
A POLE Splice Site Deletion Detected in a Patient with Biclonal CLL and Prostate Cancer: A Case Report

 , ,
, ,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
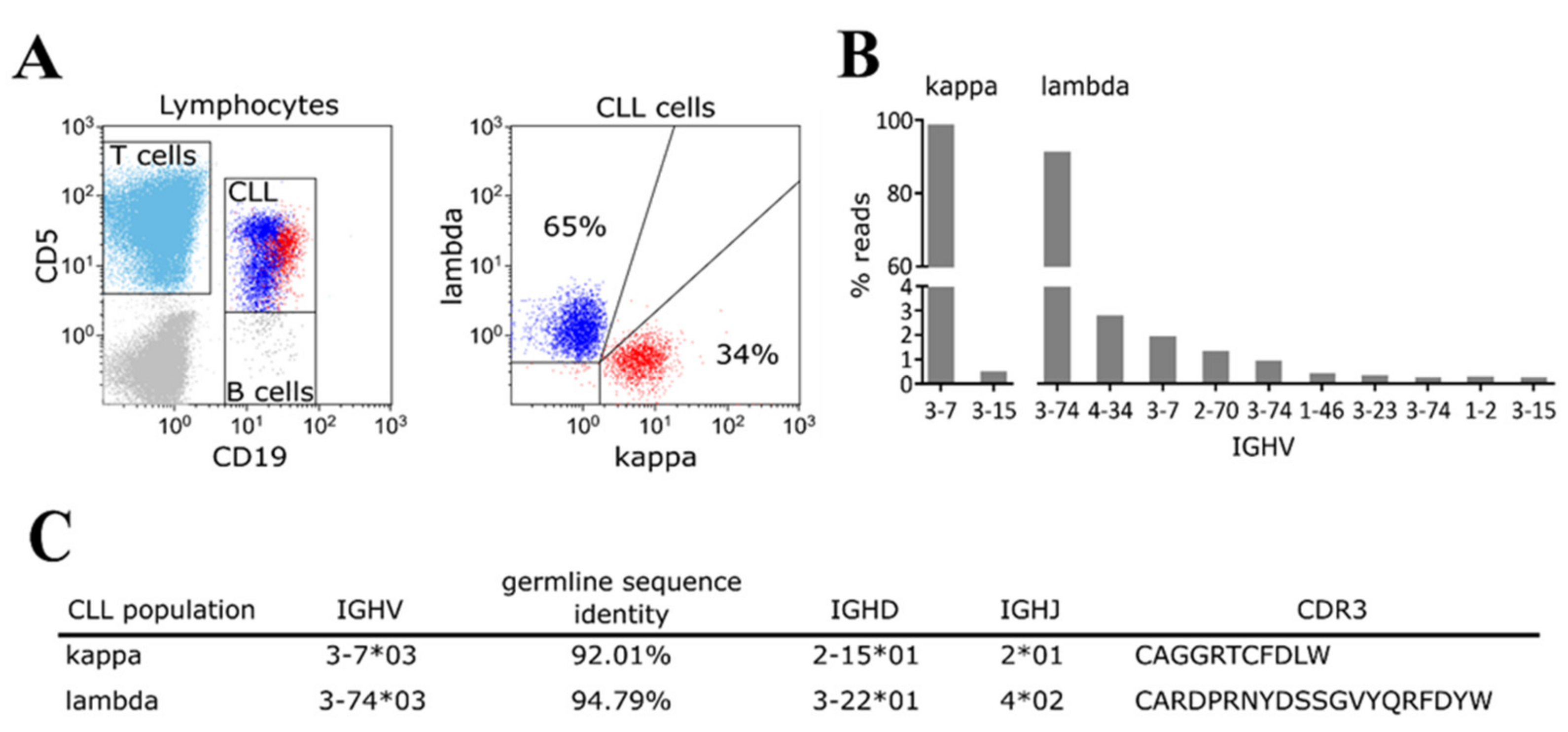
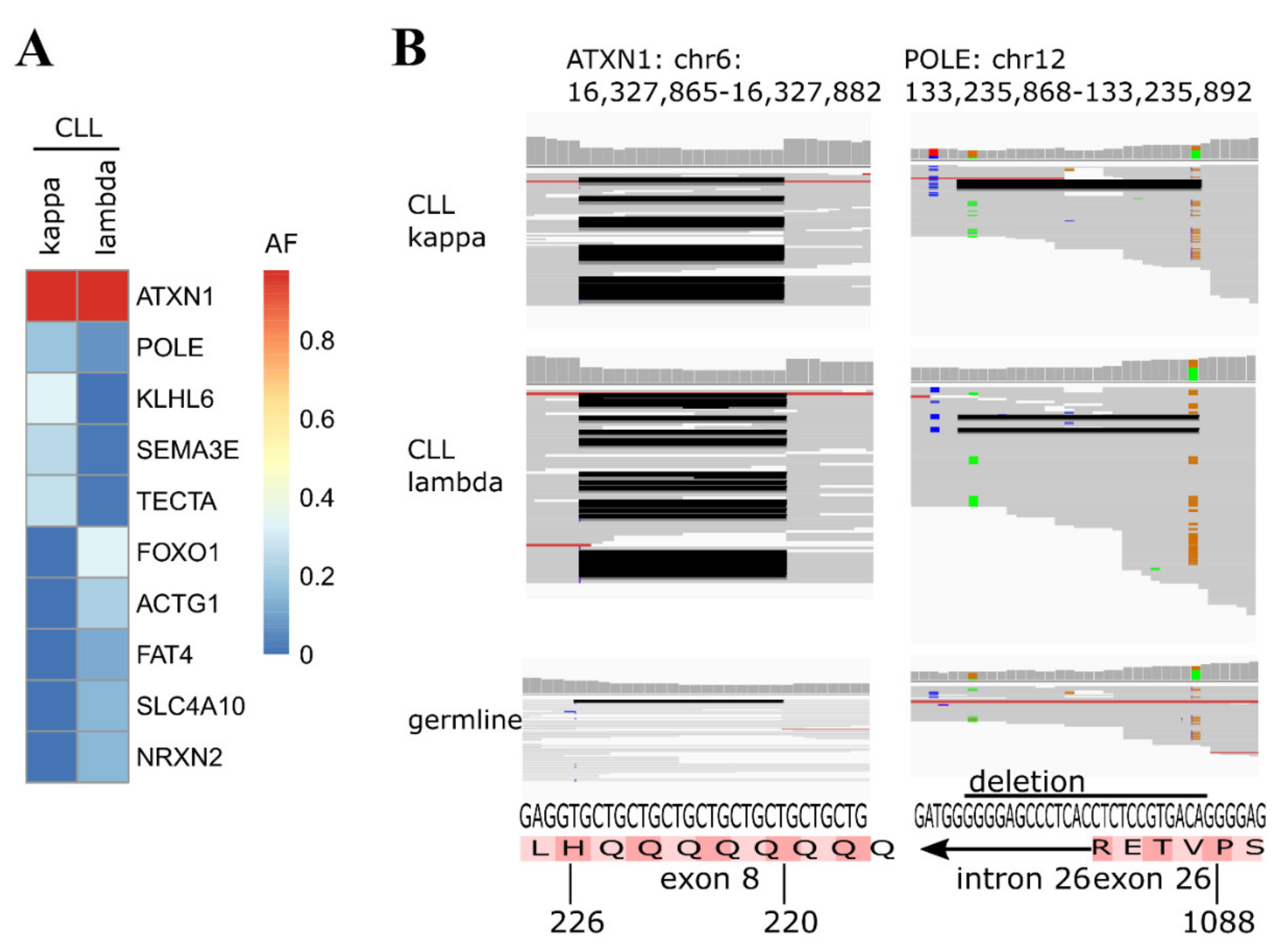
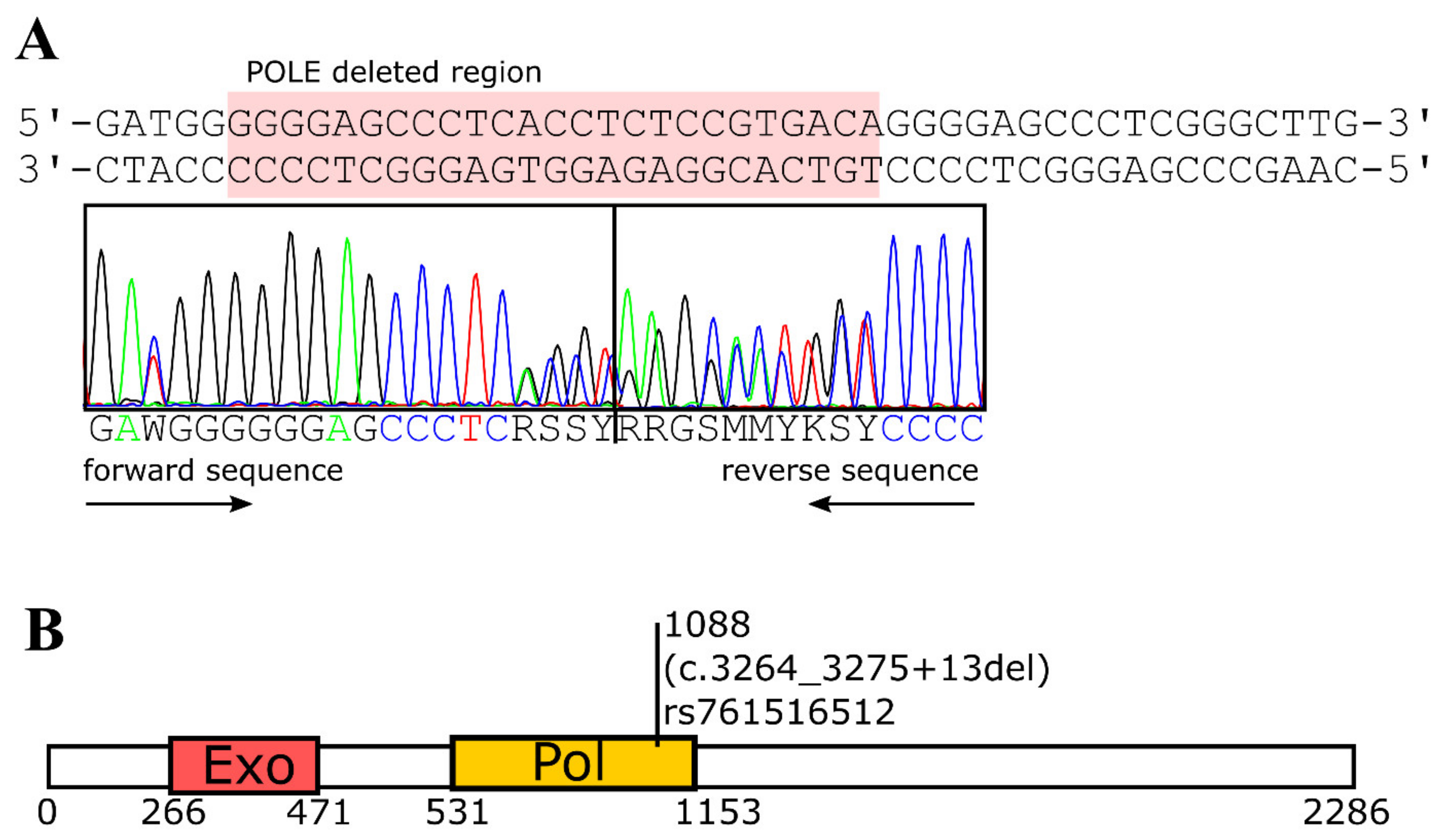
2. Results
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Flow Cytometric Kappa/Lambda Staining and Cell Sorting
4.3. IGHV Mutation/Rearrangement Analysis
4.4. Focused Exome Sequencing
4.5. Sequencing Data Analysis
4.6. Sanger Sequencing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Milne, K.; Sturrock, B.; Chevassut, T. Chronic Lymphocytic Leukaemia in 2020: The Future Has Arrived. Curr. Oncol. Rep. 2020, 22, 36. [Google Scholar] [CrossRef] [Green Version]
- Pleyer, L.; Egle, A.; Hartmann, T.N.; Greil, R. Molecular and cellular mechanisms of CLL: Novel therapeutic approaches. Nat. Rev. Clin. Oncol. 2009, 6, 405–418. [Google Scholar] [CrossRef]
- Zaborsky, N.; Gassner, F.J.; Höpner, J.P.; Schubert, M.; Hebenstreit, D.; Stark, R.; Asslaber, D.; Steiner, M.; Geisberger, R.; Greil, R.; et al. Exome sequencing of the TCL1 mouse model for CLL reveals genetic heterogeneity and dynamics during disease development. Leukemia 2018, 33, 957–968. [Google Scholar] [CrossRef]
- Langerak, A.W.; Davi, F.; Ghia, P.; Hadzidimitriou, A.; Murray, F.; Potter, K.N.; Rosenquist, R.; Stamatopoulos, K.; Belessi, C. Immunoglobulin sequence analysis and prognostication in CLL: Guidelines from the ERIC review board for reliable interpretation of problematic cases. Leukemia 2011, 25, 979–984. [Google Scholar] [CrossRef] [Green Version]
- Brazdilova, K.; Plevova, K.; Francova, H.S.; Kockova, H.; Borsky, M.; Bikos, V.; Malcikova, J.; Oltova, A.; Kotaskova, J.; Tichy, B.; et al. Multiple productive IGH rearrangements denote oligoclonality even in immunophenotypically monoclonal CLL. Leukemia 2017, 32, 234–236. [Google Scholar] [CrossRef]
- Sanchez, M.-L.; Almeida, J.; Gonzalez, D.; Gonzalez, M.; Garcia-Marcos, M.-A.; Balanzategui, A.; Lopez-Berges, M.-C.; Nomdedeu, J.; Vallespi, T.; Barbon, M.; et al. Incidence and clinicobiologic characteristics of leukemic B-cell chronic lymphoproliferative disorders with more than one B-cell clone. Blood 2003, 102, 2994–3002. [Google Scholar] [CrossRef]
- Chang, H.; Cerny, J. Molecular Characterization of Chronic Lymphocytic Leukemia with Two Distinct Cell Populations: Evidence for Separate Clonal Origins. Am. J. Clin. Pathol. 2006, 126, 23–28. [Google Scholar] [CrossRef]
- Kikushige, Y.; Ishikawa, F.; Miyamoto, T.; Shima, T.; Urata, S.; Yoshimoto, G.; Mori, Y.; Iino, T.; Yamauchi, T.; Eto, T.; et al. Self-Renewing Hematopoietic Stem Cell Is the Primary Target in Pathogenesis of Human Chronic Lymphocytic Leukemia. Cancer Cell 2011, 20, 246–259. [Google Scholar] [CrossRef] [Green Version]
- Cerhan, J.R.; Slager, S.L. Familial predisposition and genetic risk factors for lymphoma. Blood 2015, 126, 2265–2273. [Google Scholar] [CrossRef]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.-Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG–AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [Green Version]
- Tsimberidou, A.M.; Wen, S.; McLaughlin, P.; O’Brien, S.; Wierda, W.G.; Lerner, S.; Strom, S.; Freireich, E.J.; Medeiros, L.J.; Kantarjian, H.M.; et al. Other Malignancies in Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. J. Clin. Oncol. 2009, 27, 904–910. [Google Scholar] [CrossRef] [Green Version]
- COSMIC Database. Available online: https://cancer.sanger.ac.uk/census (accessed on 10 August 2021).
- Suarez-Puente, X.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martin-Subero, J.I.; Munar, M.; Rubio-Perez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef]
- SNP-Database. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 10 August 2021).
- Tazelaar, G.H.P.; Boeynaems, S.; De Decker, M.; Van Vugt, J.J.F.A.; Kool, L.; Goedee, H.S.; McLaughlin, R.L.; Sproviero, W.; Iacoangeli, A.; Moisse, M.; et al. ATXN1 repeat expansions confer risk for amyotrophic lateral sclerosis and contribute to TDP-43 mislocalization. Brain Commun. 2020, 2, fcaa064. [Google Scholar] [CrossRef]
- Bellido, F.; Pineda, M.; Aiza, G.; Valdés-Mas, R.; Navarro, M.; Puente, D.; Pons, T.; González, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2015, 18, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Briggs, S.; Tomlinson, I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol. 2013, 230, 148–153. [Google Scholar] [CrossRef] [Green Version]
- Mur, P.; García-Mulero, S.; Del Valle, J.; Magraner-Pardo, L.; Vidal, A.; Pineda, M.; Cinnirella, G.; Martín-Ramos, E.; Pons, T.; López-Doriga, A.; et al. Role of POLE and POLD1 in familial cancer. Genet. Med. 2020, 22, 2089–2100. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; Fazi, C.; Agathangelidis, A.; Villamor, N.; Letestu, R.; Nomdedeu, J.; Palacio-Garcia, C.; Stehlikova, O.; Kreuzer, K.-A.; Liptrot, S.; et al. A complementary role of multiparameter flow cytometry and high-throughput sequencing for minimal residual disease detection in chronic lymphocytic leukemia: An European Research Initiative on CLL study. Leukemia 2015, 30, 929–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brochet, X.; Lefranc, M.-P.; Giudicelli, V. IMGT/V-QUEST: The highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 2008, 36, W503–W508. [Google Scholar] [CrossRef] [Green Version]
- Giudicelli, V.; Brochet, X.; Lefranc, M.P. IMGT/V-QUEST: IMGT Standardized Analysis of the Immunoglobulin (IG) and T Cell Receptor (TR) Nucleotide Sequences. Cold Spring Harb. Protoc. 2011, 2011, 695–715. [Google Scholar] [CrossRef]
- Bystry, V.; Agathangelidis, A.; Bikos, V.; Sutton, L.A.; Baliakas, P.; Hadzidimitriou, A.; Stamatopoulos, K.; Darzentas, N. ARResT/AssignSubsets: A novel application for robust subclassification of chronic lymphocytic leukemia based on B cell receptor IG stereotypy. Bioinformatics 2015, 31, 3844–3846. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Li, H.; Handsaker, R.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Larson, D.E.; Wilson, R.K. Using VarScan 2 for Germline Variant Calling and Somatic Mutation Detection. Curr. Protoc. Bioinform. 2013, 44, 15.4.1–15.4.17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BAM-Readcount. Available online: https://github.com/genome/bam-readcount (accessed on 10 August 2021).
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steiner, M.; Gassner, F.J.; Parigger, T.; Neureiter, D.; Egle, A.; Geisberger, R.; Greil, R.; Zaborsky, N. A POLE Splice Site Deletion Detected in a Patient with Biclonal CLL and Prostate Cancer: A Case Report. Int. J. Mol. Sci. 2021, 22, 9410. https://doi.org/10.3390/ijms22179410
Steiner M, Gassner FJ, Parigger T, Neureiter D, Egle A, Geisberger R, Greil R, Zaborsky N. A POLE Splice Site Deletion Detected in a Patient with Biclonal CLL and Prostate Cancer: A Case Report. International Journal of Molecular Sciences. 2021; 22(17):9410. https://doi.org/10.3390/ijms22179410
Chicago/Turabian StyleSteiner, Markus, Franz J. Gassner, Thomas Parigger, Daniel Neureiter, Alexander Egle, Roland Geisberger, Richard Greil, and Nadja Zaborsky. 2021. "A POLE Splice Site Deletion Detected in a Patient with Biclonal CLL and Prostate Cancer: A Case Report" International Journal of Molecular Sciences 22, no. 17: 9410. https://doi.org/10.3390/ijms22179410
APA StyleSteiner, M., Gassner, F. J., Parigger, T., Neureiter, D., Egle, A., Geisberger, R., Greil, R., & Zaborsky, N. (2021). A POLE Splice Site Deletion Detected in a Patient with Biclonal CLL and Prostate Cancer: A Case Report. International Journal of Molecular Sciences, 22(17), 9410. https://doi.org/10.3390/ijms22179410