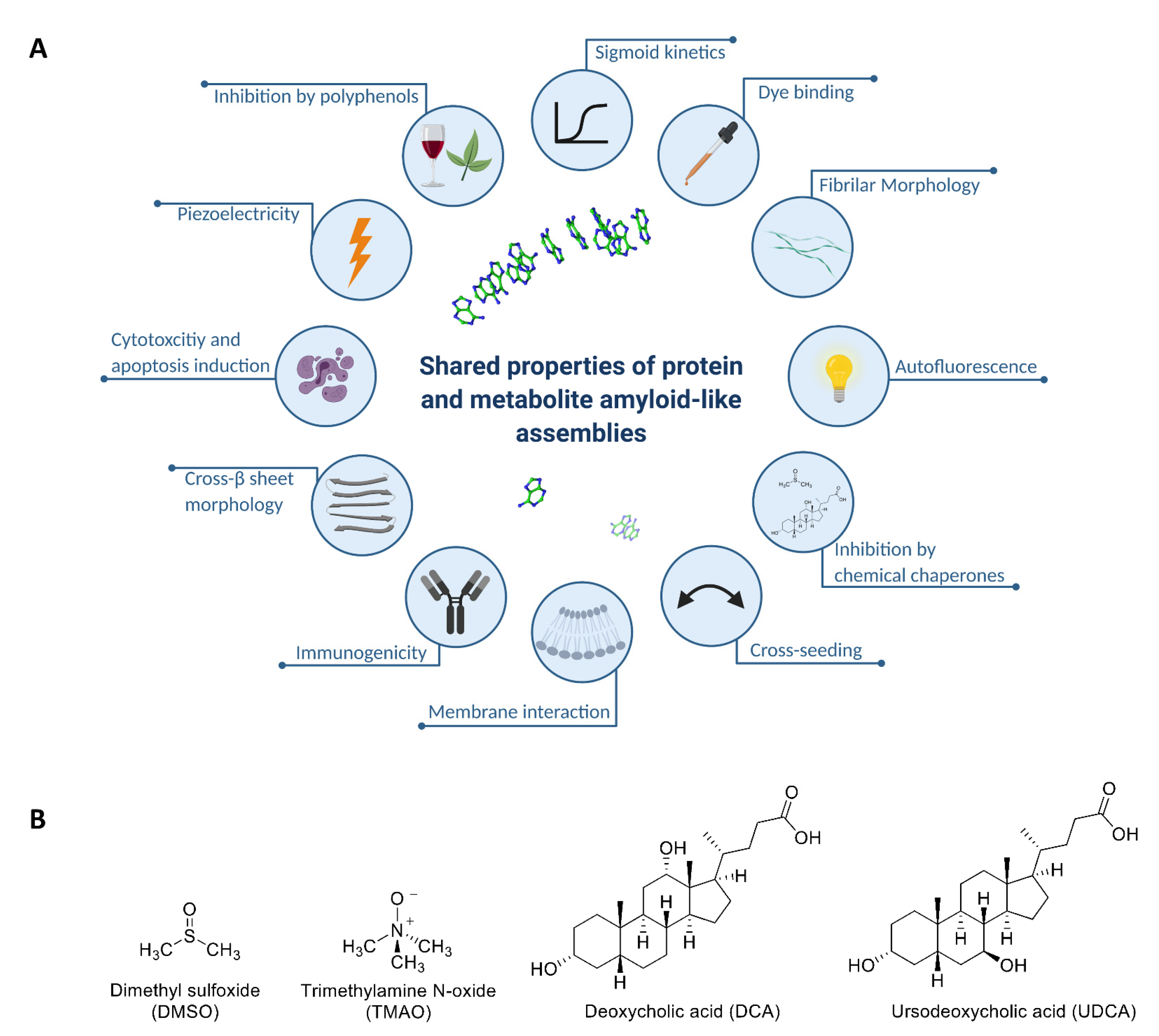
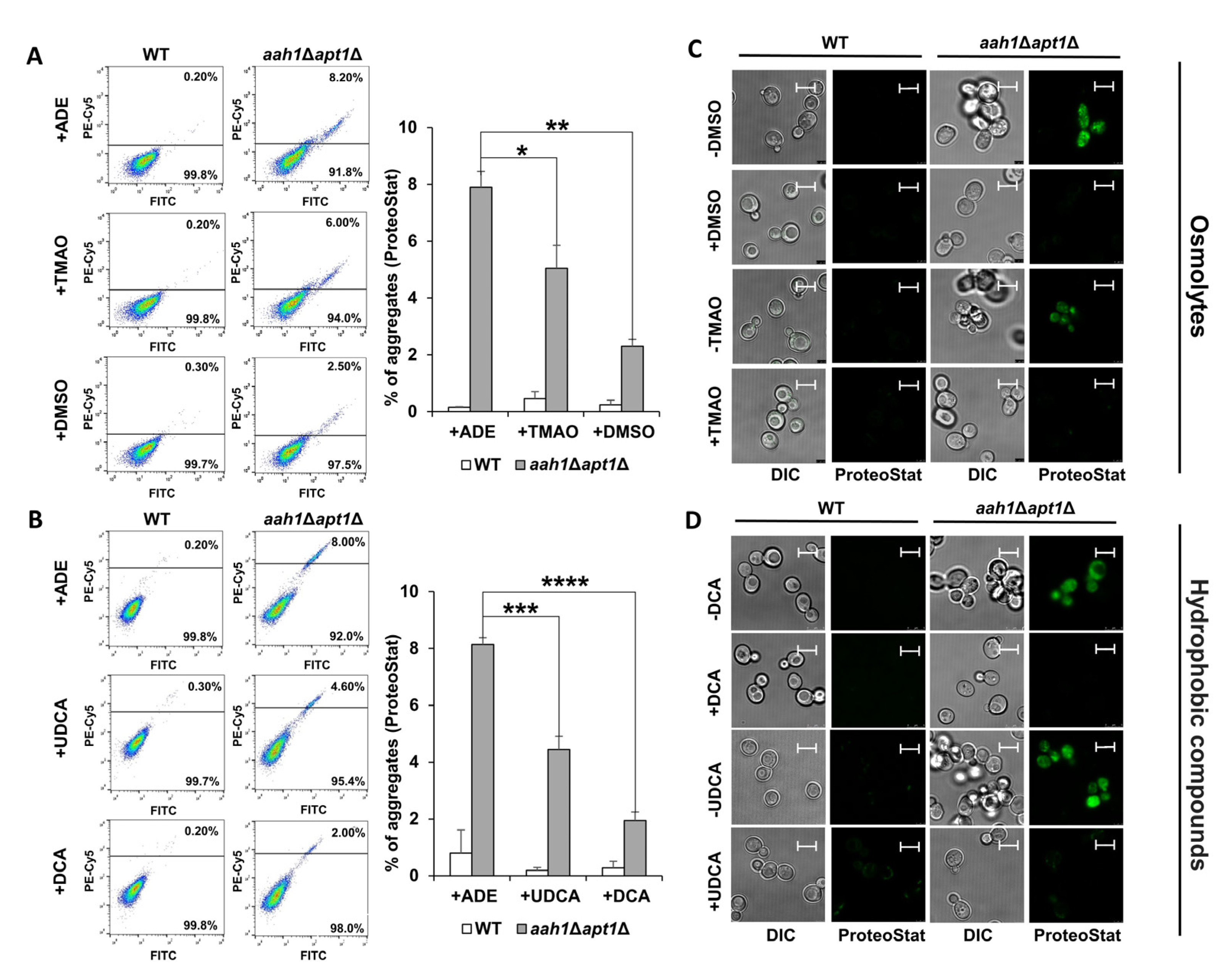
2.1. Chemical Chaperones Suppress Adenine Toxicity and Inhibit Intracellular Amyloid Formation in a Mutant Yeast Salvage Model
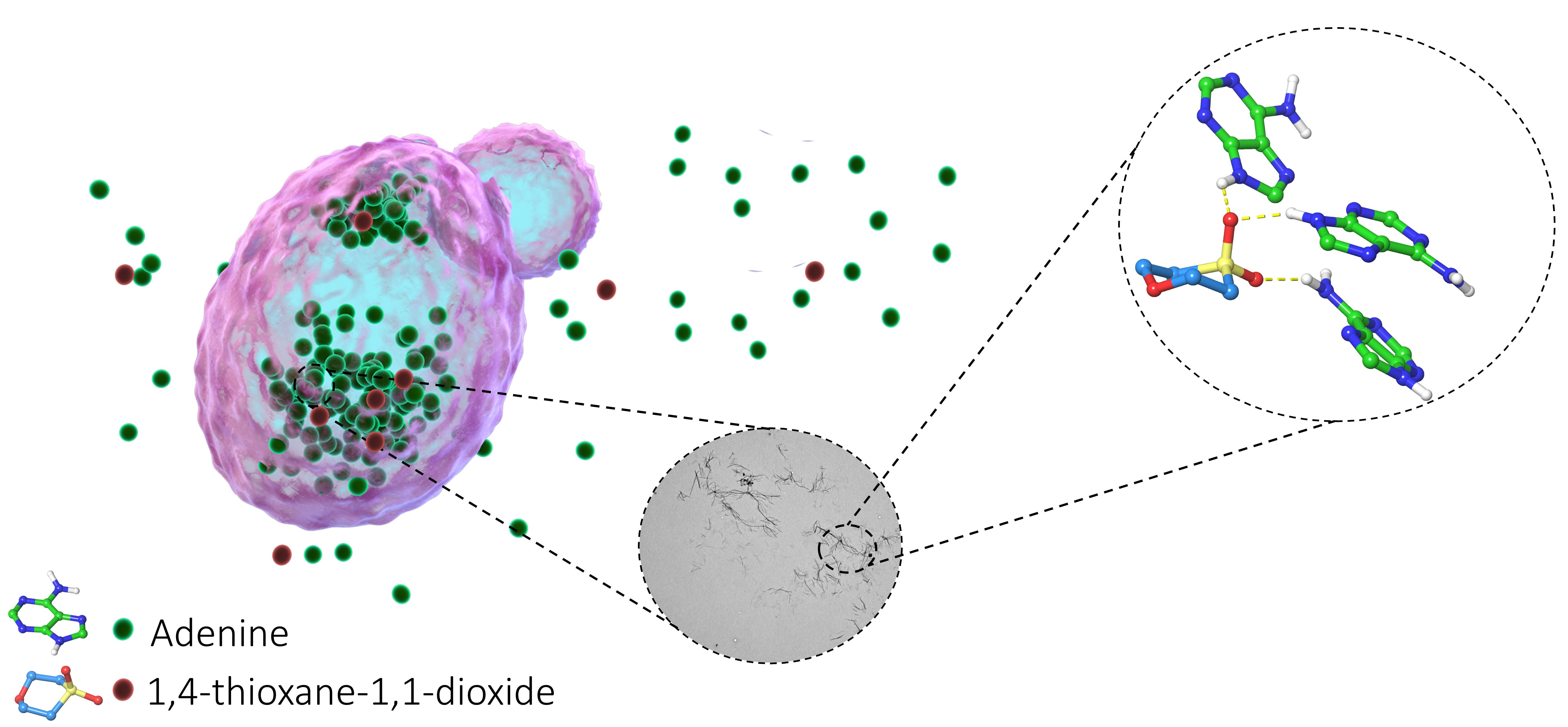
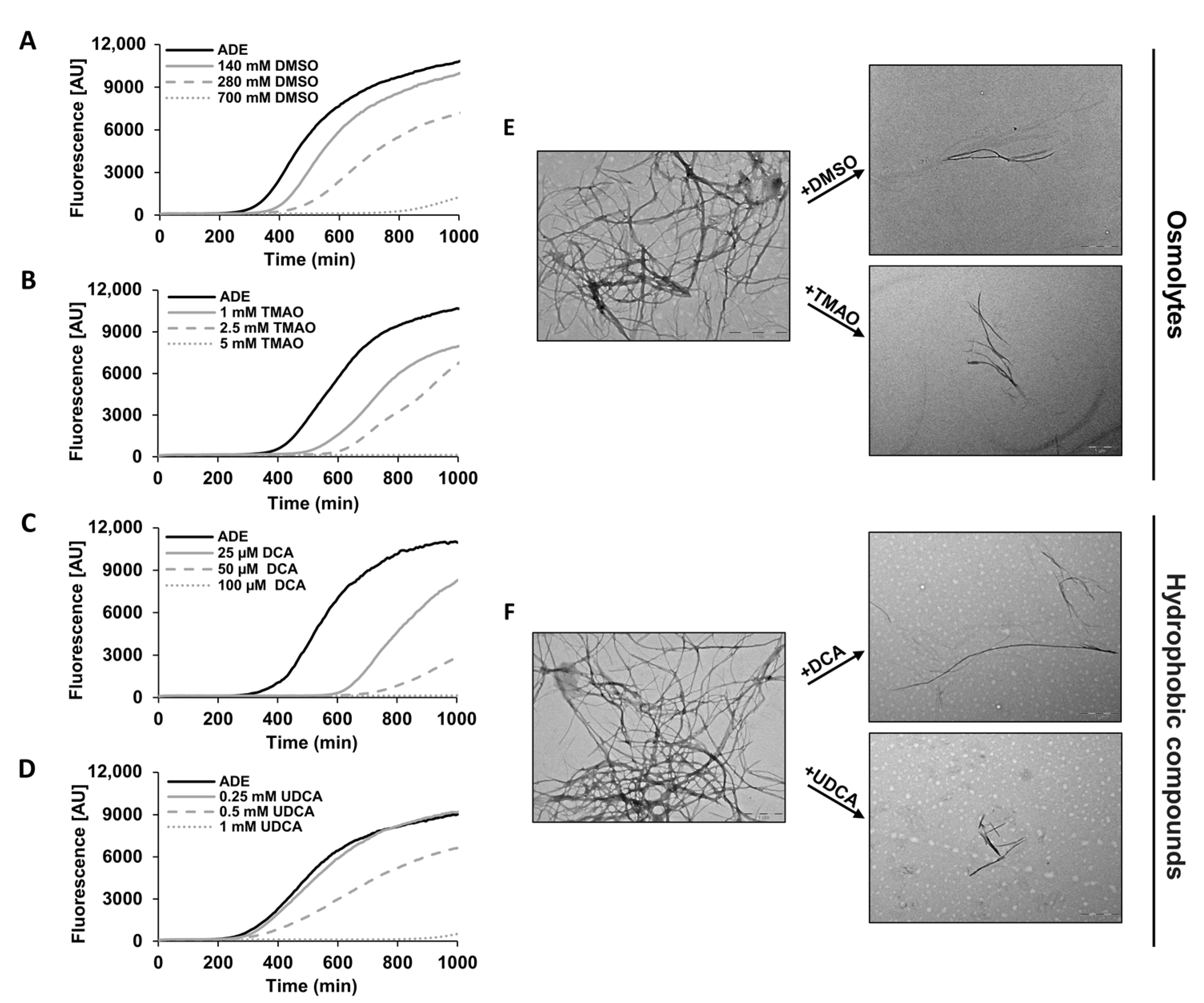
2.2. Chemical Chaperones Alter the Biophysical Properties of Adenine Amyloid-like Assemblies
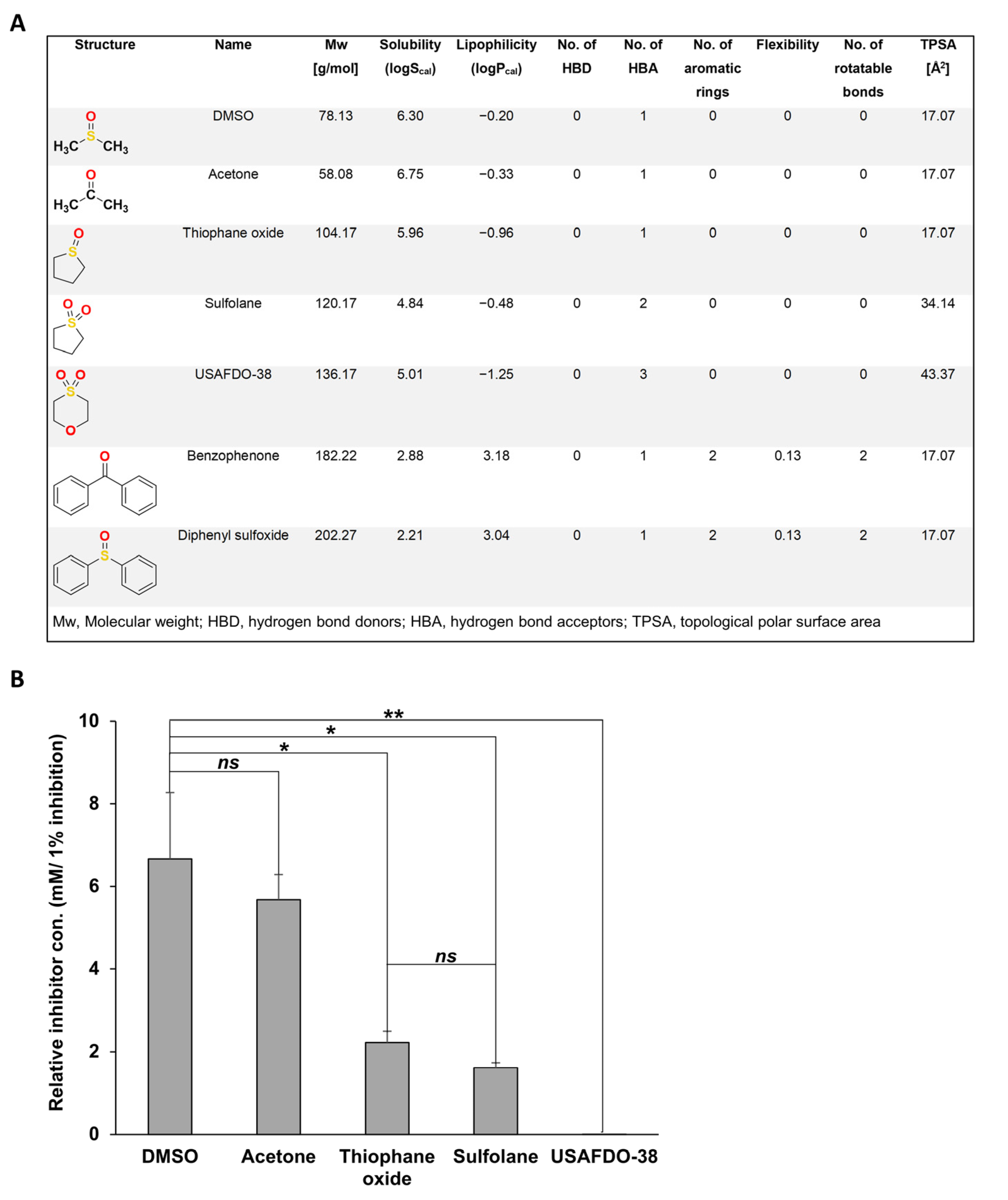
2.3. Structure–Activity and Structure–Properties Relationships of DMSO Derivatives
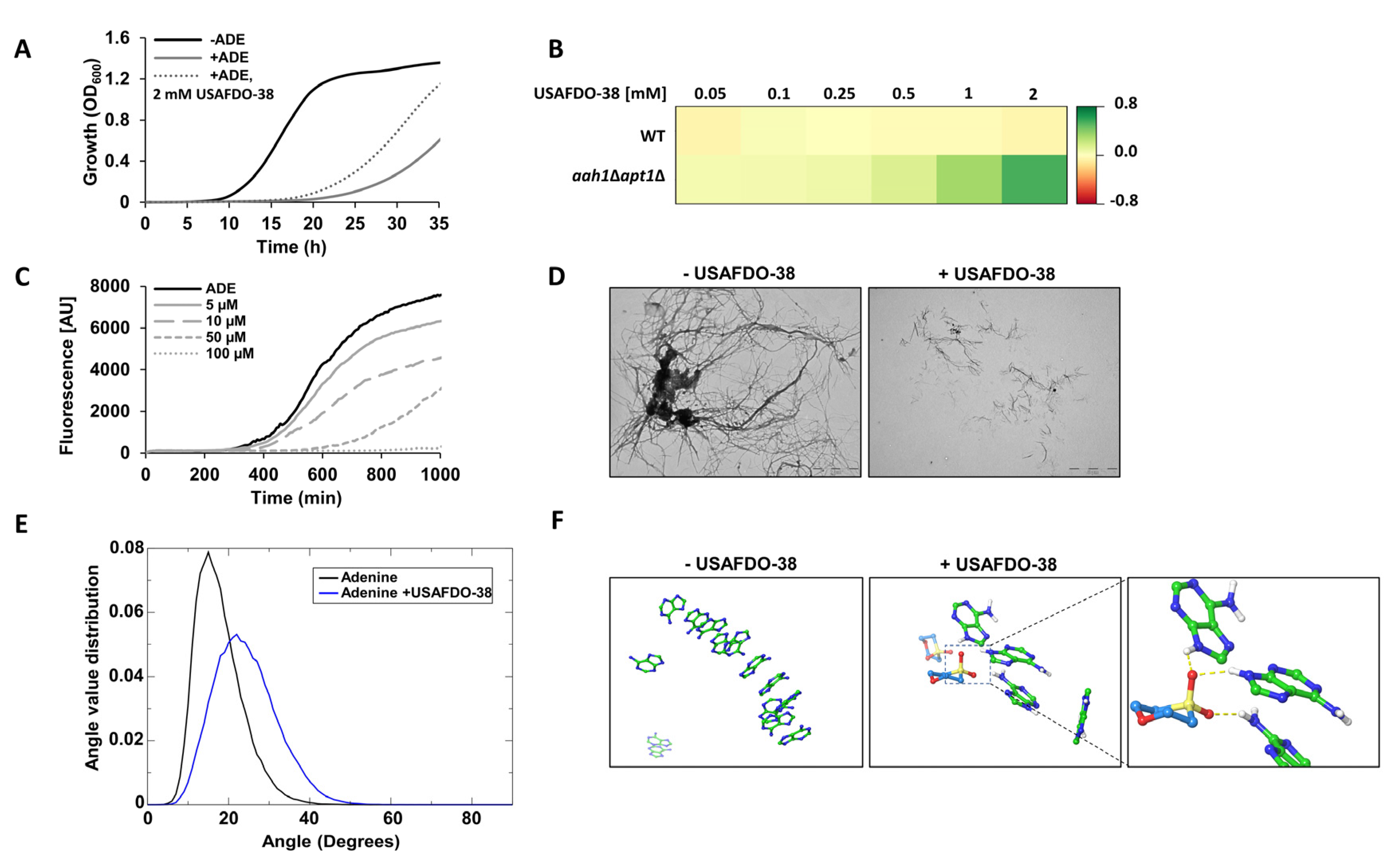
2.4. Effective Inhibition of Adenine Self-Assembly by USAFDO-38 Correlates with Improved Viability in Yeast
3. Discussion
4. Materials and Methods
4.1. Yeast Strains and Culture
4.2. Materials
4.3. Yeast Growth Assays
4.4. Flow Cytometry
4.5. Confocal Microscopy
4.6. ThT Fluorescence In Vitro Kinetic Assay
4.7. ProteoStat Fluorescence In Vitro Kinetic Assay
4.8. Transmission Electron Microscopy
4.9. MD Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vendruscolo, M.; Knowles, T.P.; Dobson, C.M. Protein Solubility and Protein Homeostasis: A Generic View of Protein Misfolding Disorders. Cold Spring Harb. Perspect. Biol. 2011, 3, a010454. [Google Scholar] [CrossRef] [Green Version]
- Gazit, E. The ”Correctly Folded” state of proteins: Is it a metastable state? Angew. Chem. Int. Ed. 2002, 41, 257–259. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Knowles, T.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.; Mossuto, M.F.; Meehan, S.; Gras, S.; et al. Metastability of Native Proteins and the Phenomenon of Amyloid Formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.C.; Zhou, R.; Serpell, L.C.; Riek, R.; Knowles, T.P.J.; Lashuel, H.A.; Gazit, E.; Hamley, I.W.; Davis, T.P.; Fändrich, M.; et al. Half a century of amyloids: Past, present and future. Chem. Soc. Rev. 2020, 49, 5473–5509. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In Vivo aspects of protein folding and quality control. Science 2016, 353, 4354. [Google Scholar] [CrossRef] [PubMed]
- Ringe, D.; Petsko, G. What are pharmacological chaperones and why are they interesting? J. Biol. 2009, 8, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nat. Cell Biol. 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The Biology of Proteostasis in Aging and Disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [Green Version]
- Bernier, V.; Lagacé, M.; Bichet, D.-G.; Bouvier, M. Pharmacological chaperones: Potential treatment for conformational diseases. Trends Endocrinol. Metab. 2004, 15, 222–228. [Google Scholar] [CrossRef]
- Cortez, L.; Sim, V. The therapeutic potential of chemical chaperones in protein folding diseases. Prion 2014, 8, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R. Role of naturally occurring osmolytes in protein folding and stability. Arch. Biochem. Biophys. 2009, 491, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Hanna, A.; Wang, H.; Dagnino-Acosta, A.; Joshi, A.D.; Knoblauch, M.; Xia, Y.; Georgiou, D.K.; Xu, J.; Long, C.; et al. A chemical chaperone improves muscle function in mice with a RyR1 mutation. Nat. Commun. 2017, 8, 14659. [Google Scholar] [CrossRef] [Green Version]
- Gong, B.; Zhang, L.Y.; Lam, D.S.C.; Pang, C.P.; Yam, G.H.F. Sodium 4-phenylbutyrate ameliorates the effects of cataract-causing mutant γD-crystallin in cultured cells. Mol. Vis. 2010, 16, 997–1003. [Google Scholar]
- Ozcan, U.; Yilmaz, E.; Özcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Perlmutter, D.H. Chemical Chaperones: A Pharmacological Strategy for Disorders of Protein Folding and Trafficking. Pediatr. Res. 2002, 52, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Basseri, S.; Lhoták, Š.; Sharma, A.M.; Austin, R.C. The chemical chaperone 4-phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. J. Lipid Res. 2009, 50, 2486–2501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Getter, T.; Zaks, I.; Barhum, Y.; Ben-Zur, T.; Böselt, S.; Gregoire, S.; Viskind, O.; Shani, T.; Gottlieb, H.; Green, O.; et al. A Chemical Chaperone-Based Drug Candidate is Effective in a Mouse Model of Amyotrophic Lateral Sclerosis (ALS). ChemMedChem 2015, 10, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Pujols, J.; Díaz, S.P.; Pallares, I.; Ventura, S. Chemical Chaperones as Novel Drugs for Parkinson’s Disease. Trends Mol. Med. 2020, 26, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Saxena, K.; Kasturia, N.; Dalal, V.; Bhatt, N.; Rajkumar, A.; Maity, S.; Sengupta, S.; Chakraborty, K. Chemical chaperones assist intracellular folding to buffer mutational variations. Nat. Chem. Biol. 2012, 8, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Ward, C.L.; Krouse, M.E.; Wine, J.J.; Kopito, R.R. Glycerol Reverses the Misfolding Phenotype of the Most Common Cystic Fibrosis Mutation. J. Biol. Chem. 1996, 271, 635–638. [Google Scholar] [CrossRef] [Green Version]
- Ip, P.; Sharda, P.R.; Cunningham, A.; Chakrabartty, S.; Pande, V.; Chakrabartty, A. Quercitrin and quercetin 3-β-d-glucoside as chemical chaperones for the A4V SOD1 ALS-causing mutant. Protein Eng. Des. Sel. 2017, 30, 431–440. [Google Scholar] [CrossRef]
- Chang, K.-H.; Chiu, Y.-J.; Chen, S.-L.; Huang, C.-H.; Lin, C.-H.; Lin, T.-H.; Lee, C.-M.; Ramesh, C.; Wu, C.-H.; Huang, C.-C.; et al. The potential of synthetic indolylquinoline derivatives for Aβ aggregation reduction by chemical chaperone activity. Neuropharmacology 2016, 101, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Bennion, B.J.; Daggett, V. Counteraction of urea-induced protein denaturation by trimethylamine N-oxide: A chemical chaperone at atomic resolution. Proc. Natl. Acad. Sci. USA 2004, 101, 6433–6438. [Google Scholar] [CrossRef] [Green Version]
- Perez-Miller, S.; Younus, H.; Vanam, R.; Chen, C.-H.; Mochly-Rosen, D.; Hurley, T.D. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat. Struct. Mol. Biol. 2010, 17, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Gazit, E. Metabolite amyloids: A new paradigm for inborn error of metabolism disorders. J. Inherit. Metab. Dis. 2016, 39, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Laor, D.; Sade, D.; Shaham-Niv, S.; Zaguri, D.; Gartner, M.; Basavalingappa, V.; Raveh, A.; Pichinuk, E.; Engel, H.; Iwasaki, K.; et al. Fibril formation and therapeutic targeting of amyloid-like structures in a yeast model of adenine accumulation. Nat. Commun. 2019, 10, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rencus-Lazar, S.; DeRowe, Y.; Adsi, H.; Gazit, E.; Laor, D. Yeast Models for the Study of Amyloid-Associated Disorders and Development of Future Therapy. Front. Mol. Biosci. 2019, 6, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leandro, P. Protein Misfolding in Conformational Disorders: Rescue of Folding Defects and Chemical Chaperoning. Mini-Rev. Med. Chem. 2008, 8, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Ulrich Hartl, F. Molecular Chaperone Functions in Protein Folding and Proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Ganguly, P.; Boserman, P.; Van Der Vegt, N.F.A.; Shea, J.-E. Trimethylamine N-oxide Counteracts Urea Denaturation by Inhibiting Protein–Urea Preferential Interaction. J. Am. Chem. Soc. 2018, 140, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.W.; Dawson, H.J.; Lackey, H.B. Naturally occurring levels of dimethyl sulfoxide in selected fruits, vegetables, grains, and beverages. J. Agric. Food Chem. 1981, 29, 1089–1091. [Google Scholar] [CrossRef] [PubMed]
- Adler-Abramovich, L.; Vaks, L.; Carny, O.; Trudler, D.; Magno, A.; Caflisch, A.; Frenkel, D.; Gazit, E. Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. Nat. Chem. Biol. 2012, 8, 701–706. [Google Scholar] [CrossRef]
- Shaham-Niv, S.; Adler-Abramovich, L.; Schnaider, L.; Gazit, E. Extension of the generic amyloid hypothesis to nonproteinaceous metabolite assemblies. Sci. Adv. 2015, 1, e1500137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaham-Niv, S.; Rehak, P.; Vuković, L.; Adler-Abramovich, L.; Král, P.; Gazit, E. Formation of Apoptosis-Inducing Amyloid Fibrils by Tryptophan. Isr. J. Chem. 2017, 57, 729–737. [Google Scholar] [CrossRef]
- Tavassoly, O.; Sade, D.; Bera, S.; Shaham-Niv, S.; Vocadlo, D.J.; Gazit, E. Quinolinic Acid Amyloid-like Fibrillar Assemblies Seed α-Synuclein Aggregation. J. Mol. Biol. 2018, 430, 3847–3862. [Google Scholar] [CrossRef] [PubMed]
- Zaguri, D.; Shaham-Niv, S.; Chakraborty, P.; Arnon, Z.A.; Makam, P.; Bera, S.; Rencus-Lazar, S.; Stoddart, P.R.; Gazit, E.; Reynolds, N.P. Nanomechanical Properties and Phase Behavior of Phenylalanine Amyloid Ribbon Assemblies and Amorphous Self-Healing Hydrogels. ACS Appl. Mater. Interfaces 2020, 12, 21992–22001. [Google Scholar] [CrossRef] [PubMed]
- Zaguri, D.; Shaham-Niv, S.; Naaman, E.; Mimouni, M.; Magen, D.; Pollack, S.; Kreiser, T.; Leibu, R.; Rencus-Lazar, S.; Adler-Abramovich, L.; et al. Induction of retinopathy by fibrillar oxalate assemblies. Commun. Chem. 2020, 3, 1–9. [Google Scholar] [CrossRef]
- De Luigi, A.; Mariani, A.; De Paola, M.; Depaolini, A.R.; Colombo, L.; Russo, L.; Rondelli, V.; Brocca, P.; Adler-Abramovich, L.; Gazit, E.; et al. Doxycycline hinders phenylalanine fibril assemblies revealing a potential novel therapeutic approach in phenylketonuria. Sci. Rep. 2015, 5, 15902. [Google Scholar] [CrossRef]
- Shaham-Niv, S.; Rehak, P.; Zaguri, D.; Kolusheva, S.; Král, P.; Gazit, E. Metabolite amyloid-like fibrils interact with model membranes. Chem. Commun. 2018, 54, 4561–4564. [Google Scholar] [CrossRef] [PubMed]
- Griffith, E.; Perkins, R.; Telesford, D.-M.; Adams, E.M.; Cwiklik, L.; Allen, H.C.; Roeselová, M.; Vaida, V. Interaction of l-Phenylalanine with a Phospholipid Monolayer at the Water–Air Interface. J. Phys. Chem. B 2015, 119, 9038–9048. [Google Scholar] [CrossRef]
- Cutró, A.; Hollmann, A.; Cejas, J.; Maturana, P.; Disalvo, E.; Frías, M. Phenylalanine interaction with lipid monolayers at different pHs. Colloids Surf. B Biointerfaces 2015, 135, 504–509. [Google Scholar] [CrossRef]
- Rosa, A.S.; Cutro, A.C.; Frías, M.A.; Disalvo, E.A. Interaction of Phenylalanine with DPPC Model Membranes: More Than a Hydrophobic Interaction. J. Phys. Chem. B 2015, 119, 15844–15847. [Google Scholar] [CrossRef]
- Sankaranarayanan, K. Fibrils of phenylalanine adsorbed to Langmuir-Blodgett films: Role of Lipids. Soft Mater. 2015, 13, 219–224. [Google Scholar] [CrossRef]
- Shaham-Niv, S.; Rehak, P.; Zaguri, D.; Levin, A.; Adler-Abramovich, L.; Vuković, L.; Král, P.; Gazit, E. Differential inhibition of metabolite amyloid formation by generic fibrillation-modifying polyphenols. Commun. Chem. 2018, 1, 25. [Google Scholar] [CrossRef] [Green Version]
- Sade, D.; Shaham-Niv, S.; Arnon, Z.A.; Tavassoly, O.; Gazit, E. Seeding of proteins into amyloid structures by metabolite assemblies may clarify certain unexplained epidemiological associations. Open Biol. 2018, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Zaguri, D.; Kreiser, T.; Shaham-Niv, S.; Gazit, E. Antibodies towards Tyrosine Amyloid-Like Fibrils Allow Toxicity Modulation and Cellular Imaging of the Assemblies. Molecules 2018, 23, 1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Luo, W.; Ma, H.; Peng, Q.; Yuan, W.Z.; Zhang, Y. Prevalent intrinsic emission from nonaromatic amino acids and poly(amino acids). Sci. China Chem. 2018, 61, 351–359. [Google Scholar] [CrossRef]
- Shaham-Niv, S.; Arnon, Z.A.; Sade, D.; Lichtenstein, A.; Shirshin, E.A.; Kolusheva, S.; Gazit, E. Intrinsic Fluorescence of Metabolite Amyloids Allows Label-Free Monitoring of Their Formation and Dynamics in Live Cells. Angew. Chem. Int. Ed. 2018, 57, 12444–12447. [Google Scholar] [CrossRef] [PubMed]
- Guerin, S.; Stapleton, A.; Chovan, D.; Mouras, R.; Gleeson, M.; McKeown, C.; Noor, M.R.; Silien, C.; Rhen, F.M.F.; Kholkin, A.; et al. Control of piezoelectricity in amino acids by supramolecular packing. Nat. Mater. 2018, 17, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Oshinbolu, S.; Shah, R.; Finka, G.; Molloy, M.; Uden, M.; Bracewell, D.G. Evaluation of fluorescent dyes to measure protein aggregation within mammalian cell culture supernatants. J. Chem. Technol. Biotechnol. 2017, 93, 909–917. [Google Scholar] [CrossRef]
- Shultz, M.D. Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J. Med. Chem. 2018, 62, 1701–1714. [Google Scholar] [CrossRef]
- Chen, H.; Engkvist, O.; Kogej, T. Compound Properties and their Influence on Drug Quality. In The Practice of Medicinal Chemistry, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 379–393. [Google Scholar] [CrossRef]
- Tong, W.; Welsh, W.J.; Shi, L.; Fang, H.; Perkins, R. Structure–Activity Relationship Approaches and Applications. Environ. Toxicol. Chem. 2003, 22, 1680. [Google Scholar] [CrossRef]
- Guha, R. On Exploring Structure–Activity Relationships. Methods Mol. Biol. 2013, 993, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; DeCrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef]
- Böhm, H.-J.; Brode, S.; Hesse, U.; Klebe, G. Oxygen and Nitrogen in Competitive Situations: Which is the Hydrogen-Bond Acceptor? Chem. A Eur. J. 1996, 2, 1509–1513. [Google Scholar] [CrossRef]
- Meli, M.; Engel, H.; Laor, D.; Gazit, E.; Colombo, G. Mechanisms of Metabolite Amyloid Formation: Computational Studies for Drug Design against Metabolic Disorders. ACS Med. Chem. Lett. 2019, 10, 666–670. [Google Scholar] [CrossRef]
- Ferreira, N.; Pereira-Henriques, A.; Almeida, M.R. Transthyretin chemical chaperoning by flavonoids: Structure–activity insights towards the design of potent amyloidosis inhibitors. Biochem. Biophys. Rep. 2015, 3, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Uncovering the Neuroprotective Mechanisms of Curcumin on Transthyretin Amyloidosis. Int. J. Mol. Sci. 2019, 20, 1287. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Epigallocatechin-3-Gallate as a Potential Therapeutic Drug for TTR-Related Amyloidosis: “In Vivo” Evidence from FAP Mice Models. PLoS ONE 2012, 7, e29933. [Google Scholar] [CrossRef]
- Porat, Y.; Abramowitz, A.; Gazit, E. Inhibition of Amyloid Fibril Formation by Polyphenols: Structural Similarity and Aromatic Interactions as a Common Inhibition Mechanism. Chem. Biol. Drug Des. 2005, 67, 27–37. [Google Scholar] [CrossRef]
- Murray, C.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef]
- Muntau, A.C.; Leandro, J.; Staudigl, M.; Mayer, F.; Gersting, S.W. Innovative strategies to treat protein misfolding in inborn errors of metabolism: Pharmacological chaperones and proteostasis regulators. J. Inherit. Metab. Dis. 2014, 37, 505–523. [Google Scholar] [CrossRef]
- Yue, W.W. From structural biology to designing therapy for inborn errors of metabolism. J. Inherit. Metab. Dis. 2016, 39, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Evers, R.; Vliet, D.; Spronsen, F.J. Tetrahydrobiopterin treatment in phenylketonuria: A repurposing approach. J. Inherit. Metab. Dis. 2020, 43, 189–199. [Google Scholar] [CrossRef]
- Melenovska, P.; Kopecká, J.; Krijt, J.; Hnizda, A.; Raková, K.; Janošík, M.; Wilcken, B.; Kožich, V. Chaperone therapy for homocystinuria: The rescue of CBS mutations by heme arginate. J. Inherit. Metab. Dis. 2015, 38, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.R.; Chen, X.; Kožich, V.; Kruger, W.D. Chemical chaperone rescue of mutant human cystathionine β-synthase. Mol. Genet. Metab. 2007, 91, 335–342. [Google Scholar] [CrossRef]
- Song, J.-L.; Chuang, D.T. Natural Osmolyte Trimethylamine N-Oxide Corrects Assembly Defects of Mutant Branched-chain α-Ketoacid Decarboxylase in Maple Syrup Urine Disease. J. Biol. Chem. 2001, 276, 40241–40246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randhawa, R.; Bhardwaj, R.; Kaur, T. Amelioration of hyperoxaluria-induced kidney dysfunction by chemical chaperone 4-phenylbutyric acid. Urolithiasis 2019, 47, 171–179. [Google Scholar] [CrossRef]
- Leandro, P.; Lechner, M.C.; De Almeida, I.T.; Konecki, D. Glycerol Increases the Yield and Activity of Human Phenylalanine Hydroxylase Mutant Enzymes Produced in a Prokaryotic Expression System. Mol. Genet. Metab. 2001, 73, 173–178. [Google Scholar] [CrossRef]
- Frisch, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 09, Revision A, Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Case, D.A.; Cerutti, D.S.; Cheatham, T.E.I.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Greene, D.; Homey-er, N.; et al. AMBER; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adsi, H.; Levkovich, S.A.; Haimov, E.; Kreiser, T.; Meli, M.; Engel, H.; Simhaev, L.; Karidi-Heller, S.; Colombo, G.; Gazit, E.; et al. Chemical Chaperones Modulate the Formation of Metabolite Assemblies. Int. J. Mol. Sci. 2021, 22, 9172. https://doi.org/10.3390/ijms22179172
Adsi H, Levkovich SA, Haimov E, Kreiser T, Meli M, Engel H, Simhaev L, Karidi-Heller S, Colombo G, Gazit E, et al. Chemical Chaperones Modulate the Formation of Metabolite Assemblies. International Journal of Molecular Sciences. 2021; 22(17):9172. https://doi.org/10.3390/ijms22179172
Chicago/Turabian StyleAdsi, Hanaa, Shon A. Levkovich, Elvira Haimov, Topaz Kreiser, Massimiliano Meli, Hamutal Engel, Luba Simhaev, Shai Karidi-Heller, Giorgio Colombo, Ehud Gazit, and et al. 2021. "Chemical Chaperones Modulate the Formation of Metabolite Assemblies" International Journal of Molecular Sciences 22, no. 17: 9172. https://doi.org/10.3390/ijms22179172