
Joint Degeneration in a Mouse Model of Pseudoachondroplasia: ER Stress, Inflammation, and Block of Autophagy

Abstract
1. Introduction
2. Results
2.1. Mutant-COMP Is Retained in the ER in Adult Articular Chondrocytes
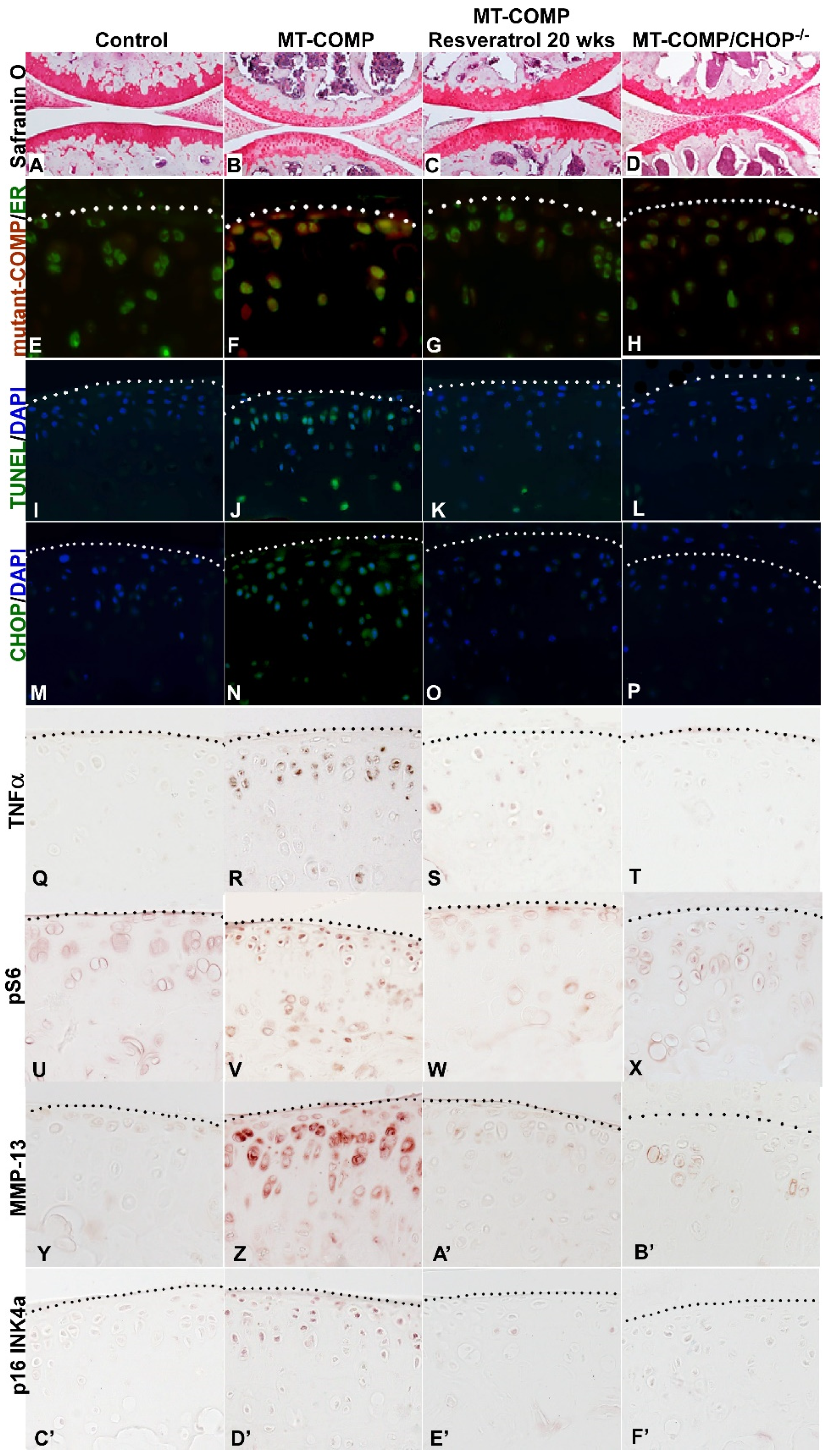
2.2. ER Stress, Inflammation, Matrix Degradation, Autophagy Repression, Senescence, and Chondrocyte Death Are Present in MT-COMP Articular Chondrocytes
2.3. MT-COMP Mice Show Signs of Pain in Early Adulthood
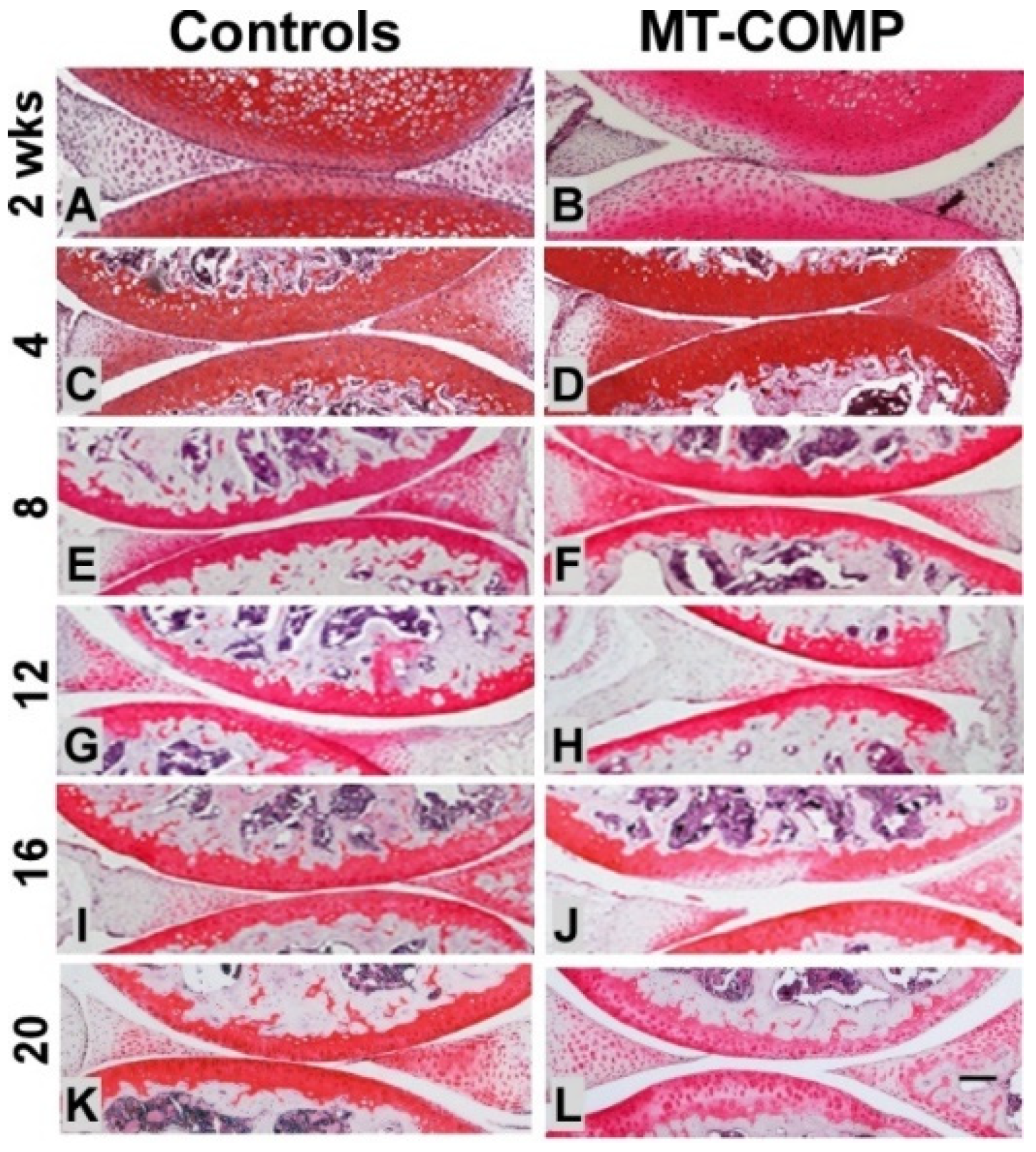
2.4. Joint Degeneration in MT-COMP Mice Is Validated by OA Scoring
2.5. Prevention of Joint Degeneration in MT-COMP Mice with Resveratrol Treatment or Ablation of CHOP
3. Discussion
4. Materials and Methods
4.1. Bigenic Mice
4.2. Generation of CHOP Null Bigenic Mice
4.3. Immunohistochemistry
4.4. Gait Analysis
4.5. OA Scoring
4.6. MMPSense
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Dicesare, P.E.; Morgelin, M.; Mann, K.; Paulsson, M. Cartilage oligomeric matrix protein and thrombospondin 1. Purification from articular cartilage, electron microscopic structure, and chondrocyte binding. JBIC J. Biol. Inorg. Chem. 1994, 223, 927–937. [Google Scholar] [CrossRef]
- Hecht, J.T.; Deere, M.; Putnam, E.; Cole, W.; Vertel, B.; Chen, H.; Lawler, J. Characterization of cartilage oligomeric matrix protein (COMP) in human normal and pseudochondroplasia musculoskeletal tissues. Matrix Biol. 1998, 17, 269–278. [Google Scholar] [CrossRef]
- Hedbom, E.; Antonsson, P.; Hjerpe, A.; Aeschlimann, D.; Paulsson, M.; Rosa-Pimentel, E.; Sommarin, Y.; Wendel, M.; Oldberg, A.; Heinegård, D. Cartilage matrix proteins. An acidic oligomeric protein (COMP) detected only in cartilage. J. Biol. Chem. 1992, 267, 6132–6136. [Google Scholar] [CrossRef]
- Urban, J.; Maroudas, A.; Bayliss, M.; Dillon, J. Swelling pressures of proteoglycans at the concentrations found in cartilaginous tissues. Biorheology 1979, 16, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Kempson, G.E.; Freeman, M.A.R.; Swanson, S.A.V. Tensile Properties of Articular Cartilage. Nat. Cell Biol. 1968, 220, 1127–1128. [Google Scholar] [CrossRef]
- Schmidt, M.B.; Mow, V.C.; Chun, L.E.; Eyre, D.R. Effects of proteoglycan extraction on the tensile behavior of articular cartilage. J. Orthop. Res. 1990, 8, 353–363. [Google Scholar] [CrossRef]
- Lawler, J.; Duquette, M.; Whittaker, C.A.; Adams, J.; McHenry, K.; DeSimone, D. Identification and characterization of thrombospondin-4, a new member of the thrombospondin gene family. J. Cell Biol. 1993, 120, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Holden, P.; Meadows, R.S.; Chapman, K.L.; Grant, M.E.; Kadler, K.E.; Briggs, M.D. Cartilage Oligomeric Matrix Protein Interacts with Type IX Collagen, and Disruptions to These Interactions Identify a Pathogenetic Mechanism in a Bone Dysplasia Family. J. Biol. Chem. 2001, 276, 6046–6055. [Google Scholar] [CrossRef]
- Thur, J.; Rosenberg, K.; Nitsche, D.P.; Pihlajamaa, T.; Ala-Kokko, L.; Heinegård, D.; Paulsson, M.; Maurer, P. Mutations in Cartilage Oligomeric Matrix Protein Causing Pseudoachondroplasia and Multiple Epiphyseal Dysplasia Affect Binding of Calcium and Collagen I, II, and IX. J. Biol. Chem. 2001, 276, 6083–6092. [Google Scholar] [CrossRef]
- Chen, F.H.; Herndon, M.E.; Patel, N.; Hecht, J.T.; Tuan, R.S.; Lawler, J. Interaction of Cartilage Oligomeric Matrix Protein/Thrombospondin 5 with Aggrecan. J. Biol. Chem. 2007, 282, 24591–24598. [Google Scholar] [CrossRef] [PubMed]
- Mann, H.H.; Özbek, S.; Engel, J.; Paulsson, M.; Wagener, R. Interactions between the Cartilage Oligomeric Matrix Protein and Matrilins. J. Biol. Chem. 2004, 279, 25294–25298. [Google Scholar] [CrossRef]
- Di Cesare, P.E.; Chen, F.S.; Moergelin, M.; Carlson, C.S.; Leslie, M.P.; Perris, R.; Fang, C. Matrix–matrix interaction of cartilage oligomeric matrix protein and fibronectin. Matrix Biol. 2002, 21, 461–470. [Google Scholar] [CrossRef]
- Smith, R.K.W.; Gerard, M.; Dowling, B.; Dart, A.J.; Birch, H.; Goodship, A.E. Correlation of cartilage oligomeric matrix protein (COMP) levels in equine tendon with mechanical properties: A proposed role for COMP in determining function-specific mechanical characteristics of locomotor tendons. Equine Vet. J. 2010, 34, 241–244. [Google Scholar] [CrossRef]
- Kipnes, J.; Carlberg, A.; Loredo, G.; Lawler, J.; Tuan, R.; Hall, D. Effect of cartilage oligomeric matrix protein on mesenchymal chondrogenesis in vitro. Osteoarthr. Cartil. 2003, 11, 442–454. [Google Scholar] [CrossRef][Green Version]
- Xu, K.K.; Zhang, Y.; Ilalov, K.; Carlson, C.S.; Feng, J.Q.; Di Cesare, P.E.; Liu, C. Cartilage Oligomeric Matrix Protein Associates with Granulin-Epithelin Precursor (GEP) and Potentiates GEP-stimulated Chondrocyte Proliferation. J. Biol. Chem. 2007, 282, 11347–11355. [Google Scholar] [CrossRef]
- Briggs, M.D.; Chapman, K.L. Pseudoachondroplasia and multiple epiphyseal dysplasia: Mutation review, molecular interactions, and genotype to phenotype correlations. Hum. Mutat. 2002, 19, 465–478. [Google Scholar] [CrossRef]
- Briggs, M.; Hoffman, S.; King, L.; Olsen, A.; Mohrenweiser, H.; Leroy, J.; Mortier, G.; Rimoin, D.; Lachman, R.; Gaines, E.; et al. Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nat. Genet. 1995, 10, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Briggs, M.D.; Mortier, G.; Cole, W.G.; King, L.M.; Golik, S.S.; Bonaventure, J.; Nuytinck, L.; De Paepe, A.; Leroy, J.G.; Biesecker, L.; et al. Diverse Mutations in the Gene for Cartilage Oligomeric Matrix Protein in the Pseudoachondroplasia–Multiple Epiphyseal Dysplasia Disease Spectrum. Am. J. Hum. Genet. 1998, 62, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Deere, M.; Hecht, J.T.; Lawler, J. Cartilage Oligomeric Matrix Protein Is a Calcium-binding Protein, and a Mutation in Its Type 3 Repeats Causes Conformational Changes. J. Biol. Chem. 2000, 275, 26538–26544. [Google Scholar] [CrossRef]
- Chen, T.-L.L.; Stevens, J.W.; Cole, W.G.; Hecht, J.T.; Vertel, B.M. Cell-type specific trafficking of expressed mutant COMP in a cell culture model for PSACH. Matrix Biol. 2004, 23, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.R.; Ponseti, I.V.; Maynard, J.A. Pseudoachondroplastic dwarfism. A rough-surfaced endoplasmic reticulum storage disorder. JBJS 1973, 55, 475–484. [Google Scholar] [CrossRef]
- Délot, E.; Brodie, S.G.; King, L.M.; Wilcox, W.R.; Cohn, D.H. Physiological and Pathological Secretion of Cartilage Oligomeric Matrix Protein by Cells in Culture. J. Biol. Chem. 1998, 273, 26692–26697. [Google Scholar] [CrossRef] [PubMed]
- Dicesare, P.E.; Mörgelin, M.; Carlson, C.S.; Pasumarti, S.; Paulsson, M. Cartilage oligomeric matrix protein: Isolation and characterization from human articular cartilage. J. Orthop. Res. 1995, 13, 422–428. [Google Scholar] [CrossRef]
- Dinser, R.; Zaucke, F.; Kreppel, F.; Hultenby, K.; Kochanek, S.; Paulsson, M.; Maurer, P. Pseudoachondroplasia is caused through both intra- and extracellular pathogenic pathways. J. Clin. Investig. 2002, 110, 505–513. [Google Scholar] [CrossRef]
- Duke, J.; Montufar-Solis, D.; Underwood, S.; Lalani, Z.; Hecht, J.T. Apoptosis staining in cultured pseudoachondroplasia chondrocytes. Apoptosis 2003, 8, 191–197. [Google Scholar] [CrossRef]
- Ikegawa, S.; Ohashi, H.; Nishimura, G.; Kim, K.C.; Sannohe, A.; Kimizuka, M.; Fukushima, Y.; Nagai, T.; Nakamura, Y. Novel and recurrent COMP (cartilage oligomeric matrix protein) mutations in pseudoachondroplasia and multiple epiphyseal dysplasia. Qual. Life Res. 1998, 103, 633–638. [Google Scholar] [CrossRef]
- Kleerekoper, Q.; Hecht, J.T.; Putkey, J.A. Disease-causing Mutations in Cartilage Oligomeric Matrix Protein Cause an Unstructured Ca2+ Binding Domain. J. Biol. Chem. 2002, 277, 10581–10589. [Google Scholar] [CrossRef] [PubMed]
- Maddox, B.; Mokashi, A.; Keene, D.R.; Bächinger, H.P. A Cartilage Oligomeric Matrix Protein Mutation Associated with Pseudoachondroplasia Changes the Structural and Functional Properties of the Type 3 Domain. J. Biol. Chem. 2000, 275, 11412–11417. [Google Scholar] [CrossRef] [PubMed]
- McKeand, J.; Rotta, J.; Hecht, J.T. Natural history study of pseudoachondroplasia. Am. J. Med. Genet. 1996, 63, 406–410. [Google Scholar] [CrossRef]
- Merritt, T.M.; Alcorn, J.L.; Haynes, R.; Hecht, J.T. Expression of mutant cartilage oligomeric matrix protein in human chondrocytes induces the pseudoachondroplasia phenotype. J. Orthop. Res. 2006, 24, 700–707. [Google Scholar] [CrossRef]
- Merritt, T.M.; Bick, R.; Poindexter, B.J.; Alcorn, J.L.; Hecht, J.T. Unique Matrix Structure in the Rough Endoplasmic Reticulum Cisternae of Pseudoachondroplasia Chondrocytes. Am. J. Pathol. 2007, 170, 293–300. [Google Scholar] [CrossRef]
- Unger, S.; Hecht, J.T. Pseudoachondroplasia and multiple epiphyseal dysplasia: New etiologic developments. Am. J. Med Genet. 2001, 106, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.T.; Montufar-Solis, D.; Decker, G.; Lawler, J.; Daniels, K.; Duke, P. Retention of cartilage oligomeric matrix protein (COMP) and cell death in redifferentiated pseudoachondroplasia chondrocytes. Matrix Biol. 1998, 17, 625–633. [Google Scholar] [CrossRef]
- Hecht, J.T.; Hayes, E.; Haynes, R.; Cole, W.G. COMP mutations, chondrocyte function and cartilage matrix. Matrix Biol. 2005, 23, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.T.; Hayes, E.; Snuggs, M.; Decker, G.; Montufar-Solis, D.; Doege, K.; Mwalle, F.; Poole, R.; Stevens, J.; Duke, P. Calreticulin, PDI, Grp94 and BiP chaperone proteins are associated with retained COMP in pseudoachondroplasia chondrocytes. Matrix Biol. 2001, 20, 251–262. [Google Scholar] [CrossRef]
- Hecht, J.T.; Nelson, L.D.; Crowder, E.; Wang, Y.; Elder, F.F.B.; Harrison, W.R.; Francomano, C.A.; Prange, C.K.; Lennon, G.G.; Deere, M.; et al. Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nat. Genet. 1995, 10, 325–329. [Google Scholar] [CrossRef]
- Posey, K.L.; Hayes, E.; Haynes, R.; Hecht, J.T. Role of TSP-5/COMP in Pseudoachondroplasia. Int. J. Biochem. Cell Biol. 2004, 36, 1005–1012. [Google Scholar] [CrossRef]
- Bonafe, L.; Cormier-Daire, V.; Hall, C.; Lachman, R.; Mortier, G.; Mundlos, S.; Nishimura, G.; Sangiorgi, L.; Savarirayan, R.; Sillence, D.; et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am. J. Med Genet. Part A 2015, 167, 2869–2892. [Google Scholar] [CrossRef]
- Kvansakul, M.; Adams, J.; Hohenester, E. Structure of a thrombospondin C-terminal fragment reveals a novel calcium core in the type 3 repeats. EMBO J. 2004, 23, 1223–1233. [Google Scholar] [CrossRef]
- Posey, K.L.; Veerisetty, A.C.; Liu, P.; Wang, H.R.; Poindexter, B.J.; Bick, R.; Alcorn, J.L.; Hecht, J.T. An Inducible Cartilage Oligomeric Matrix Protein Mouse Model Recapitulates Human Pseudoachondroplasia Phenotype. Am. J. Pathol. 2009, 175, 1555–1563. [Google Scholar] [CrossRef]
- Coustry, F.; Posey, K.L.; Maerz, T.; Baker, K.; Abraham, A.M.; Ambrose, C.G.; Nobakhti, S.; Shefelbine, S.J.; Bi, X.; Newton, M.; et al. Mutant cartilage oligomeric matrix protein (COMP) compromises bone integrity, joint function and the balance between adipogenesis and osteogenesis. Matrix Biol. 2018, 67, 75–89. [Google Scholar] [CrossRef]
- Posey, K.L.; Coustry, F.; Veerisetty, A.C.; Hossain, M.G.; Gambello, M.J.; Hecht, J.T. Novel mTORC1 Mechanism Suggests Therapeutic Targets for COMPopathies. Am. J. Pathol. 2019, 189, 132–146. [Google Scholar] [CrossRef]
- Posey, K.L.; Coustry, F.; Veerisetty, A.C.; Liu, P.; Alcorn, J.L.; Hecht, J.T. Chop (Ddit3) Is Essential for D469del-COMP Retention and Cell Death in Chondrocytes in an Inducible Transgenic Mouse Model of Pseudoachondroplasia. Am. J. Pathol. 2012, 180, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Coustry, F.; Posey, K.L.; Liu, P.; Alcorn, J.L.; Hecht, J.T. D469del-COMP Retention in Chondrocytes Stimulates Caspase-Independent Necroptosis. Am. J. Pathol. 2012, 180, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Posey, K.L.; Coustry, F.; Veerisetty, A.C.; Liu, P.; Alcorn, J.L.; Hecht, J.T. Chondrocyte-specific pathology during skeletal growth and therapeutics in a murine model of pseudoachondroplasia. J. Bone Miner. Res. 2014, 29, 1258–1268. [Google Scholar] [CrossRef]
- Decker, R.S.; Koyama, E.; Pacifici, M. Articular Cartilage: Structural and Developmental Intricacies and Questions. Curr. Osteoporos. Rep. 2015, 13, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Mangiavini, L.; Merceron, C.; Schipani, E. Analysis of Mouse Growth Plate Development. Curr. Protoc. Mouse Biol. 2016, 6, 67–130. [Google Scholar] [CrossRef]
- Marciniak, S.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Peter, S.B.; Mix, K.S.; Brinckerhoff, C.E. Matrix Metalloproteinases: Role In Arthritis. Front. Biosci. 2006, 11, 529–543. [Google Scholar] [CrossRef]
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 471–478. [Google Scholar] [CrossRef]
- Goldring, M.B. Articular Cartilage Degradation in Osteoarthritis. HSS J. 2012, 8, 7–9. [Google Scholar] [CrossRef]
- Jeon, H.; Im, G.-I. Autophagy in osteoarthritis. Connect. Tissue Res. 2016, 58, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, Y.R.; Otten, E.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef]
- Rim, Y.A.; Nam, Y.; Ju, J.H. The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. Int. J. Mol. Sci. 2020, 21, 2358. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, J.A.; Collins, J.A.; Loeser, R.F. Reactive oxygen species, aging and articular cartilage homeostasis. Free. Radic. Biol. Med. 2019, 132, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Coryell, P.R.; Diekman, B.O.; Loeser, R.F. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat. Rev. Rheumatol. 2021, 17, 47–57. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 2009, 17, 971–979. [Google Scholar] [CrossRef]
- Deuis, J.R.; Dvorakova, L.S.; Vetter, I. Methods Used to Evaluate Pain Behaviors in Rodents. Front. Mol. Neurosci. 2017, 10, 284. [Google Scholar] [CrossRef]
- Sheahan, T.; Copits, B.A.; Golden, J.; Iv, R.W.G. Voluntary Exercise Training: Analysis of Mice in Uninjured, Inflammatory, and Nerve-Injured Pain States. PLoS ONE 2015, 10, e0133191. [Google Scholar] [CrossRef]
- Cobos, E.J.; Portillo-Salido, E. “Bedside-to-Bench” Behavioral Outcomes in Animal Models of Pain: Beyond the Evaluation of Reflexes. Curr. Neuropharmacol. 2013, 11, 560–591. [Google Scholar] [CrossRef] [PubMed]
- Lakes, E.; Allen, K. Gait analysis methods for rodent models of arthritic disorders: Reviews and recommendations. Osteoarthr. Cartil. 2016, 24, 1837–1849. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.Y.; Kloefkorn, H.E.; Allen, K.D. Gait Analysis Methods for Rodent Models of Osteoarthritis. Curr. Pain Headache Rep. 2014, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kwok, J.; Onuma, H.; Olmer, M.; Lotz, M.; Grogan, S.; D’Lima, D. Histopathological analyses of murine menisci: Implications for joint aging and osteoarthritis. Osteoarthr. Cartil. 2016, 24, 709–718. [Google Scholar] [CrossRef]
- Hecht, J.T.; Coustry, F.; Veerisetty, A.C.; Hossain, M.G.; Posey, K.L. Resveratrol Reduces COMPopathy in Mice Through Activation of Autophagy. JBMR Plus 2021, 5. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nat. Cell Biol. 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Posey, K.L.; Alcorn, J.L.; Hecht, J.T. Pseudoachondroplasia/COMP—Translating from the bench to the bedside. Matrix Biol. 2014, 37, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Posey, K.L.; Coustry, F.; Hecht, J.T. Cartilage oligomeric matrix protein: COMPopathies and beyond. Matrix Biol. 2018, 71–72, 161–173. [Google Scholar] [CrossRef]
- Posey, K.L.; Coustry, F.; Veerisetty, A.C.; Hossain, M.; Alcorn, J.L.; Hecht, J.T. Antioxidant and anti-inflammatory agents mitigate pathology in a mouse model of pseudoachondroplasia. Hum. Mol. Genet. 2015, 24, 3918–3928. [Google Scholar] [CrossRef] [PubMed]
- Posey, K.L.; Hecht, J.T. Novel therapeutic interventions for pseudoachondroplasia. Bone 2017, 102, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Mehana, E.-S.E.; Khafaga, A.F.; El-Blehi, S.S. The role of matrix metalloproteinases in osteoarthritis pathogenesis: An updated review. Life Sci. 2019, 234, 116786. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | Genotype | Pro. Femur | Pro. Tibia | Bone/Cartilage | Synovitis | Total |
|---|---|---|---|---|---|---|
| 4 weeks | Control | 0.78 ± 0.63 | 0.56 ± 0.50 | 0.11 ± 0.31 | 0.33 ± 0.47 | 1.78 ± 1.13 |
| MT-COMP | 0.70 ± 0.64 | 0.30 ± 0.46 | 0.40 ± 0.66 | 1.0 ± 0.89 | 2.4 ± 1.85 | |
| p value | - | - | - | - | - | |
| Control | 0.80 ± 0.60 | 0.60 ± 0.66 | 0.10 ± 0.30 | 0.40 ± 0.66 | 1.90 ± 1.76 | |
| 8 weeks | MT-COMP | 1.3 ± 0.90 | 0.63 ± 0.14 | 0.40 ± 0.49 | 0.60 ± 0.92 | 2.28 ± 0.11 |
| p value | - | - | - | - | - | |
| 12 weeks | Control | 0.77 ± 0.32 | 0.57 ± 0.26 | 0.30 ± 0.20 | 0.52 ± 0.30 | 1.48 ± 1.06 |
| MT-COM | 1.10 ± 0.83 | 1.20 ± 1.10 | 0.90 ± 0.94 | 1.00 ± 0.89 | 4.20 ± 3.25 | |
| p value | - | - | - | - | - | |
| 16 weeks | Control | 1.00 ± 0.63 | 0.80 ± 0.60 | 0.50 ± 0.50 | 0.90 ± 0.83 | 3.20 ± 2.27 |
| MT-COMP | 1.60 ± 0.49 | 1.10 ± 0.54 | 1.10 ± 0.83 | 1.50 ± 0.81 | 5.30 ± 2.19 | |
| p value | 0.0372 * | - | 0.0798 | - | 0.0614 | |
| 20 weeks | Control | 1.00 ± 0.78 | 0.56 ± 0.65 | 0.11 ± 0.32 | 0.89 ± 0.83 | 2.56 ± 2.15 |
| MT-COMP | 1.80 ± 0.87 | 1.20 ± 0.60 | 0.90 ± 0.70 | 1.80 ± 0.60 | 5.70 ± 2.14 | |
| p value | 0.0683 | 0.0542 | 0.0091 ** | 0.0218 * | 0.0133 * |
| Mutant-COMP Pathology | 2 | 4 | 8 | 12 | 16 | 20 Weeks |
|---|---|---|---|---|---|---|
| Mutant-COMP intracellular retention | X | X | X | X | X | |
| Proteoglycan loss (Safranin O) | X | X | ||||
| ER stress (CHOP) | X | X | X | |||
| Chondrocyte death (TUNEL) | X | X | X | X | ||
| Inflammation (TNFα) | * | X | X | X | X | X |
| Autophagy block (pS6) | * | X | X | X | ||
| Enzymatic degradation (MMP-13) | * | X | X | X | ||
| Senescent chondrocytes (p16 INK4a) | * | X | X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hecht, J.T.; Veerisetty, A.C.; Hossain, M.G.; Patra, D.; Chiu, F.; Coustry, F.; Posey, K.L. Joint Degeneration in a Mouse Model of Pseudoachondroplasia: ER Stress, Inflammation, and Block of Autophagy. Int. J. Mol. Sci. 2021, 22, 9239. https://doi.org/10.3390/ijms22179239
Hecht JT, Veerisetty AC, Hossain MG, Patra D, Chiu F, Coustry F, Posey KL. Joint Degeneration in a Mouse Model of Pseudoachondroplasia: ER Stress, Inflammation, and Block of Autophagy. International Journal of Molecular Sciences. 2021; 22(17):9239. https://doi.org/10.3390/ijms22179239
Chicago/Turabian StyleHecht, Jacqueline T., Alka C. Veerisetty, Mohammad G. Hossain, Debabrata Patra, Frankie Chiu, Francoise Coustry, and Karen L. Posey. 2021. "Joint Degeneration in a Mouse Model of Pseudoachondroplasia: ER Stress, Inflammation, and Block of Autophagy" International Journal of Molecular Sciences 22, no. 17: 9239. https://doi.org/10.3390/ijms22179239
APA StyleHecht, J. T., Veerisetty, A. C., Hossain, M. G., Patra, D., Chiu, F., Coustry, F., & Posey, K. L. (2021). Joint Degeneration in a Mouse Model of Pseudoachondroplasia: ER Stress, Inflammation, and Block of Autophagy. International Journal of Molecular Sciences, 22(17), 9239. https://doi.org/10.3390/ijms22179239