
The Role of ER Stress-Related Phenomena in the Biology of Malignant Peripheral Nerve Sheath Tumors


{kind=link}
Abstract
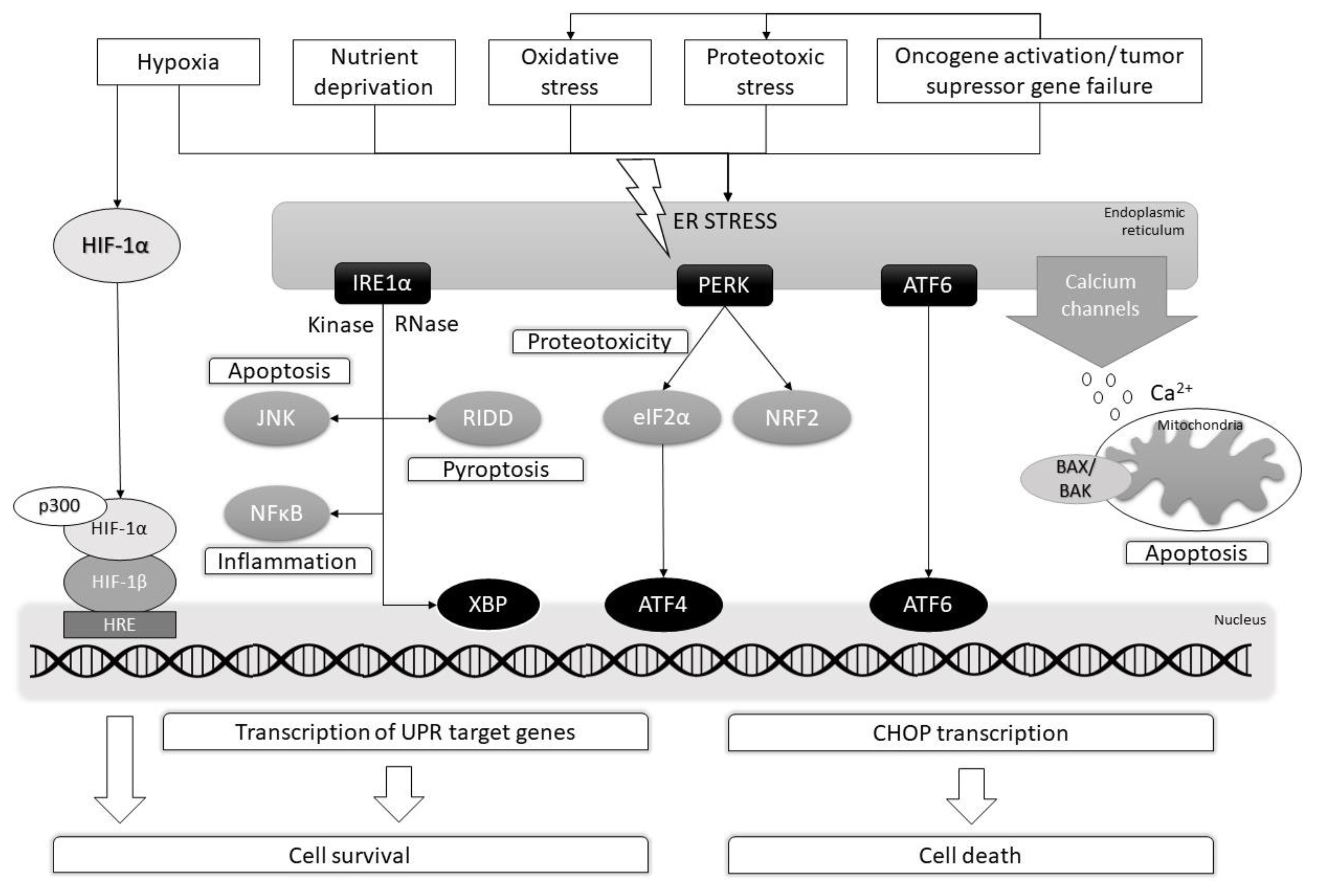
:1. Introduction
2. MPNST Pathogenesis
3. Proteotoxic Stress
4. Hypoxia
5. Oxidative Stress
6. The Role of Nutrient Deprivation
7. ER-Stress and Its Regulators
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hirbe, A.C.; Gutmann, D.H. The management of neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors: Challenges, progress, and future prospects. Expert Opin. Orphan Drugs 2017, 5, 623–631. [Google Scholar] [CrossRef]
- Bottillo, I.; Ahlquist, T.; Brekke, H.; Danielsen, S.A.; van den Berg, E.; Mertens, F.; Lothe, R.A.; Dallapiccola, B. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J. Pathol. 2009, 217, 693–701. [Google Scholar] [CrossRef]
- Farid, M.; Demicco, E.G.; Garcia, R.; Ahn, L.; Merola, P.R.; Cioffi, A.; Maki, R.G. Malignant peripheral nerve sheath tumors. Oncologist 2014, 19, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Pala-Sadza, A.; Zajączkiewicz, H.; Banaś-Samson, R.; Glinianowicz, I.; Ożóg, J.; Gnacik-Stawarczyk, E.; Dubrawska, M.; Rauch, B. Rzadki przypadek złośliwego nerwiaka osłonkowego jamy nosa. Pol. Przegląd Otorynolaryngologiczny 2014, 3, 210–213. [Google Scholar] [CrossRef]
- Ralli, M.; Singh, S.; Hasija, S.; Verma, R. Intrathoracic Malignant Peripheral Nerve Sheath Tumor: Histopathological and Immunohistochemical Features. Iran. J. Pathol. 2015, 10, 74–78. [Google Scholar] [PubMed]
- Watson, K.L.; Al Sannaa, G.A.; Kivlin, C.M.; Ingram, D.R.; Landers, S.M.; Roland, C.L.; Cormier, J.N.; Hunt, K.K.; Feig, B.W.; Ashleigh Guadagnolo, B.; et al. Patterns of recurrence and survival in sporadic, neurofibromatosis Type 1-associated, and radiation-associated malignant peripheral nerve sheath tumors. J. Neurosurg. 2017, 126, 319–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Tang, X.; Liang, H.; Yang, R.; Yan, T.; Guo, W. Prognosis and risk factors for malignant peripheral nerve sheath tumor: A systematic review and meta-analysis. World J. Surg Oncol. 2020, 18, 257. [Google Scholar] [CrossRef]
- Maane, L.A.; Bouzidi, A.A.L.; Damou, M.; Ismaili, N. Primary intrapulmonary malignant peripheral nerve sheath tumor: A rare case. Cancer Treat. Res. Commun. 2020, 25, 100243. [Google Scholar] [CrossRef]
- Morimoto, A.; Asai, J.; Wakabayashi, Y.; Tashima, S.; Wada, M.; Iida, S.; Komori, S.; Hanada, K.; Takenaka, H.; Katoh, N. Lymph node metastasis of a malignant peripheral nerve sheath tumor without distant metastasis. Indian J. Dermatol. 2014, 59, 635. [Google Scholar]
- Meng, Z.H.; Yang, Y.S.; Cheng, K.L.; Chen, G.Q.; Wang, L.P.; Li, W. A huge malignant peripheral nerve sheath tumor with hepatic metastasis arising from retroperitoneal ganglioneuroma. Oncol. Lett. 2013, 5, 123–126. [Google Scholar] [CrossRef]
- Malone, C.F.; Emerson, C.; Ingraham, R.; Barbosa, W.; Guerra, S.; Yoon, H.; Liu, L.L.; Michor, F.; Haigis, M.; Macleod, K.F.; et al. mTOR and HDAC inhibitors converge on the TXNIP/thioredoxin pathway to cause catastrophic oxidative stress and regression of RAS-driven tumors. Cancer Discov. 2017, 7, 1450–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, S.; Endo, M.; Matsumoto, Y.; Fukushi, J.I.; Matsunobu, T.; Kawaguchi, K.I.; Setsu, N.; IIda, K.; Yokoyama, N.; Nakagawa, M.; et al. Hypoxia-inducible factor 1 alpha is a poor prognostic factor and potential therapeutic target in malignant peripheral nerve sheath tumor. PLoS ONE 2017, 12, e0178064. [Google Scholar] [CrossRef]
- Castorina, A.; Giunta, S.; Scuderi, S.; D’Agata, V. Involvement of PACAP/ADNP signaling in the resistance to cell death in malignant peripheral nerve sheath tumor (MPNST) cells. J. Mol. Neurosci. 2012, 48, 674–683. [Google Scholar] [CrossRef]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Reviews. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Chipurupalli, S.; Kannan, E.; Tergaonkar, V.; D’Andrea, R.; Robinson, N. Hypoxia induced ER stress response as an adaptive mechanism in cancer. Int. J. Mol. Sci. 2019, 20, 749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blazanin, N.; Son, J.; Craig-Lucas, A.B.; John, C.L.; Breech, K.J.; Podolsky, M.A.; Glick, A.B. ER stress and distinct outputs of the IRE1α RNase control proliferation and senescence in response to oncogenic Ras. Proc. Natl. Acad. Sci. USA 2017, 114, 9900–9905. [Google Scholar] [CrossRef] [Green Version]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar]
- Tang, Y.C.; Williams, B.R.; Siegel, J.J.; Amon, A. Identification of aneuploidy-selective antiproliferation compounds. Cell 2011, 144, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Limonta, P.; Moretti, R.M.; Marzagalli, M.; Fontana, F.; Raimondi, M.; Montagnani Marelli, M. Role of endoplasmic reticulum stress in the anticancer activity of natural compounds. Int. J. Mol. Sci. 2019, 20, 961. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.K.; Chae, S.-W.; Kim, H.-R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, Y.; Jones, S.; Sausen, M.; McMahon, K.; Sharma, R.; Wang, Q.; Belzberg, A.J.; Chaichana, K.; Gallia, G.L.; et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1170–1172. [Google Scholar] [CrossRef] [Green Version]
- Beert, E.; Brems, H.; Daniëls, B.; De Wever, I.; Van Calenbergh, F.; Schoenaers, J.; Debiec-Rychter, M.; Gevaert, O.; De Raedt, T.; Van Den Bruel, A.; et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer 2011, 50, 1021–1032. [Google Scholar] [CrossRef]
- Hirbe, A.C.; Dahiya, S.; Miller, C.A.; Li, T.; Fulton, R.S.; Zhang, X.; McDonald, S.; DeSchryver, K.; Duncavage, E.J.; Walrath, J.; et al. Whole exome sequencing reveals the order of genetic changes during malignant transformation and metastasis in a single patient with NF1-plexiform neurofibroma. Clin. Cancer Res. 2015, 21, 4201–4211. [Google Scholar] [CrossRef] [Green Version]
- Carroll, S.L. The challenge of cancer genomics in rare nervous system neoplasms: Malignant peripheral nerve sheath tumors as a paradigm for cross-species comparative oncogenomics. Am. J. Pathol. 2016, 186, 464–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Liu, W.; Williams, J.P.; Ratner, N. EGFR-Stat3 signalling in nerve glial cells modifies neurofibroma initiation. Oncogene 2017, 36, 1669–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Velasco-Miguel, S.; Vass, W.C.; Parada, L.F.; DeClue, J.E. Epidermal growth factor receptor signaling pathways are associated with tumorigenesis in the Nf1:p53 mouse tumor model. Cancer Res. 2002, 62, 4507–4513. [Google Scholar]
- Wiles, E.T.; Selker, E.U. H3K27 methylation: A promiscuous repressive chromatin mark. Curr. Opin. Genet. Dev. 2017, 43, 31–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Raedt, T.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014, 514, 247–251. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Granada, C.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.; Fan, X.; Lei, Y.; Chen, R.; Wang, J.; Fu, C.; Yi, S.; Luo, J.; Zhang, S.; Yang, Q.; et al. A novel de novo microdeletion at 17q11.2 adjacent to NF1 gene associated with developmental delay, short stature, microcephaly and dysmorphic features. Mol. Cytogenet. 2016, 9, 41. [Google Scholar] [CrossRef] [Green Version]
- Cleven, A.H.; Sannaa, G.A.; Briaire-de Bruijn, I.; Ingram, D.R.; van de Rijn, M.; Rubin, B.P.; de Vries, M.W.; Watson, K.L.; Torres, K.E.; Wang, W.L.; et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod. Pathol. 2016, 29, 582–590. [Google Scholar] [CrossRef] [Green Version]
- Meany, H.; Dombi, E.; Reynolds, J.; Whatley, M.; Kurwa, A.; Tsokos, M.; Salzer, W.; Gillespie, A.; Baldwin, A.; Derdak, J.; et al. 18-fluorodeoxyglucose-positron emission tomography (FDG-PET) evaluation of nodular lesions in patients with Neurofibromatosis type 1 and plexiform neurofibromas (PN) or malignant peripheral nerve sheath tumors (MPNST). Pediatric Blood Cancer 2013, 60, 59–64. [Google Scholar] [CrossRef]
- Vasudevan, A.; Schukken, K.M.; Sausville, E.L.; Girish, V.; Adebambo, O.A.; Sheltzer, J.M. Aneuploidy as a promoter and suppressor of malignant growth. Nat. Rev. Cancer 2021, 21, 89–103. [Google Scholar] [CrossRef]
- Oromendia, A.B.; Amon, A. Aneuploidy: Implications for protein homeostasis and disease. Dis. Models Mech. 2014, 7, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheltzer, J.M.; Torres, E.M.; Dunham, M.J.; Amon, A. Transcriptional consequences of aneuploidy. Proc. Natl. Acad. Sci. USA 2012, 109, 12644–12649. [Google Scholar] [CrossRef] [Green Version]
- Oromendia, A.B.; Dodgson, S.E.; Amon, A. Aneuploidy causes proteotoxic stress in yeast. Genes Dev. 2012, 26, 2696–2708. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, A.; Ohori, M.; Iwai, K.; Nakayama, Y.; Nambu, T.; Morishita, D.; Kawamoto, T.; Miyamoto, M.; Hirayama, T.; Okaniwa, M.; et al. Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53-mediated post-mitotic apoptosis in SAC-impaired cells. Nat. Commun. 2015, 6, 7668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridge, R.S., Jr.; Bridge, J.A.; Neff, J.R.; Naumann, S.; Althof, P.; Bruch, L.A. Recurrent chromosomal imbalances and structurally abnormal breakpoints within complex karyotypes of malignant peripheral nerve sheath tumour and malignant triton tumour: A cytogenetic and molecular cytogenetic study. J. Clin. Pathol. 2004, 57, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.M.; Dephoure, N.; Panneerselvam, A.; Tucker, C.M.; Whittaker, C.A.; Gygi, S.P.; Dunham, M.J.; Amon, A. Identification of aneuploidy-tolerating mutations. Cell 2010, 143, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Santagata, S.; Tang, Z.; Shi, J.; Cao, J.; Kwon, H.; Bronson, R.T.; Whitesell, L.; Lindquist, S. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J. Clin. Investig. 2012, 122, 3742–3754. [Google Scholar] [CrossRef] [Green Version]
- Pobre, K.F.R.; Poet, G.J. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef] [Green Version]
- De Raedt, T.; Walton, Z.; Yecies, J.L.; Li, D.; Chen, Y.; Malone, C.F.; Maertens, O.; Jeong, S.M.; Bronson, R.T.; Lebleu, V.; et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell 2011, 20, 400–413. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Xing, Y.; Liu, Y. Emerging roles for the ER stress sensor IRE1α in metabolic regulation and disease. J. Biol. Chem. 2019, 294, 18726–18741. [Google Scholar] [CrossRef] [Green Version]
- Carrara, M.; Prischi, F.; Ali, M.M.U. UPR signal activation by luminal sensor domains. Int. J. Mol. Sci. 2013, 14, 6454–6466. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.Y.; Hurwitz, B.S.; Bolyard, C.; Yu, J.G.; Zhang, J.; Selvendiran, K.; Rath, K.S.; He, S.; Bailey, Z.; Eaves, D.; et al. Bortezomib-induced unfolded protein response increases oncolytic HSV-1 replication resulting in synergistic antitumor effects. Clin. Cancer Res. 2014, 20, 3787–3798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, M.; Yamamoto, H.; Setsu, N.; Kohashi, K.; Takahashi, Y.; Ishii, T.; Iida, K.; Matsumoto, Y.; Hakozaki, M.; Aoki, M.; et al. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin. Cancer Res. 2013, 19, 450–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rad, E.; Dodd, K.; Thomas, L.; Upadhyaya, M.; Tee, A. STAT3 and HIF1α signaling drives oncogenic cellular phenotypes in malignant peripheral nerve sheath tumors. Mol. Cancer Res. 2015, 13, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-modified cancer cell metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, C.; Fontana, S. Hypoxia and HIF signaling: One axis with divergent effects. Int. J. Mol. Sci. 2020, 21, 5611. [Google Scholar] [CrossRef] [PubMed]
- Sendoel, A.; Hengartner, M.O. Apoptotic cell death under hypoxia. Physiology 2014, 29, 168–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasa, J.; Nishida, Y.; Suzuki, Y.; Tsukushi, S.; Shido, Y.; Hosono, K.; Shimoyama, Y.; Nakamura, S.; Ishiguro, N. Differential expression of angiogenic factors in peripheral nerve sheath tumors. Clin. Exp. Metastasis 2008, 25, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Gampala, S.; Shah, F.; Zhang, C.; Rhodes, S.D.; Babb, O.; Grimard, M.; Wireman, R.S.; Rad, E.; Calver, B.; Bai, R.-Y.; et al. Exploring transcriptional regulators Ref-1 and STAT3 as therapeutic targets in malignant peripheral nerve sheath tumours. Br. J. Cancer 2021, 124, 1566–1580. [Google Scholar] [CrossRef]
- Krawczyk, M.A.; Kunc, M. High expression of solute carrier family 2 member 1 (SLC2A1) in cancer cells is an independent unfavorable prognostic factor in pediatric malignant peripheral nerve sheath tumor. Diagnostics 2021, 11, 598. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free. Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NavaneethaKrishnan, S.; Rosales, J.L.; Lee, K.Y. ROS-mediated cancer cell killing through dietary phytochemicals. Oxidative Med. Cell. Longev. 2019, 2019, 9051542. [Google Scholar] [CrossRef]
- Brewer, T.F.; Garcia, F.J.; Onak, C.S.; Carroll, K.S.; Chang, C.J. Chemical approaches to discovery and study of sources and targets of hydrogen peroxide redox signaling through NADPH oxidase proteins. Annu. Rev. Biochem. 2015, 84, 765–790. [Google Scholar] [CrossRef]
- Maertens, O.; Johnson, B.; Hollstein, P.; Frederick, D.T.; Cooper, Z.A.; Messiaen, L.; Bronson, R.T.; McMahon, M.; Granter, S.; Flaherty, K.; et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013, 3, 338–349. [Google Scholar] [CrossRef] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- TRPA1-mediated Ca2+ influx protects tumor cells from oxidative stress. Cancer Discov. 2018, 8, 790.
- Miller, S.J.; Jessen, W.J.; Mehta, T.; Hardiman, A.; Sites, E.; Kaiser, S.; Jegga, A.G.; Li, H.; Upadhyaya, M.; Giovannini, M. Integrative genomic analyses of neurofibromatosis tumours identify SOX9 as a biomarker and survival gene. EMBO Mol. Med. 2009, 1, 236–248. [Google Scholar] [CrossRef]
- Takahashi, N.; Chen, H.-Y.; Harris, I.S.; Stover, D.G.; Selfors, L.M.; Bronson, R.T.; Deraedt, T.; Cichowski, K.; Welm, A.L.; Mori, Y.; et al. Cancer cells Co-opt the neuronal redox-sensing channel TRPA1 to promote oxidative-stress tolerance. Cancer Cell 2018, 33, 985–1003.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Chen, C.; Chen, L.; Yu, C.; Kondo, T. Decorin as a prognostic biomarker in patients with malignant peripheral nerve sheath tumors. Oncol. Lett. 2019, 17, 3517–3522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.R.; Cohen, J.E.; Tendi, E.A.; Farrer, R.; De Vries, G.H.; Becker, K.G.; Fields, R.D. Transcriptional profiling in an MPNST-derived cell line and normal human Schwann cells. Neuron Glia Biol. 2004, 1, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Mellier, G.; Pervaiz, S. The three Rs along the TRAIL: Resistance, re-sensitization and reactive oxygen species (ROS). Free. Radic. Res. 2012, 46, 996–1003. [Google Scholar] [CrossRef]
- Reuss, D.E.; Mucha, J.; Hagenlocher, C.; Ehemann, V.; Kluwe, L.; Mautner, V.; von Deimling, A. Sensitivity of malignant peripheral nerve sheath tumor cells to TRAIL is augmented by loss of NF1 through modulation of MYC/MAD and is potentiated by curcumin through induction of ROS. PLoS ONE 2013, 8, e57152. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Hung, S.H.; Huang, M.C.; Tsai, T.; Chen, C.T. Doxycycline potentiates antitumor effect of 5-aminolevulinic acid-mediated photodynamic therapy in malignant peripheral nerve sheath tumor cells. PLoS ONE 2017, 12, e0178493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberti, M.V.; Locasale, J.W. The warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Bolaños, J.P.; Almeida, A.; Moncada, S. Glycolysis: A bioenergetic or a survival pathway? Trends Biochem. Sci. 2010, 35, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Badana, A.K.; G, M.M.; G, S.; Malla, R. Reactive oxygen species: A key constituent in cancer survival. Biomark Insights 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Ruiz, R.; Rigoulet, M.; Devin, A. The warburg and crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim. Biophys. Acta 2011, 1807, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Linke, C.; Wösle, M.; Harder, A. Anti-cancer agent 3-bromopyruvate reduces growth of MPNST and inhibits metabolic pathways in a representative in-vitro model. BMC Cancer 2020, 20, 896. [Google Scholar] [CrossRef] [PubMed]
- Finicle, B.T.; Jayashankar, V.; Edinger, A.L. Nutrient scavenging in cancer. Nat. Rev. Cancer 2018, 18, 619–633. [Google Scholar] [CrossRef]
- Macchioni, L.; Davidescu, M.; Sciaccaluga, M.; Marchetti, C.; Migliorati, G.; Coaccioli, S.; Roberti, R.; Corazzi, L.; Castigli, E. Mitochondrial dysfunction and effect of antiglycolytic bromopyruvic acid in GL15 glioblastoma cells. J. Bioenerg. Biomembr. 2011, 43, 507. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, H.J.; Shin, J.; Kang, S.T.; Kang, S.; Yoo, Y.D. Bcl-XL prevents serum deprivation-induced oxidative stress mediated by Romo1. Oncol. Rep. 2011, 25, 1337–1342. [Google Scholar]
- Moncsek, A.; Gruner, M.; Meyer, H.; Lehmann, A.; Kloetzel, P.M.; Stohwasser, R. Evidence for anti-apoptotic roles of proteasome activator 28γ via inhibiting caspase activity. Apoptosis 2015, 20, 1211–1228. [Google Scholar] [CrossRef]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the warburg effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [Green Version]
- Mishra, D.; Banerjee, D. Lactate Dehydrogenases as metabolic links between tumor and stroma in the tumor microenvironment. Cancers 2019, 11, 750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin structure, functions and regulation. Cells 2020, 9, 2365. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Sun, D.; Dong, L.; Zhu, H.; Hou, H. Advancement in research and therapy of NF1 mutant malignant tumors. Cancer Cell Int. 2020, 20, 492. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Reczek, E.E.; James, M.F.; Brems, H.; Legius, E.; Cichowski, K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc. Natl. Acad. Sci. USA 2005, 102, 8573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannessen, C.M.; Johnson, B.W.; Williams, S.M.; Chan, A.W.; Reczek, E.E.; Lynch, R.C.; Rioth, M.J.; McClatchey, A.; Ryeom, S.; Cichowski, K. TORC1 is essential for NF1-associated malignancies. Curr. Biol. CB 2008, 18, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Denoyelle, C.; Abou-Rjaily, G.; Bezrookove, V.; Verhaegen, M.; Johnson, T.M.; Fullen, D.R.; Pointer, J.N.; Gruber, S.B.; Su, L.D.; Nikiforov, M.A.; et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat. Cell Biol. 2006, 8, 1053–1063. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Marcu, M.G.; Doyle, M.; Bertolotti, A.; Ron, D.; Hendershot, L.; Neckers, L. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1α. Mol. Cell. Biol. 2002, 22, 8506–8513. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Lu, Y.; Okuno, S.H.; Reinke, D.; Maertens, O.; Perentesis, J.; Basu, M.; Wolters, P.L.; De Raedt, T.; Chawla, S.; et al. Targeting refractory sarcomas and malignant peripheral nerve sheath tumors in a Phase I/II study of sirolimus in combination with ganetespib (SARC023). Sarcoma 2020, 2020, 5784876. [Google Scholar] [CrossRef] [PubMed]
- Karpel-Massler, G.; Shu, C.; Chau, L.; Banu, M.; Halatsch, M.E.; Westhoff, M.A.; Ramirez, Y.; Ross, A.H.; Bruce, J.N.; Canoll, P.; et al. Combined inhibition of Bcl-2/Bcl-xL and Usp9X/Bag3 overcomes apoptotic resistance in glioblastoma in vitro and in vivo. Oncotarget 2015, 6, 14507–14521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtaza, M.; Jolly, L.A.; Gecz, J.; Wood, S.A. La FAM fatale: USP9X in development and disease. Cell. Mol. Life Sci. 2015, 72, 2075–2089. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Zhang, S.; Wang, Z.; Yang, C.; Ouyang, W.; Zhou, F.; Zhou, Y.; Xie, C. Deubiquitinase USP9X deubiquitinates β-catenin and promotes high grade glioma cell growth. Oncotarget 2016, 7, 79515–79525. [Google Scholar] [CrossRef] [Green Version]
- Engel, K.; Rudelius, M.; Slawska, J.; Jacobs, L.; Ahangarian Abhari, B.; Altmann, B.; Kurutz, J.; Rathakrishnan, A.; Fernández-Sáiz, V.; Brunner, A.; et al. USP9X stabilizes XIAP to regulate mitotic cell death and chemoresistance in aggressive B-cell lymphoma. EMBO Mol. Med. 2016, 8, 851–862. [Google Scholar] [CrossRef]
- Kapuria, V.; Peterson, L.F.; Fang, D.; Bornmann, W.G.; Talpaz, M.; Donato, N.J. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 2010, 70, 9265–9276. [Google Scholar] [CrossRef] [Green Version]
- Ishida, C.T.; Bianchetti, E.; Shu, C.; Halatsch, M.E.; Westhoff, M.A.; Karpel-Massler, G.; Siegelin, M.D. BH3-mimetics and BET-inhibitors elicit enhanced lethality in malignant glioma. Oncotarget 2017, 8, 29558–29573. [Google Scholar] [CrossRef] [Green Version]
- Singleton, D.C.; Harris, A.L. Targeting the ATF4 pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhong, N.; Liu, G.; Chen, K.; Liu, X.; Su, L.; Singhal, S. Usp9x- and Noxa-mediated Mcl-1 downregulation contributes to pemetrexed-induced apoptosis in human non-small-cell lung cancer cells. Cell Death Dis. 2014, 5, e1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS sources in physiological and pathological conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Eletto, D.; Chevet, E.; Argon, Y.; Appenzeller-Herzog, C. Redox controls UPR to control redox. J. Cell Sci. 2014, 127, 3649–3658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walczak, A.; Radek, M.; Majsterek, I. The Role of ER Stress-Related Phenomena in the Biology of Malignant Peripheral Nerve Sheath Tumors. Int. J. Mol. Sci. 2021, 22, 9405. https://doi.org/10.3390/ijms22179405
Walczak A, Radek M, Majsterek I. The Role of ER Stress-Related Phenomena in the Biology of Malignant Peripheral Nerve Sheath Tumors. International Journal of Molecular Sciences. 2021; 22(17):9405. https://doi.org/10.3390/ijms22179405
Chicago/Turabian StyleWalczak, Anna, Maciej Radek, and Ireneusz Majsterek. 2021. "The Role of ER Stress-Related Phenomena in the Biology of Malignant Peripheral Nerve Sheath Tumors" International Journal of Molecular Sciences 22, no. 17: 9405. https://doi.org/10.3390/ijms22179405