
Application of Molecular Dynamics Simulations in the Analysis of Cyclodextrin Complexes




Abstract
:1. Introduction
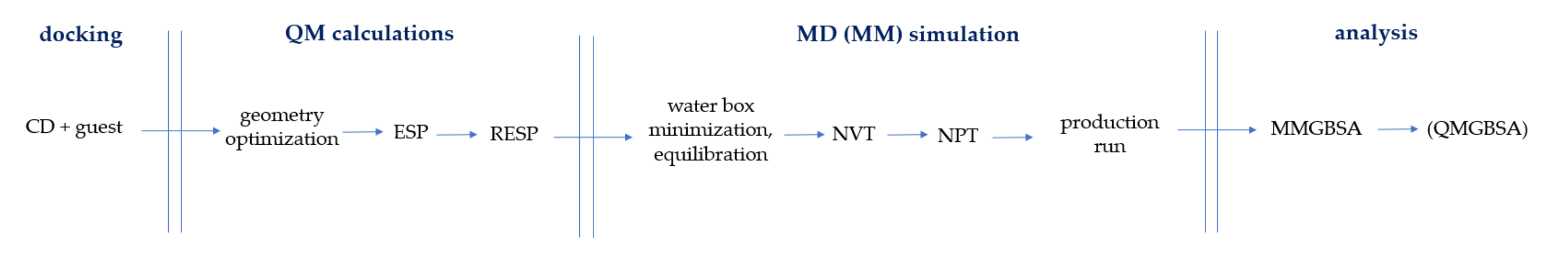
2. Molecular Modeling of CD Host–Guest Complexes—Theoretical and Practical Aspects
2.1. MD Simulations—A Perfect Choice to Study CD Complexes
2.2. Force Fields Dedicated to Cyclodextrins
2.3. MD Simulations of CD Complexes in Water Environment
2.4. Post-MD Simulation Analysis: GBSA, PBSA
2.5. Umbrella Sampling (US) and Steered (Biased) MD
2.6. Coarse-Grained MD
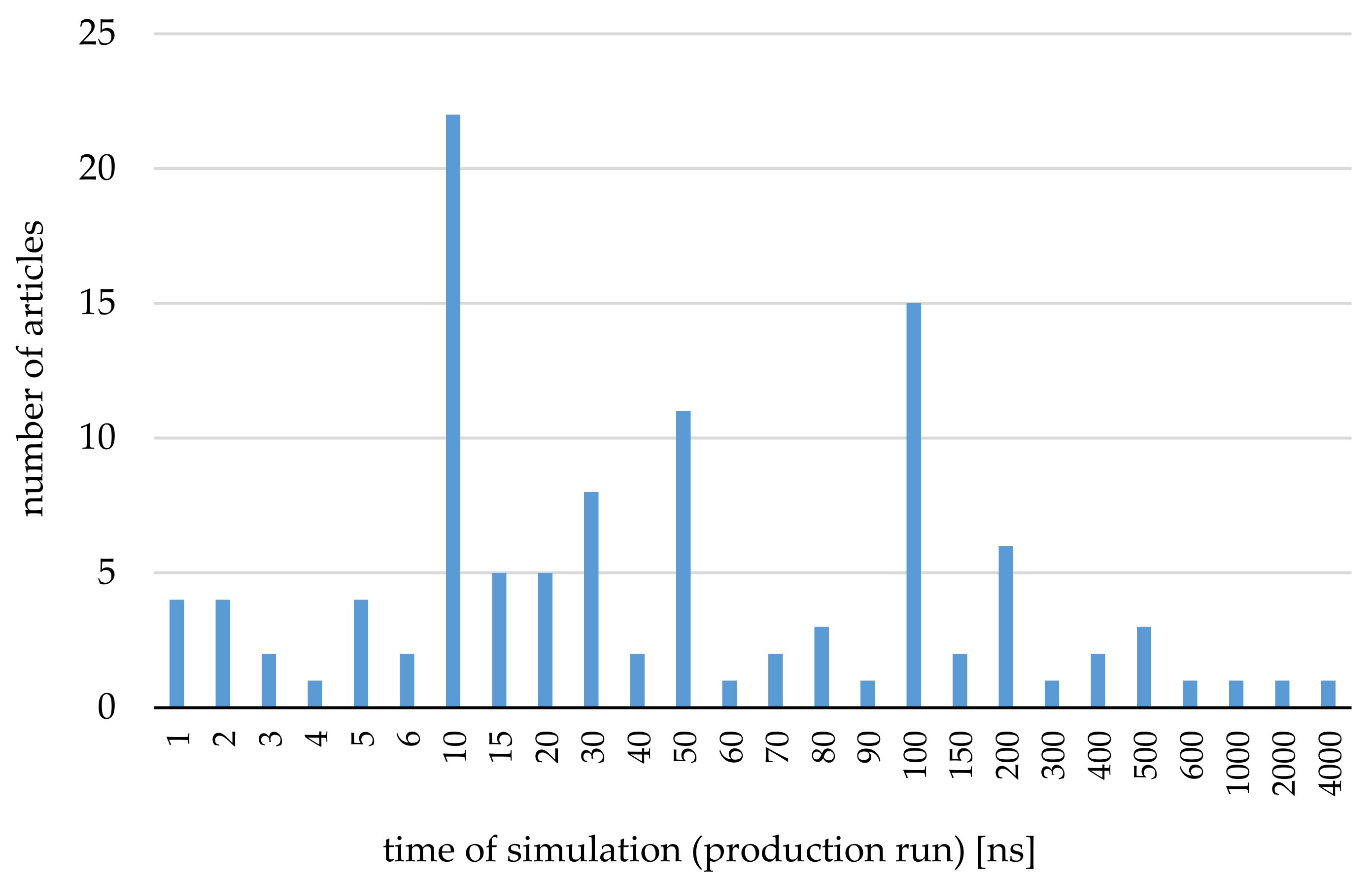
3. Application of the Molecular Dynamics Simulations for Systems Including Cyclodextrins—The Most Important and Interesting Cases
3.1. CDs Used as Drug Carriers (Water Environment)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N° | Reference | CD | Guest | Software Used for MD | Force Field | Equilibration Time and Conditions | Production Run Time and Conditions | Time Step | Water Model | Temperature (K) |
|---|---|---|---|---|---|---|---|---|---|---|
| I. CDs used as drug carriers (water environment) | ||||||||||
| A. 2:1 host–guest CD complexes | ||||||||||
| 1 | [77] | β-CD | piroxicam | Materials Studio and Insight II/Discover packages | CVFF | no i.p. | 10 ns (1:1 host–guest), 100 ns (2:1 host–guest) | 1 fs | implicit | 300 |
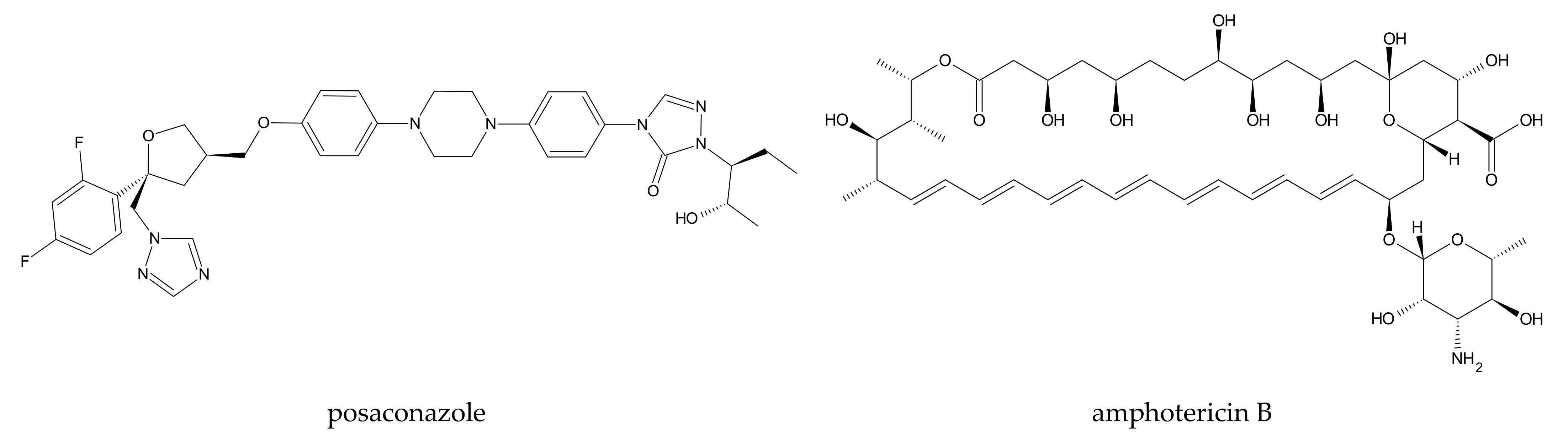
| 2 | [78] | β-CD | posaconazole | GROMACS | GROMOS53A6 | no i.p. | 100 ns, NPT (p = 1 bar) | no i.p. | implicit; water and hydrogen peroxide | 298 |
| 3 | [79] | β- and HP-β-CD | sulfamethoxazole | Desmond | OPLS2005 | 1 ns | 15–20 ns, NPT (p = 1 bar) | no i.p. | TIP3P | 300 |
| 4 | [80] | β-CD | 17-α-methyltestosterone | AMBER 12 | Glycam06h, GAFF | 200 ps NVT, 200 ps NPT | 50 ns, NPT (p = 1 bar) | no i.p. | TIP3P | 298 |
| 5 | [81] | α-, β-, 2,6-DM-β-, and 2,3,6-TM-β-CD | citral isomers | AMBER 12 | Clycam06 (native CD), q4md-CD (CD-derivatives), GAFF (guest) | no i.p. | 12 ns | 1 fs | TIP3P | 300 |
| 6 | [82] | 2-HP-β- and 2-HP-γ-CD | imazapyr | Desmond | OPLS2005 | no i.p. | 30 ns, NPT (p = 1 atm) | TIP3P | 300 | |
| B. NSAIDs* | ||||||||||
| 1 | [83] | HP-β-CD | etodolac and L-arginine | Desmond | OPLS2005 | no i.p. | 5 ns, NPT (p = 1.013 bar) | 2 fs | TIP4P | 300 |
| 2 | [84] | β- and HP-β-CD derivatives | flurbiprofen, ibuprofen, ketoprofen, and naproxen | GROMACS | ffgmx (derivative of GROMOS87) | no i.p. | 500 ps | no i.p. | no i.p. | 300 |
| 3 | [85] | β-CD | ketoprofen | GROMACS | GROMOS | 20 ps | 10 ns, 20 ns, NVT | 2 fs | no i.p. | 298 |
| 4 | [86] | α-, β-, γ-, HP-β-, M-β-, and SBE-β-CD | ketoprofen | AMBER 14 | GAFF | NVT, NPT | 100 ns, NPT (p = 1 bar) | 2 fs | TIP3P | 310 |
| 5 | [87] | SBE-β-CD | celecoxib | YASARA | AMBER ff14SB | no i.p. | 600 ns | 1.25 (intramolecular forces), 2.5 fs (intermolecular forces) | explicit | 298 |
| 6 | [88] | α-, β-, and γ-CD | etoricoxib | AMBER 11 | Glycam06h (CD), GAFF (guest) | 60 ps NVT, 1000 ps NPT | 20 ns, NPT (p = 1 bar) | TIP3P | 298 | |
| 7 | [89] | α-, β-, and γ-CD | nabumetone | AMBER 14 | GLYCAM-06j (CD), GAFF (guest) | 120 ps NVT, 2 ns NPT | 5 µs, NVT | 2 fs | TIP3P | 300 |
| 8 | [90] | β-CD | R- and S-ketoprofen | Desmond | OPLS2005 | 12 ps NVT (10 K), 12 ps NPT (10 K), 24 ps NPT (300 Km 1 atm), 24 ps (300 K, 1 atm) | 50 ns, NPT (p = 1.01325 bar) | no i.p. | TIP4P | 300 |
| 9 | [91] | α-, β-, and γ-CD | antipyrine | AMBER 12 | FF99SB | 50 ps NVT, 500 ps NPT | 10 ns, NPT (p = 1 bar) | no i.p. | TIP3P | 300 |
| C. Anti-fungal drugs and antibiotics | ||||||||||
| 1 | [92] | 2,6-DM-β-CD | natamycin | GROMACS | GROMOS96 | no i.p. | 30 ns, NPT (p = 1 bar) | no i.p. | TIP3P | 300 |
| 2 | [93] | α-, β-, γ-, and 2-HP-β-CD | cefuroxime axetil | GROMACS | GROMOS 56A6 | 1 ns NPT | 500 ns NPT (p = 1 bar) | 2 fs | SPC | 298 |
| 3 | [94] | γ-CD | alamethicin | CHARMM | CHARMM36 | 5 ns NVT | 1000 ns, NPT (p = 1 atm) | no i.p. | TIP3P | 303 |
| 4 | [95] | α-, β-, and γ-CD | chloramphenicol | AMBER 14 | no i.p. | heating up to 300 K, 50 ps; NVT 500 ps | 10 ns, NPT (p = 1 bar) | no i.p. | TIP3P | 300 |
| 5 | [96] | β- and γ-CD | amphotericin B | NAMD | CSFF, CHARMM27 | no i.p. | 10 ns | 2 fs (short-range interactions), 4 fs (long-range interactions) | TIP3P | 300 |
| D. Plant-derived substances | ||||||||||
| 1 | [97] | β-, 2-HP-β-, 6-HP-β-, 2,6-DHP-β-, 2,6-DM-β-, and RM-β-CD | 2-acetyl-1-pyrroline | AMBER 16 | Glycam06 (CD), GAFF2 (guest) | 500 ps (heating up) | 500 ns, NPT (p = 1 atm) | 2 fs | TIP3P | 298 |
| 2 | [98] | β- and γ-CD | polydatin | AMBER 14 | GAFF | 200 ps (heating up), 300 ps NVT | 55 ns, NPT (p = 1 bar) | 2 fs | TIP3P, 2545 ±29 water molecules | 300 |
| 3 | [99] | γ-CD | 3-hydroxyflavone | AMBER 16 | Glycam06 (CD), GAFF (guest) | 100 ps NVT | 300 ns, NPT (p = 1 atm) | no i.p. | TIP3P | 298 |
| 4 | [100] | β- and HP-β-CD | borneol | GROMACS | GROMOS54a7 | NVT, NPT (2 fs time step) | 100 ns, NPT | 1 fs | no i.p. | 300 |
| 5 | [101] | β-, 2,6-DM-β-, 2-HP-β-, 6-HP-β-, and 2,6-DHP-β-CD | eucalyptol | AMBER 14 | Glycam06-h (CD), GAFF (guest) | 100 ns, NVT | 70 ns NPT (p = 1 atm) | 2 fs | SPC, 2000 water molecules | 298 |
| 6 | [102] | β- and γ-CD | triterpene glycoside and glycyrrhizic acid | PRESTO | GAFF | 10,000 steps (heating up), 200,000 NVT | 0.8 ns | 1 fs | TIP3P | 300 |
| 7 | [103] | β-, 2,6-DM-β-, 2-HP-β-, 6-HP-β-, 2,6-DHP-β-, and RM-β-CD | luteolin and pinocembrin | AMBER 16 | Glycam-06 (CD), GAFF (guest) | 60 ps (heating up) | 100 ns, NPT (p = 1 atm) | 2 fs | TIP3P | 298 |
| 8 | [104] | β-, 2,6-DM-β-, and HP-β-CD | mansonone G | AMBER 16 | Glycam-06 (CD), GAFF (guest) | 60 ps (heating up) | 90 ns, NPT (p = 1 atm) | 2 fs | TIP3P | 303 |
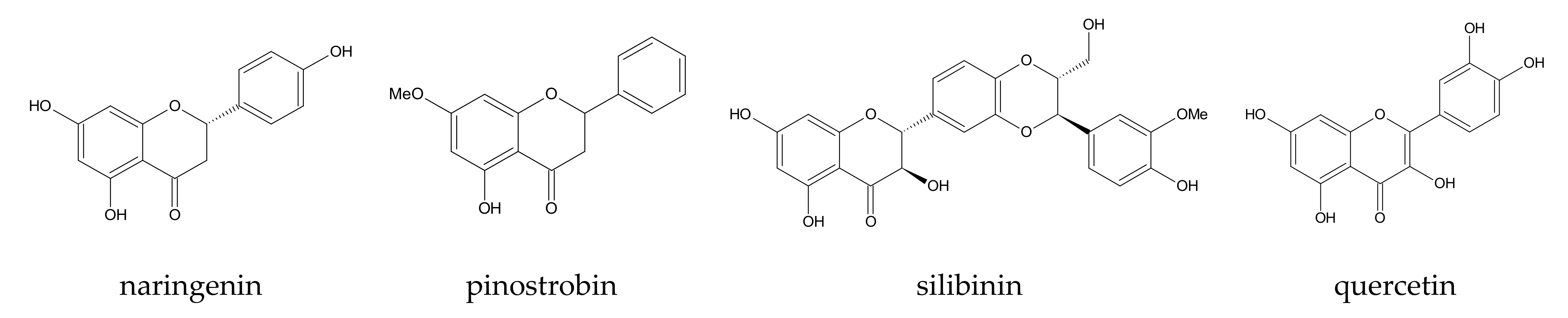
| 9 | [105] | β-, 2,6-DM-β-, 2-HP-β-, 6-HP-β-, and 2,6-DHP-β-CD | pinostrobin | AMBER 12 | Glycam06 (CD), partial charges of guest: standard parametrization procedures | 100 ps (heating up) | 80 ns | 2 fs | explicit, 1400+- 42 water molecules | 298 |
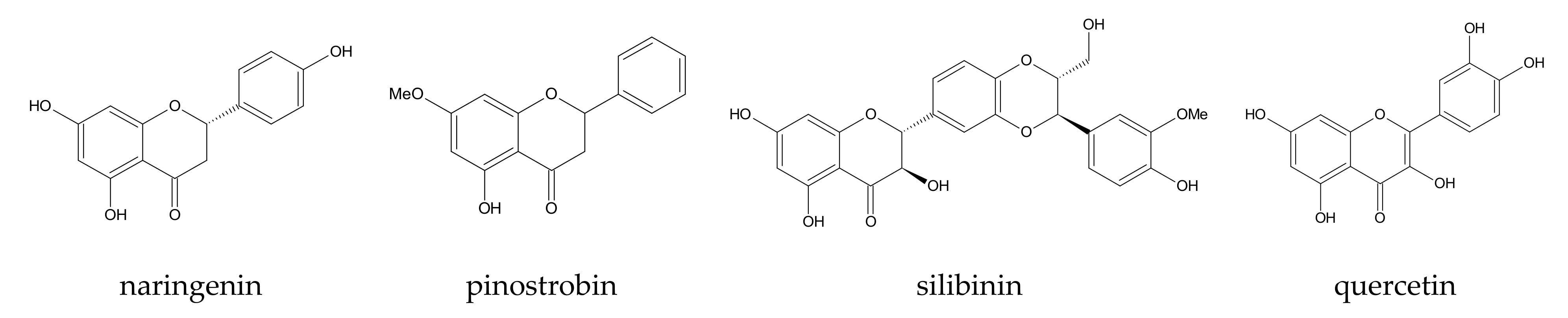
| 10 | [64] | β-, 2,6-DM-β-, DM-β-, and randomly methylated β-CD | hesperetin and naringenin | AMBER 12 | Glycam06 (CD), partial charges of guest: standard parametrization procedures | 100 ps (heating up) | 80 ns, NPT (p = 1 atm) | 2 fs | SPC | 298 |
| 11 | [65] | β- and 2,6-DM-β-CD | naringenin | AMBER 12 | Glycam06 (CD), partial charges of guest: standard parametrization procedures | 100 ps | 80 ns, NPT | 2 fs | SPC, 1480 ± 10 and 1750 ± 3 water molecules | 298 |
| 12 | [106] | β-CD | daidzin | GROMACS | GROMOS96 | NPT | 12 ns, NPT (p = 1 atm) | 0.002 ps | explicit, 3100 water molecules | 300 |
| 13 | [107] | γ-CD | glycyrrhizin | CHARMM | added from cff | no i.p. | 1 ns | 1 fs | explicit, 2969 water molecules for β-CD and 5718 for γ-CD | 300 |
| 14 | [108] | β-CD | eriocitrin (flavanone) | AMBER 19 | Glycam06j (carbohydrates, 2-hydroxypropyl units), missing parameters and atom types: GAFF2 | 1 ns (heating and cooling: 0 K <-> 300 K); 5 ns, NPT | 200 ns, NVT | 2 ps | explicit, 8527–9303 water molecules | 300 |
| 15 | [109] | α-, β-, and γ-CD | cannabidiol | GROMACS | OPLS-AA | no i.p. | 250 ns, NPT (p = 1 bar) | 2 ps | no i.p. | 298, 310, 322, 334 |
| 16 | [110] | β- and γ-CD | rosmarinic acid | AMBER | q4md-CD (CD), GAFF (guest) | no i.p. | 50 ns, NPT | no i.p. | TIP3P | 300 |
| 17 | [111] | β-CD | harman (alkaloid) | GROMACS | GROMOS 54a7 | NVT, NPT, 100 ps | 50 ns, NPT (p = 1 bar) | 2 fs | SPC | 300 |
| 18 | [112] | β,- γ-, HP-β-, and DM-β-CD | myricetin | Desmond 2018.4 | OPLS3 | no i.p. | 30 ns, NPT (p = 1.013 bar) | no i.p. | TIP3P | 300 |
| 19 | [113] | HP-β-CD | capsaicin | AMBER 16 | GAFF | NVT 50 ps, NPT 50 ps | 5000 ps, NPT (p = 1 atm) | 2 fs | TIP3P | 300 |
| 20 | [58] | β- and γ-CD | pseudoginsenoside PF11 | YASARA | AMBER 14 | no i.p. | 100/150 ns, NPT (p = 1 bar) | no i.p. | COSMO | 298 |
| 21 | [114] | α- and β-CD | thymol | Desmond | OPLS 2005 | no i.p. | 48 ns | no i.p. | SPC | |
| 22 | [115] | α-, β-, and γ-CD | daidzein (isoflavone) | AMBER 12 | q4md-CD (CD), GAFF (guest) | no i.p. | 50 ns, NPT (p = 1 atm) | no i.p. | TIP3P | 300 |
| 23 | [116] | α-, β-, and γ-CD | cathinone | AMBER | GAFF | 200 ps NVT, 20000 ps NPT | 30 ns, NPT (p = 1 bar) | 2 fs | TIP3P | 298 |
| 24 | [117] | 2,6-DM-β- and 2,3,6-TM-β-CD | β-citronellol | AMBER | CLYCAM (β-CD), q4md-CD (methylated β-CD) | NVT, 250 ps NPT | 11 ns, NPT | no i.p. | explicit | 300 |
| 25 | [118] | β- and HP-β-CD | naringin | AMBER 14 | GAFF | no i.p. | 100 ps, NPT (p = 1 bar) | no i.p. | TIP3P | no i.p. |
| 26 | [119] | γ- and HP-γ-CD | naringin | Desmond | OPLS2005 | no i.p. | 100 ps, NPT (p = 1.0325 bar) | 2 fs | VSGB 2.0 (implicit) | 310 |
| 27 | [120] | 2-HP-β-CD | quercetin | AMBER 14 | GLYCAM_06j-1 (CD part of molecule), GAFF (2-HP groups of CD and guest) | no i.p. | 400 ns | no i.p. | TIP3P | 300 |
| 28 | [121] | HP-β-CD | silibinin | AMBER 12 | GLYCAM_06i-12SB (CD-part of molecule), GAFF (2-HP groups of CD) | 100 ps NVT, 100 ps NPT | 190 ns and 250 ns | no i.p. | TIP3P, 3841 water molecules | 300 |
| 29 | [122] | β-CD | cyanidin-3-O-glucoside | AMBER 10 | GLYCAM_04 (CD), GAFF (guest) | 100 ps NPT | 30 ns, NPT | 2 fs | TIP3P | 303.15 |
| 30 | [123] | β-CD | resveratrol | AMBER 11 | GLYCAM_06 (CD), GAFF (guest) | 100 ps NVT | 20 ns, NPT (p = 1 atm) | 2 fs | no i.p. | 300 |
| 31,32 | [124,125] | β-, 2,6-DM-β-, and 2-HP-β-CD | α-mangostin | AMBER (PMEMD module) | Glycam06j (CD) | 10 ns NVT | 100 ns, NPT (p = 1 atm) | 2 fs | TIP3P | 298 |
| E. Others | ||||||||||
| 1 | [126] | 3-mono-amino-β-LHRH (luteinizing hormone releasing hormone) conjugate | MacroModel (implicit water model), Desmond (explicit water model) | OPLS2005 | 1 ns for implicit water model | 20 ns for implicit water model; 40 ns, NPT (p = 1.01325 bar) for explicit water model | 2 fs (explicit water model) | implicit and explicit (SPC, 2618 water molecules) | 298.1 | |
| 2 | [127] | HP-β-CD | efavirenz and L-arginine | Desmond | OPLS2005 | no i.p. | 5 ns, NPT (p = 1.013 bar) | TIP4P | 300 | |
| 3 | [128] | β- and M-β-CD | omeprazole and L-arginine | GROMACS | ffgmx | 3 ns | 15 ns (L-arginine: drug, 1:1), 6 ns (other L-arginine-drug ratio), NPT | 1.5 fs | explicit, more than 1000 water molecules | 300 |
| 4 | [129] | β- and γ-CD | pyrazoline dye | MOPAC2012 | Amber99 | 50 ps NVT, 2000 ps NPT | 2000 ps | 2 fs | TIP3P | 298 |
| 5 | [130] | β-CD | cyanine dye | SYBYL-X | Tripos | 500 fs per each 20 K gain; then 25 ps NVT | 2 ns | 2 fs | Molecular Silverware algorithm | 300 |
| 6 | [131] | β-CD | carbazole-based dyes | Chem3D Pro | MM2 | no i.p. | no i.p. | 2 fs | no i.p. | no i.p. |
| 7 | [132] | α-, β-, γ-, and 6-HP-β-CD | lutein | AMBER 14 | GAFF | 10000 steps (heating up) | 100 ns, NPT (p = 1 bar) | 2 fs | TIP3P | 310 |
| 8 | [133] | β-CD | maltogenic amylase | GROMACS | GROMOS96 | 50 ns | 10 ns | 2 fs | SPC | 343 |
| 9 | [134] | sulfated β- and M-β-CD | levosulpiride | AMBER 9 | GAFF | 10 ps (heating up) | 3 ns | 2 fs | 300 | |
| 10 | [135] | HP-β- and 2,6-DM-β-CD | bisacodyl | Forcite | COMPASS | 50 ps, 298 K, NVT | 40 ps | 1 fs | explicit, 20 water molecules | 500 → 300 |
| 11 | [136] | α-, β-, γ-, and differently substituted β- and γ-CD | chlorpromazine | Amber 16 | q4md-CD (CD), GAFF (guest) | heating up by 25 ps periods, 200 ps relaxation | 50 ns, NPT (p = 1 atm) | 2 fs | TIP3P | 300 |
| 12 | [137] | α- and β-CD | ambroxol hydrochloride | MOE | MMFF94x. | 100 ps | 500 ps | no i.p. | no i.p. | 300 |
| 13 | [45] | β- and 2-HP-β-CD | sertraline | Tinker code v8.4 | MM3 | no i.p. | no i.p. | no i.p. | COSMO | 298 |
| 14 | [138] | γ-, HP-γ-, and HP17-γ-CD | lopinavir | GROMACS | GROMOS-96 54a7 | NVT (1 ns, 300 K), NPT (2 ps, 300 K, 1 bar) | 100 ns, NPT (p = 1 bar) | no i.p. | SPC | 300 |
| 15 | [113] | β-, γ-, HP-β-, and M-β-CD | glipizide | AMBER 14 | GAFF | no i.p. | 70 ns | 2 fs | TIP3P | 310.15 |
| 16 | [139] | β-CD | metyrapone | YASARA | AMBER14 | no i.p. | 136 ns, NPT (p = 1 bar) | no i.p. | explicit | 298 |
| 17 | [140] | β-CD | calixarene sulfonates with 4-aminoazobenzene | LAMMPS | AMBER | 1 ns | 20 ns, NPT (p = 1 bar) | 2 fs | TIP4P2005, 2000 water molecules | 300 |
| 18 | [141] | 2,3,6-TM-β-CD | temoporfin | AMBER 14 | q4md-CD (CD), GAFF (guest) | 500 ps, NPT | 10 ns, NPT (p = 1 atm) | 1 fs | TIP3P | 300 |
| 19 | [142] | β-CD | theophylline | GROMACS | amber99sbildn | 0.1 ns NVT, 1 ns NPT | 50 ns | no i.p. | TIP3P, 1353 water molecules | 300 |
| 20 | [143] | 2,6-DM-β- and 2,3,6-TM-β-CD | α-naphthaleneacetic acid | AMBER 12 | q4md-CD (CD), GAFF (guest) | 250 ps NPT | 11 ns, NPT | no i.p. | TIP3P | 300 |
| 21 | [144] | β-, γ-, and randomly sulfated β- and 6-S-β-CD | medetomidine | AMBER | parm10 and ff14SB | 120 ns | 100 ns, NPT (p = 1 atm) | 2 fs | explicit | 300 |
| 22 | [145] | β-, DM-β-, TM-β-, and randomly methylated β-CD | glycocholate | AMBER 12 | GAFF, q4md-CD (CD-derivatives) | 400 ps (heating up) | 2 ns, NPT (p = 1 atm) | 2 fs | TIP3P | 300 |
| 23 | [146] | β- and 2,3-di-O-acetyl-β-CD | clenpenterol | no i.p. | Amber | 40 ns | 100 ns | no i.p. | explicit | no i.p. |
| 24 | [147] | β-CD | norepinephrine | Desmond | OPLS2005 | no i.p. | 15 ns, NPT (p = 1bar) | no i.p. | TIP3P | 300 |
| 25 | [67] | RM-β- and HP-β-CD | triamcinolone | GROMACS | GROMOS 54a7 | NVT 5 ns, NPT 5 ns | 200 ns | no i.p. | TIP3P | 298 |
| 26 | [148] | HP-β-CD | 1-indanone thiosemicarbazones | GROMACS | GROMOS96 53a6 | no i.p. | 100 ns | 1 fs | SPC | 300 |
| 27 | [149] | α-, β-, γ-, and 2-HP-β-CD | fentanyl | AMBER | literature source (CD), GAFF (guest) | 200 ns (heating up), 2.5 ns (equilibrium) | 10 ns or 30 ns (depending on guest) | 2 fs | TIP3P | 300 |
| 28 | [150] | HP-β-CD | clonidine | GROMACS | GROMOS-96 53a6 | no i.p. | 100 ns | 1 fs | SPC | 300 |
| 29 | [151] | sulfobutylether-β-, sulfated β-, and monochlorotriazinyl-β-CD | propiconazole nitrate | GROMACS | q4-MD (CD), GAFF (guest) | no i.p. | 50 ns, NPT (p = 1 atm) | no i.p. | TIP3P | 293,15 |
| 30 | [152] | β-CD | mammea A/AA | AMBER 12 | ff99SB | no i.p. | 10 ns, NPT (p = 1 atm) | 2 fs | TIP3P, 1452 water molecules | 300 |
| 31 | [153] | HP-β-CD | carbamazepine | NAMD | CHARMM | 5 ps heating up, 50 ps equilibration | 2 ns | 1 fs for covalent, 2 fs for vdW, 4 fs for electrostatic atom interactions | no i.p. | 300 |
| 32 | [154] | β-CD | methotrexate | AMBER 12 | GLYCAM_06 (CD), GAFF (guest) | 1 ns | 10 ns, NPT | 2 fs | TIP3P | 298 |
| 33 | [155] | α-, β-, and γ-CD | cumene hydroperoxide | GROMACS | GROMOS96 | no i.p. | 16 ns | no i.p. | 298 | |
| 34 | [156] | β-CD | N-methyl carbamates | AMBER 7 | GAFF | no i.p. | 3 ns, NPT (p = 1 atm) | 2 fs | TIP3P, 9000 water molecules | 300 |
| 35 | [157] | β- and randomly methylated β-CD | isosorbide diesters | AMBER 10 | GAFF | no i.p. | 10 ns, NPT (p = 1 bar) | 2 fs | TIP3P | 300 |
| 36 | [158] | β-CD | adamantane derivatives | NAMD | CHARMM (β-CD), CGenFF program (bond, angle and dihedral parameters of guest) and CHARMM GFF (atomic charges of guest) | 10 ns | 20 ns | 2 fs | TIP3P | no i.p. |
| 37 | [159] | amino-β-CD (protonated and non-protonated) | doxorubicin | NAMD | CHARMM FF | no i.p. | 30 ns, NPT (p = 1 atm) | 0.5 fs | TIP3P | 298.15 |
| 38 | [160] | β-CD | caffeine | GROMACS | GROMOS 56A | no i.p. | 4000 ps | 1 fs | explicit, 1000 water molecules | 333.15 |
| 39 | [161] | β-CD | zwitterionic phenylalanine | PINY-MD code | GROMOS | NVT; NPT (p = 0 bar) 500 ps | 30 ns, NVT | 4 fs | explicit, 2903 water molecules | 300 |
| F. Umbrella sampling and steered (biased) molecular dynamics | ||||||||||
| 1 | [162] | β-CD | trimethylammonium adamantane salt | AMBER 16 | Glycam06 (CD), GAFF2 (guest) | unbiased MD: 100 ps (1 fs time step, heating up); 500 ps NVT density equilibration; NPT | NPT (p = 100 kPa); SMD | 1 μs (unbiased MD), 4.4 μs (biased MD) | TIP3P | 300 |
| 2 | [163] | β-CD | adamantane-doxorubicin | GROMACS | AMBER99SB-ILDN | NVT 2 ns, NPT 2 ns | 100 ns, NPT (p = 1 bar); umbrella sampling: 10 ns | no i.p. | TIP3P, 15,000 water molecules (for 6 drugs in one water box) | 310 |
| 3 | [164] | α-, β-, and γ-CD | adamantane-terminated gold nanoclusters | NAMD | potential model based on CHARMM (CD, guest), GolP (interactions with gold atoms) | no. i.p. | 10 ns, NPT (p = 1 atm); SMD; umbrella sampling for each window: 500 ps | 2 fs | SPC | 300 |
| 4 | [165] | 2-HP-β-CD | cilexetil | GROMACS | MMFF (guest), CHARMM (water) | NVT, NPT | umbrella sampling for each window: 500 ps | 2 fs | no i.p. | 310 |
| 5 | [66] | β-CD | chalcone and 2′,4′-dihydroxychalcone | GROMACS | GROMOS 53a6 | 10 ns for each window | 100 ns for each window, total time: 4290 ns | 2 ps | SPC water model, 1200 water molecules | 310 |
| 6 | [63] | β-CD | genistein | AMBER, umbrella sampling: GROMACS; DFTB-MD | Glycam06 (CD), partial atomic charges of guest: standard parameterization procedures | 2 ns (SMD) | unbiased MD: 100 ns, NPT (p = 1 atm); SMD: 8 ns; DFTB+: 1000 ps | 2 fs, DFTB+: 1 fs | SPC, 1400 water molecules | 298, DFTB+: 400 |
| 7 | [166] | β-, 2,6-DM-β-, and 2-HP-β-CD | pinostrobin | GROMACS | Glycam06 (CD), GAFF (guest) | no i.p. | 1 ns, NPT | no i.p. | SPC, 3200 water molecules | 289 |
| 8 | [167] | β-CD | cinnamaldehyde and eugenol | NAMD 2.6 | Charmm33b | 50 ps heating up | 1.2 ns | no i.p. | no i.p. | 298 |
| 9 | [168] | α-, β-, and γ-CD | umbelliferone | GROMACS | q4md (CD), GAFF (solvents, guest) | 2 ns | NPT (p = 1 bar); total time: 400 ns; 10 ns for each window | no i.p. | water (TIP3P model, 2:1 complex); other solvents (2:2 complex): methanol, ethanol, dimethyl sulfoxide, N,N-dimethylacetamide, N,N-dimethylformamide, acetone, tetrahydrofuran, acetonitrile, chloroform | 300 |
| 10 | [69] | β-CD | 1-butanol and aspirin | AMBER 14 | no i.p. | umbrella sampling: 1 ns NVT; heating up at 200 K, 250 K, 298 K for 150 ps | 100 ns: conventional MD; 10 ns: SMD (NPT, p = 1 bar); 2.5 ns: umbrella sampling | no i.p. | TIP3P | 300 (SMD), 298 (umbrella sampling) |
| II. CDs used as extracting agents (different solvents) | ||||||||||
| 1 | [169] | β-CD | 2,3,7,8-tetrachlorodibenzo-p-dioxin | GROMACS | GROMOS96 | no i.p. | overall 12 ns (equilibrium + run), NPT (p = 1 bar) | 0.001 ps | SPC, 2500 water molecules | 298 |
| 2 | [170] | α-, β-, and γ-CD | 2,2′,5,5′-tetrachlorobiphenyl | Discover Model of Materials Studio | COMPASS | no i.p. | NPT (p = 1 atm) | 0.5 fs | COSMO, 800 water molecules | 298 |
| 3 | [171] | α-, β-, and γ-CD | DDT | NAMD | CHARMM27 | no i.p. | NPT (p = 1 atm) | 2 fs | TIP3P | 298 |
| 4 | [172] | quaternary ammonium β-CD | ochratoxin A | HyperChem | Amber | no i.p. | a few hundreds of ps time | TIP3P | 298 | |
| 5 | [173] | β-CD | PCB126 | GROMACS | GROMOS96 | no i.p. | 15 ns, NPT (p = 1 bar) | 1 fs | SPC, 2500 water molecules | 300 |
| 6 | [174] | β-CD | ibuprofen (racemic mixture) | GROMACS | GROMOS54a7 | 1 ns NVT, 10 ns NPT (P = 1 bar) | 100 ns, NVT | 1 fs | methanol, 2000 molecules | 260–380 |
| 7 | [76] | β-CD | ibuprofen (racemic mixture) | GROMACS | GROMOS54a7 | 1000 ps NPT (P = 1 bar), 500 ps NVT | 10 ns | 1 fs | methanol, 2000 molecules | 273.15 |
| 8 | [175] | HP-β-CD | (E)-piceatannol | GROMACS | GROMOS53A6 | no i.p. | 2 ns, NPT (p = 1 atm) | 2 fs | SPC/E (water), methanol + water, ethanol + water, n-propanol + water, glycerol + water cosolvents | 298.2 |
| 9 | [176] | β-, 2,6-DM-β-, and 2-HP-β-CD | UC781 | AMBER 10 | Amber parm03 | 400 ps NVT, 11 ns NPT | 10 ns, NPT (p = 1 atm) | 2 fs | 308 water molecules (TIP3P), 1670 ethanol molecules | 300 |
| 10 | [177] | γ-CD | gold nanoparticles | GROMACS | CHARMM36 | 100 ps; NPT 100 ps | 200 ns, 250 ns (depending on the number of CD molecules), NPT (p = 1 bar) | 2 fs | explicit, 10,500–12,620 water molecules | 298.15 |
| 11 | [178] | β- and 2,3-di-O-acetyl-β-CD | terbutaline enantiomers | AMBER 12 | q4md-CD and Glycam04 and Amber99SB (CD), GAFF (guest molecules with one positive charge) | 400 ps NVT | 6 ns, NPT (p = 1 atm) | 2 fs | TIP3P | 300 |
| 12 | [179] | γ-CD | regioisomers of bis-N-methylfulleropyrrolidines | NAMD | CHARMM | 20 ns | 60 ns, NPT (p = 1 bar) | 2 fs | DMSO, water (TIP3P) | 298 |
| 13 | [180] | permethylated β-CD | phenylazetidin derivatives | GROMACS | GROMOS | no i.p. | 4 ns, NPT (p = 1 bar) | no i.p. | no i.p. | 300 |
| 14 | [181] | β-CD | isoleucine enantiomers | no i.p. | AMBER ff99SB | no i.p. | 5 ns | 1 fs | 2021 | 293 |
| 15 | [182] | β-CD | terminally blocked phenylalanine dipeptide (Ace-Phe-Nme), | AMBER 9 | Amber 03 | no i.p. | 8 ns, NPT (p = 1 atm) | no i.p. | TIP3P | 300 |
3.2. Host–Guest Stoichiometry of CD Complexes
3.3. NSAIDs
3.4. Anti-Fungal Drugs and Antibiotics
3.5. Plant-Derived Substances
3.6. Others
3.7. Umbrella Sampling and SMD Used for CD Simulations
3.8. CDs Used as Extracting Agents (Different Solvents)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Crini, G. Review: A history of cyclodextrins. Chem. Rev. 2014, 114, 10940–10975. [Google Scholar] [CrossRef]
- French, D. The schardinger dextrins. In Advances in Carbohydrate Chemistry; Wolfrom, M.L., Tipson, R.S., Eds.; Academic Press: Cambridge, MA, USA, 1957; Volume 12, pp. 189–260. [Google Scholar]
- Szejtli, J. Introduction and general overview of cyclodextrin chemistry. Chem. Rev. 1998, 98, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Irie, T.; Uekama, K. Pharmaceutical applications of cyclodextrins. III. Toxicological issues and safety evaluation. J. Pharm. Sci. 1997, 86, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Beta Cyclodextrin Price. Available online: https://www.alibaba.com/product-detail/Best-Quality-and-Price-from-factory_1600280714089.html?spm=a2700.7724857.normal_offer.d_title.2a9c5ad6kDODQB (accessed on 25 July 2021).
- Jambhekar, S.S.; Breen, P. Cyclodextrins in pharmaceutical formulations II: Solubilization, binding constant, and complexation efficiency. Drug Discov. Today 2016, 21, 363–368. [Google Scholar] [CrossRef]
- European Medicines Agency. Available online: https://www.ema.europa.eu/en (accessed on 21 July 2021).
- U.S. Food & Drug Administration (FDA). Available online: https://www.fda.gov (accessed on 22 July 2021).
- Pharmaceutical and Medical Devices Agencty. Available online: https://www.pmda.go.jp/english/index.html (accessed on 23 July 2021).
- Jicsinszky, L.; Martina, K.; Cravotto, G. Cyclodextrins in the antiviral therapy. J. Drug Deliv. Sci. Technol. 2021, 64, 102589. [Google Scholar] [CrossRef] [PubMed]
- Johnson & Johnson COVID-19 Vaccine Authorized by U.S. FDA For Emergency Use-First Single-Shot Vaccine in Fight against Global Pandemic. Available online: https://www.jnj.com/johnson-johnson-covid-19-vaccine-authorized-by-u-s-fda-for-emergency-usefirst-single-shot-vaccine-in-fight-against-global-pandemic (accessed on 19 July 2021).
- Available online: https://www.innovationintextiles.com/seamless-supply-chain-meets-high-demand-for-antiviral-heiq-viroblock-npj03/ (accessed on 15 June 2021).
- Jambhekar, S.S.; Breen, P. Cyclodextrins in pharmaceutical formulations I: Structure and physicochemical properties, formation of complexes, and types of complex. Drug Discov. Today 2016, 21, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Szmeja, S.; Gubica, T.; Ostrowski, A.; Zalewska, A.; Szeleszczuk, Ł.; Zawada, K.; Zielińska-Pisklak, M.; Skowronek, K.; Wiweger, M. Caffeine-cyclodextrin complexes as solids: Synthesis, biological and physicochemical characterization. Int. J. Mol. Sci. 2021, 22, 4191. [Google Scholar] [CrossRef]
- Abdolmaleki, A.; Ghasemi, F.; Ghasemi, J.B. Computer-aided drug design to explore cyclodextrin therapeutics and biomedical applications. Chem. Biol. Drug Des. 2017, 89, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quevedo, M.A.; Zoppi, A. Current trends in molecular modeling methods applied to the study of cyclodextrin complexes. J. Incl. Phenom. Macrocycl. Chem. 2018, 90, 1–14. [Google Scholar] [CrossRef]
- Aiassa, V.; Garnero, C.; Longhi, M.R.; Zoppi, A. Cyclodextrin multicomponent complexes: Pharmaceutical applications. Pharmaceutics 2021, 13, 1099. [Google Scholar] [CrossRef]
- Braga, S.S.; Barbosa, J.S.; Santos, N.E.; El-Saleh, F.; Paz, F.A.A. Cyclodextrins in antiviral therapeutics and vaccines. Pharmaceutics 2021, 13, 409. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, F.M.; Lis, M.J.; Firmino, H.B.; Dias da Silva, J.G.; Curto Valle, R.D.C.S.; Borges Valle, J.A.; Scacchetti, F.A.P.; Tessaro, A.L. The role of β-cyclodextrin in the textile industry—Review. Molecules 2020, 25, 3624. [Google Scholar] [CrossRef] [PubMed]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perilla, J.R.; Goh, B.C.; Cassidy, C.K.; Liu, B.; Bernardi, R.C.; Rudack, T.; Yu, H.; Wu, Z.; Schulten, K. Molecular dynamics simulations of large macromolecular complexes. Curr. Opin. Struct. Biol. 2015, 31, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Chen, J.; Selvam, B.; Shukla, D. Computational microscopy: Revealing molecular mechanisms in plants using molecular dynamics simulations. Plant Cell 2019, 31, 1319. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, A.H.; Szeleszczuk, Ł.; Pisklak, D.M. A review on combination of Ab initio molecular dynamics and NMR parameters calculations. Int. J. Mol. Sci. 2021, 22, 4378. [Google Scholar] [CrossRef]
- Ghoufi, A.; Malfreyt, P. Entropy and enthalpy calculations from perturbation and integration thermodynamics methods using molecular dynamics simulations: Applications to the calculation of hydration and association thermodynamic properties. Mol. Phys. 2006, 104, 2929–2943. [Google Scholar] [CrossRef]
- Lee, J.-U.; Lee, S.-S.; Lee, S.; Oh, H.B. Noncovalent complexes of cyclodextrin with small organic molecules: Applications and insights into host–guest Interactions in the gas phase and condensed phase. Molecules 2020, 25, 4048. [Google Scholar] [CrossRef]
- Amber Molecular Dynamics. Available online: http://ambermd.org/antechamber/gaff.html (accessed on 1 August 2021).
- GROMOS. Available online: https://www.gromacs.org/Documentation_of_outdated_versions/Terminology/Force_Fields/GROMOS (accessed on 2 August 2021).
- The CHARMM Force Field. Available online: https://www.ks.uiuc.edu/Training/Tutorials/science/forcefield-tutorial/forcefield-html/node5.html (accessed on 4 August 2021).
- NAMD and MD Simulations. Available online: https://www.ks.uiuc.edu/Research/namd/2.11/ug/node5.html (accessed on 3 August 2021).
- Schrodinger. Available online: https://www.schrodinger.com/science-articles/force-field (accessed on 31 July 2021).
- Available online: https://www.3ds.com/fileadmin/PRODUCTS-SERVICES/BIOVIA/PDF/MATERIALS-SCIENCE-MODELING-SIMULATION-BIOVIA-MATERIALS-STUDIO-PRODUCT-DESCRIPTIONS.pdf (accessed on 30 June 2021).
- GROMACS. Available online: https://www.gromacs.org/ (accessed on 20 July 2021).
- Amber Molecular Dynamics. Available online: https://ambermd.org/ (accessed on 19 June 2021).
- CHARMM. Available online: https://www.charmm.org/ (accessed on 20 June 2021).
- DESMOND. Available online: https://www.schrodinger.com/products/desmond (accessed on 21 June 2021).
- NAMD. Available online: https://www.ks.uiuc.edu/Research/namd/ (accessed on 22 June 2021).
- FORCITE. Available online: https://www.3ds.com/search/?wockw=forcite (accessed on 23 June 2021).
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [Green Version]
- Cézard, C.; Trivelli, X.; Aubry, F.; Djedaïni-Pilard, F.; Dupradeau, F.-Y. Molecular dynamics studies of native and substituted cyclodextrins in different media: 1. Charge derivation and force field performances. Phys. Chem. Chem. Phys. 2011, 13, 15103–15121. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hübschle, C.B.; van Smaalen, S. The electrostatic potential of dynamic charge densities. J. Appl. Cryst. 2017, 50, 1627–1636. [Google Scholar] [CrossRef] [Green Version]
- Woods, R.J.; Chappelle, R. Restrained electrostatic potential atomic partial charges for condensed-phase simulations of carbohydrates. Theochem 2000, 527, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Allinger, N.L.; Yuh, Y.H.; Lii, J.H. Molecular mechanics. The MM3 force field for hydrocarbons. 1. J. Am. Chem. Soc. 1989, 111, 8551–8566. [Google Scholar] [CrossRef]
- Ramraj, A.; Raju, R.K.; Wang, Q.; Hillier, I.H.; Bryce, R.A.; Vincent, M.A. An evaluation of the GLYCAM06 and MM3 force fields, and the PM3-D* molecular orbital method for modelling prototype carbohydrate–aromatic interactions. J. Mol. Graph. Model. 2010, 29, 321–325. [Google Scholar] [CrossRef]
- Bautista-Renedo, J.-M.; Cuevas-Yañez, E.; Reyes-Pérez, H.; Vargas, R.; Garza, J.; González-Rivas, N. Non-covalent interactions between sertraline stereoisomers and 2-hydroxypropyl-β-cyclodextrin: A quantum chemistry analysis. RSC Adv. 2020, 10, 20202–20210. [Google Scholar] [CrossRef]
- Review of Current Force Fields. Available online: http://cmt.dur.ac.uk/sjc/thesis_dlc/node79.html (accessed on 5 August 2021).
- Tinker Molecular Modeling. Available online: https://dasher.wustl.edu/tinker/ (accessed on 22 July 2021).
- Lagardère, L.; Jolly, L.-H.; Lipparini, F.; Aviat, F.; Stamm, B.; Jing, Z.F.; Harger, M.; Torabifard, H.; Cisneros, G.A.; Schnieders, M.J.; et al. Tinker-HP: A massively parallel molecular dynamics package for multiscale simulations of large complex systems with advanced point dipole polarizable force fields. Chem. Sci. 2018, 9, 956–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AutoDock Vina. Available online: http://vina.scripps.edu/ (accessed on 6 June 2021).
- BIOVIA Discovery Studio. Available online: https://www.3ds.com/products-services/biovia/ (accessed on 23 June 2021).
- Kleinjung, J.; Fraternali, F. Design and application of implicit solvent models in biomolecular simulations. Curr. Opin. Struct. Biol. 2014, 25, 126–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhang, H.; Wu, T.; Wang, Q.; van der Spoel, D. Comparison of implicit and explicit solvent models for the calculation of solvation free energy in organic solvents. J. Chem. Theory Comput. 2017, 13, 1034–1043. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J.; Cammi, R.; Cheeseman, J.R.; Frisch, M.J.; Devlin, F.J.; Gabriel, S.; Stephens, P.J. Polarizable continuum model (PCM) calculations of solvent effects on optical rotations of chiral molecules. J. Phys. Chem. A 2002, 106, 6102–6113. [Google Scholar] [CrossRef]
- Mongan, J.; Case, D.A.; McCammon, J.A. Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem. 2004, 25, 2038–2048. [Google Scholar] [CrossRef] [PubMed]
- Fennell, C.J.; Li, L.; Dill, K.A. Simple liquid models with corrected dielectric constants. J. Phys. Chem. B 2012, 116, 6936–6944. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Xia, Z.; Zhang, J.; Best, R.; Wu, C.; Ponder, J.W.; Ren, P. Polarizable atomic multipole-based AMOEBA force field for proteins. J. Chem. Theory Comput. 2013, 9, 4046–4063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floresta, G.; Punzo, F.; Rescifina, A. Supramolecular host-guest interactions of pseudoginsenoside F11 with β- and γ-cyclodextrin: Spectroscopic/spectrometric and computational studies. J. Mol. Struct. 2019, 1195, 387–394. [Google Scholar] [CrossRef]
- Ylilauri, M.; Pentikäinen, O.T. MMGBSA as a tool to understand the binding affinities of filamin–peptide interactions. J. Chem. Inf. Model. 2013, 53, 2626–2633. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Visual Molecular Dynamics (VMD). Available online: https://www.ks.uiuc.edu/Research/vmd/ (accessed on 6 August 2021).
- Mishra, S.K.; Koča, J. Assessing the performance of MM/PBSA, MM/GBSA, and QM–MM/GBSA approaches on protein/carbohydrate complexes: Effect of implicit solvent models, QM methods, and entropic contributions. J. Phys. Chem. B 2018, 122, 8113–8121. [Google Scholar] [CrossRef]
- Hanpaibool, C.; Chakcharoensap, T.A.; Hijikata, Y.; Irle, S.; Wolschann, P.; Kungwan, N.; Pongsawasdi, P.; Ounjai, P.; Rungrotmongkol, T. Theoretical analysis of orientations and tautomerization of genistein in β-cyclodextrin. J. Mol. Liq. 2018, 265, 16–23. [Google Scholar] [CrossRef]
- Sangpheak, W.; Kicuntod, J.; Schuster, R.; Rungrotmongkol, T.; Wolschann, P.; Kungwan, N.; Viernstein, H.; Mueller, M.; Pongsawasdi, P. Physical properties and biological activities of hesperetin and naringenin in complex with methylated β-cyclodextrin. Beilstein J. Org. Chem. 2015, 11, 2763–2773. [Google Scholar] [CrossRef] [Green Version]
- Sangpheak, W.; Khuntawee, W.; Wolschann, P.; Pongsawasdi, P.; Rungrotmongkol, T. Enhanced stability of a naringenin/2,6-dimethyl β-cyclodextrin inclusion complex: Molecular dynamics and free energy calculations based on MM- and QM-PBSA/GBSA. J. Mol. Graph. Model. 2014, 50, 10–15. [Google Scholar] [CrossRef]
- Sancho, M.I.; Andujar, S.; Porasso, R.D.; Enriz, R.D. Theoretical and Experimental Study of Inclusion Complexes of β-Cyclodextrins with Chalcone and 2′,4′-Dihydroxychalcone. J. Phys. Chem. B 2016, 120, 3000–3011. [Google Scholar] [CrossRef]
- De Medeiros, A.S.A.; Zoppi, A.; Barbosa, E.G.; Oliveira, J.I.N.; Fernandes-Pedrosa, M.F.; Longhi, M.R.; da Silva-Júnior, A.A. Supramolecular aggregates of oligosaccharides with co-solvents in ternary systems for the solubilizing approach of triamcinolone. Carbohydr. Polym. 2016, 151, 1040–1051. [Google Scholar] [CrossRef]
- Park, S.; Khalili-Araghi, F.; Tajkhorshid, E.; Schulten, K. Free energy calculation from steered molecular dynamics simulations using Jarzynski’s equality. J. Chem. Phys. 2003, 119, 3559–3566. [Google Scholar] [CrossRef]
- You, W.; Tang, Z.; Chang, C.-E.A. Potential mean force from umbrella sampling simulations: What can we learn and what is missed? J. Chem. Theory Comput. 2019, 15, 2433–2443. [Google Scholar] [CrossRef]
- Kästner, J. Umbrella sampling. Wires Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Methods for Calculating Potentials of Mean Force. Available online: https://www.ks.uiuc.edu/Training/Tutorials/science/channel/channel-tut.pdf (accessed on 6 August 2021).
- Takahashi, K.; Oda, T.; Naruse, K. Coarse-grained molecular dynamics simulations of biomolecules. Aims Biophys. 2014, 1, 1–15. [Google Scholar] [CrossRef]
- Wang, W.; Zhu, Y.-L.; Qian, H.-J.; Lu, Z.-Y. Synthesize multiblock copolymers via complex formations between β-cyclodextrin and adamantane groups terminated at diblock copolymer ends: A Brownian dynamics simulation study. J. Phys. Chem. B 2013, 117, 16283–16291. [Google Scholar] [CrossRef] [PubMed]
- López, C.A.; de Vries, A.H.; Marrink, S.J. Computational microscopy of cyclodextrin mediated cholesterol extraction from lipid model membranes. Sci. Rep. 2013, 3, 2071. [Google Scholar] [CrossRef] [Green Version]
- Cieplak, M.; Thompson, D. Coarse-grained molecular dynamics simulations of nanopatterning with multivalent inks. J. Chem. Phys. 2008, 128, 234906. [Google Scholar] [CrossRef]
- Škvára, J.; Nezbeda, I. Molecular dynamics study of racemic mixtures: Solutions of ibuprofen and β-cyclodextrin in methanol. J. Mol. Liq. 2018, 265, 791–796. [Google Scholar] [CrossRef]
- Raffaini, G.; Ganazzoli, F. Understanding surface interaction and inclusion complexes between piroxicam and native or crosslinked β-cyclodextrins: The role of drug concentration. Molecules 2020, 25, 2848. [Google Scholar] [CrossRef]
- Santana, A.C.S.G.V.; Nadvorny, D.; da Rocha Passos, T.D.; de La Roca Soares, M.F.; Soares-Sobrinho, J.L. Influence of cyclodextrin on posaconazole stability, release and activity: Improve the utility of the drug. J. Drug Deliv. Sci. Technol. 2019, 53, 101153. [Google Scholar] [CrossRef]
- Varghese, B.; Suliman, F.O.; Al-Hajri, A.; Al Bishri, N.S.S.; Al-Rwashda, N. Spectral and theoretical study on complexation of sulfamethoxazole with β- and HPβ-cyclodextrins in binary and ternary systems. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 190, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.B.D.; Burusco, K.K.; Jaime, C.; Venâncio, T.; Carvalho, A.F.S.D.; Murgas, L.D.S.; Pinto, L.D.M.A. Complexes between methyltestosterone and β-cyclodextrin for application in aquaculture production. Carbohydr. Polym. 2018, 179, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Fourtaka, K.; Christoforides, E.; Tzamalis, P.; Bethanis, K. Inclusion of citral isomers in native and methylated cyclodextrins: Structural insights by X-ray crystallography and molecular dynamics simulation analysis. J. Mol. Struct. 2021, 1234, 130169. [Google Scholar] [CrossRef]
- Mokhtar, M.S.; Suliman, F.O.; Elbashir, A.A. Experimental and molecular modeling investigations of inclusion complexes of imazapyr with 2-hydroxypropyl(β/γ) cyclodextrin. J. Mol. Liq. 2018, 262, 504–513. [Google Scholar] [CrossRef]
- Sherje, A.P.; Kulkarni, V.; Murahari, M.; Nayak, U.Y.; Bhat, P.; Suvarna, V.; Dravyakar, B. Inclusion complexation of etodolac with hydroxypropyl-beta-cyclodextrin and auxiliary agents: Formulation characterization and molecular modeling studies. Mol. Pharm. 2017, 14, 1231–1242. [Google Scholar] [CrossRef]
- Felton, L.A.; Popescu, C.; Wiley, C.; Esposito, E.X.; Lefevre, P.; Hopfinger, A.J. Experimental and computational studies of physicochemical properties influence NSAID-cyclodextrin complexation. AAPS PharmSciTech. 2014, 15, 872–881. [Google Scholar] [CrossRef] [Green Version]
- Guzzo, T.; Mandaliti, W.; Nepravishta, R.; Aramini, A.; Bodo, E.; Daidone, I.; Allegretti, M.; Topai, A.; Paci, M. Conformational change in the mechanism of inclusion of ketoprofen in β-cyclodextrin: NMR spectroscopy, Ab initio calculations, molecular dynamics simulations, and photoreactivity. J. Phys. Chem. B 2016, 120, 10668–10678. [Google Scholar] [CrossRef]
- Zhao, Q.; Gao, H.; Su, Y.; Huang, T.; Lu, J.; Yu, H.; Ouyang, D. Experimental characterization and molecular dynamic simulation of ketoprofen-cyclodextrin complexes. Chem. Phys. Lett. 2019, 736, 136802. [Google Scholar] [CrossRef]
- Rescifina, A.; Surdo, E.; Cardile, V.; Avola, R.; Eleonora Graziano, A.C.; Stancanelli, R.; Tommasini, S.; Pistarà, V.; Ventura, C.A. Gemcitabine anticancer activity enhancement by water soluble celecoxib/sulfobutyl ether-β-cyclodextrin inclusion complex. Carbohydr. Polym. 2019, 206, 792–800. [Google Scholar] [CrossRef]
- Yousef, F.O.; Ghanem, R.; Alshraa, N.H.; Al Omari, N.M.; Bodoor, K.; El-Barghouthi, M.I. Effect of pH and α-, β- and γ-cyclodextrin on the spectral properties of etoricoxib: Spectroscopic and molecular dynamics study. J. Incl. Phenom. Macrocycl. Chem. 2017, 88, 171–180. [Google Scholar] [CrossRef]
- Suárez, D.; Díaz, N. Conformational and entropy analyses of extended molecular dynamics simulations of α-, β- and γ-cyclodextrins and of the β-cyclodextrin/nabumetone complex. Phys. Chem. Chem. Phys. 2017, 19, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Albertini, B.; Iraci, N.; Schoubben, A.; Giovagnoli, S.; Ricci, M.; Blasi, P.; Rossi, C. β-cyclodextrin hinders PLGA plasticization during microparticle manufacturing. J. Drug Deliv. Sci. Technol. 2015, 30, 375–383. [Google Scholar] [CrossRef]
- Gannimani, R.; Perumal, A.; Ramesh, M.; Pillay, K.; Soliman, M.E.; Govender, P. Antipyrine–gamma cyclodextrin inclusion complex: Molecular modeling, preparation, characterization and cytotoxicity studies. J. Mol. Struct. 2015, 1089, 38–47. [Google Scholar] [CrossRef]
- Fang, S.; Peng, X.; Liang, X.; Shen, J.; Wang, J.; Chen, J.; Meng, Y. Enhancing water solubility and stability of natamycin by molecular encapsulation in methyl-β-cyclodextrin and its mechanisms by molecular dynamics simulations. Food Biophys. 2020, 15, 188–195. [Google Scholar] [CrossRef]
- Gieroba, B.; Kalisz, G.; Sroka-Bartnicka, A.; Płazińska, A.; Płaziński, W.; Starek, M.; Dąbrowska, M. Molecular structure of cefuroxime axetil complexes with α-, β-, γ-, and 2-hydroxypropyl-β-cyclodextrins: Molecular simulations and raman spectroscopic and imaging studies. Int. J. Mol. Sci. 2021, 22, 5238. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, J.; Lyu, Y.; Fitriyanti, M.; Hou, H.; Jin, Z.; Zhu, X.; Narsimhan, G. Understanding the antimicrobial activity of water soluble γ-cyclodextrin/alamethicin complex. Colloids Surf. B Biointerfaces 2018, 172, 451–458. [Google Scholar] [CrossRef]
- Gannimani, R.; Ramesh, M.; Mtambo, S.; Pillay, K.; Soliman, M.E.; Govender, P. γ-Cyclodextrin capped silver nanoparticles for molecular recognition and enhancement of antibacterial activity of chloramphenicol. J. Inorg. Biochem. 2016, 157, 15–24. [Google Scholar] [CrossRef]
- He, J.; Chipot, C.; Shao, X.; Cai, W. Cyclodextrin-mediated recruitment and delivery of amphotericin B. J. Phys. Chem. C 2013, 117, 11750–11756. [Google Scholar] [CrossRef]
- Mahalapbutr, P.; Charoenwongpaiboon, T.; Phongern, C.; Kongtaworn, N.; Hannongbua, S.; Rungrotmongkol, T. Molecular encapsulation of a key odor-active 2-acetyl-1-pyrroline in aromatic rice with β-cyclodextrin derivatives. J. Mol. Liq. 2021, 337, 116394. [Google Scholar] [CrossRef]
- Chen, M.; Li, Y.-H.; Li, Y.-G.; Li, X.-L.; Zhao, S.-Y.; Yang, L.-J.; Liu, X.-Y.; Zhang, J.-Q. Molecular dynamics simulations and theoretical calculations of cyclodextrin-polydatin inclusion complexes. J. Mol. Struct. 2021, 1230, 129840. [Google Scholar] [CrossRef]
- Kerdpol, K.; Daengngern, R.; Sattayanon, C.; Namuangruk, S.; Rungrotmongkol, T.; Wolschann, P.; Kungwan, N.; Hannongbua, S. Effect of water microsolvation on the excited-state proton transfer of 3-hydroxyflavone enclosed in γ-cyclodextrin. Molecules 2021, 26, 843. [Google Scholar] [CrossRef] [PubMed]
- Santana, D.V.S.; Trindade, I.A.S.; Carvalho, Y.M.B.G.; Carvalho-Neto, A.G.; Silva, E.C.D.; Silva-Júnior, E.F.; Leite, R.F.S.; Quintans-Júnior, L.J.; Aquino, T.M.; Serafini, M.R.; et al. Analytical techniques to recognize inclusion complexes formation involving monoterpenes and cyclodextrins: A study case with (–) borneol, a food ingredient. Food Chem. 2021, 339, 127791. [Google Scholar] [CrossRef]
- Nutho, B.; Nunthaboot, N.; Wolschann, P.; Kungwan, N.; Rungrotmongkol, T. Metadynamics supports molecular dynamics simulation-based binding affinities of eucalyptol and beta-cyclodextrin inclusion complexes. RSC Adv. 2017, 7, 50899–50911. [Google Scholar] [CrossRef] [Green Version]
- Oda, M.; Kuroda, M. Molecular dynamics simulations of inclusion complexation of glycyrrhizic acid and cyclodextrins (1:1) in water. J. Incl. Phenom. Macrocycl. Chem. 2016, 85, 271–279. [Google Scholar] [CrossRef]
- Mahalapbutr, P.; Thitinanthavet, K.; Kedkham, T.; Nguyen, H.; Theu, L.t.h.; Dokmaisrijan, S.; Huynh, L.; Kungwan, N.; Rungrotmongkol, T. A theoretical study on the molecular encapsulation of luteolin and pinocembrin with various derivatized beta-cyclodextrins. J. Mol. Struct. 2019, 1180, 480–490. [Google Scholar] [CrossRef]
- Mahalapbutr, P.; Wonganan, P.; Charoenwongpaiboon, T.; Prousoontorn, M.; Chavasiri, W.; Rungrotmongkol, T. Enhanced solubility and anticancer potential of mansonone G by β-cyclodextrin-based host-guest complexation: A computational and experimental study. Biomolecules 2019, 9, 545. [Google Scholar] [CrossRef] [Green Version]
- Kicuntod, J.; Khuntawee, W.; Wolschann, P.; Pongsawasdi, P.; Chavasiri, W.; Kungwan, N.; Rungrotmongkol, T. Inclusion complexation of pinostrobin with various cyclodextrin derivatives. J. Mol. Graph. Model. 2016, 63, 91–98. [Google Scholar] [CrossRef]
- Zhang, H.; Feng, W.; Li, C.; Tan, T. Investigation of the inclusions of puerarin and daidzin with β-cyclodextrin by molecular dynamics simulation. J. Phys. Chem. B 2010, 114, 4876–4883. [Google Scholar] [CrossRef] [PubMed]
- Kamigauchi, M.; Kawanishi, K.; Sugiura, M.; Ohishi, H.; Ishida, T. γ-cyclodextrin as inhibitor of the precipitation reaction between berberine and glycyrrhizin in decoctions of natural medicines: Interaction studies of cyclodextrins with glycyrrhizin and glycyrrhetic acid by 1H-NMR spectroscopy and molecular-dynamics calculation. Helv. Chim. Acta 2008, 91, 1614–1624. [Google Scholar] [CrossRef]
- Cesari, A.; Uccello Barretta, G.; Kirschner, K.N.; Pappalardo, M.; Basile, L.; Guccione, S.; Russotto, C.; Lauro, M.R.; Cavaliere, F.; Balzano, F. Interaction of natural flavonoid eriocitrin with β-cyclodextrin and hydroxypropyl-β-cyclodextrin: An NMR and molecular dynamics investigation. New J. Chem. 2020, 44, 16431–16441. [Google Scholar] [CrossRef]
- Da Silva, A.J.; dos Santos, E.S. Energetic and thermodynamical aspects of the cyclodextrins-cannabidiol complex in aqueous solution: A molecular-dynamics study. Eur. Biophys. J. 2020, 49, 571–589. [Google Scholar] [CrossRef] [PubMed]
- Fateminasab, F.; Bordbar, A.K.; Shityakov, S.; Saboury, A.A. Molecular insights into inclusion complex formation between β- and γ-cyclodextrins and rosmarinic acid. J. Mol. Liq. 2020, 314, 113802. [Google Scholar] [CrossRef]
- Ferraz, C.A.A.; de Oliveira Júnior, R.G.; de Oliveira, A.P.; Groult, H.; Beaugeard, L.; Picot, L.; de Alencar Filho, E.B.; Almeida, J.R.G.D.S.; Nunes, X.P. Complexation with β-cyclodextrin enhances apoptosis-mediated cytotoxic effect of harman in chemoresistant BRAF-mutated melanoma cells. Eur. J. Pharm. Sci. 2020, 150, 105353. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Han, Z.; Liu, L.; Wang, Y.; Xin, S.; Zhang, H.; Yu, Z. Solubility enhancement of myricetin by inclusion complexation with heptakis-O-(2-hydroxypropyl)-β-cyclodextrin: A joint experimental and theoretical study. Int. J. Mol. Sci. 2020, 21, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Xu, R.; Ge, X.; Cheng, J. Complexation of capsaicin with hydroxypropyl-β-cyclodextrin and its analytical application. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 223, 117278. [Google Scholar] [CrossRef]
- Bose, A.; Sengupta, P.; Pal, U.; Senapati, S.; Ahsan, M.; Roy, S.; Das, U.; Sen, K. Encapsulation of Thymol in cyclodextrin nano-cavities: A multi spectroscopic and theoretical study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 208, 339–348. [Google Scholar] [CrossRef]
- Fateminasab, F.; Bordbar, A.K.; Shityakov, S.; Gholami, S. Diadzein complexation with unmodified cyclodextrins: A detailed experimental and theoretical study. J. Mol. Liq. 2018, 271, 80–95. [Google Scholar] [CrossRef]
- Khavani, M.; Kalantarinezhad, R.; Izadyar, M. A joint QM/MD study on α-, β- and γ-cyclodextrins in selective complexation with cathinone. Supramol. Chem. 2018, 30, 687–696. [Google Scholar] [CrossRef]
- Fourtaka, K.; Christoforides, E.; Mentzafos, D.; Bethanis, K. Crystal structures and molecular dynamics studies of the inclusion compounds of β-citronellol in β-cyclodextrin, heptakis(2,6-di-O-methyl)-β-cyclodextrin and heptakis(2,3,6-tri-O-methyl)-β-cyclodextrin. J. Mol. Struct. 2018, 1161, 1–8. [Google Scholar] [CrossRef]
- Yan, H.-H.; Zhang, J.-Q.; Ren, S.-H.; Xie, X.-G.; Huang, R.; Jin, Y.; Lin, J. Experimental and computational studies of naringin/cyclodextrin inclusion complexation. J. Incl. Phenom. Macrocycl. Chem. 2017, 88, 15–26. [Google Scholar] [CrossRef]
- Shityakov, S.; Salmas, R.E.; Durdagi, S.; Roewer, N.; Förster, C.; Broscheit, J. Solubility profiles, hydration and desolvation of curcumin complexed with γ-cyclodextrin and hydroxypropyl-γ-cyclodextrin. J. Mol. Struct. 2017, 1134, 91–98. [Google Scholar] [CrossRef]
- Kellici, T.F.; Chatziathanasiadou, M.V.; Diamantis, D.; Chatzikonstantinou, A.V.; Andreadelis, I.; Christodoulou, E.; Valsami, G.; Mavromoustakos, T.; Tzakos, A.G. Mapping the interactions and bioactivity of quercetin (2-hydroxypropyl)-β-cyclodextrin complex. Int. J. Pharm. 2016, 511, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Kellici, T.F.; Ntountaniotis, D.; Leonis, G.; Chatziathanasiadou, M.; Chatzikonstantinou, A.V.; Becker-Baldus, J.; Glaubitz, C.; Tzakos, A.G.; Viras, K.; Chatzigeorgiou, P.; et al. Investigation of the interactions of silibinin with 2-hydroxypropyl-β-cyclodextrin through biophysical techniques and computational methods. Mol. Pharm. 2015, 12, 954–965. [Google Scholar] [CrossRef]
- Fernandes, A.; Ivanova, G.; Brás, N.F.; Mateus, N.; Ramos, M.J.; Rangel, M.; de Freitas, V. Structural characterization of inclusion complexes between cyanidin-3-O-glucoside and β-cyclodextrin. Carbohydr. Polym. 2014, 102, 269–277. [Google Scholar] [CrossRef]
- Troche-Pesqueira, E.; Pérez-Juste, I.; Navarro-Vázquez, A.; Cid, M.M. A β-cyclodextrin–resveratrol inclusion complex and the role of geometrical and electronic effects on its electronic induced circular dichroism. RSC Adv. 2013, 3, 10242–10250. [Google Scholar] [CrossRef]
- Hotarat, W.; Phunpee, S.; Rungnim, C.; Wolschann, P.; Kungwan, N.; Ruktanonchai, U.; Rungrotmongkol, T.; Hannongbua, S. Encapsulation of alpha-mangostin and hydrophilic beta-cyclodextrins revealed by all-atom molecular dynamics simulations. J. Mol. Liq. 2019, 288, 110965. [Google Scholar] [CrossRef]
- Rungnim, C.; Phunpee, S.; Kunaseth, M.; Namuangruk, S.; Rungsardthong, K.; Rungrotmongkol, T.; Ruktanonchai, U. Co-solvation effect on the binding mode of the α-mangostin/β-cyclodextrin inclusion complex. Beilstein J. Org. Chem. 2015, 11, 2306–2317. [Google Scholar] [CrossRef] [Green Version]
- Kordopati, G.G.; Tselios, T.V.; Kellici, T.; Merzel, F.; Mavromoustakos, T.; Grdadolnik, S.G.; Tsivgoulis, G.M. A novel synthetic luteinizing hormone-releasing hormone (LHRH) analogue coupled with modified β-cyclodextrin: Insight into its intramolecular interactions. Biochim. Biophys. Acta—Gen. Subj. 2015, 1850, 159–168. [Google Scholar] [CrossRef]
- Suvarna, V.; Thorat, S.; Nayak, U.; Sherje, A.; Murahari, M. Host-guest interaction study of Efavirenz with hydroxypropyl-β-cyclodextrin and l-arginine by computational simulation studies: Preparation and characterization of supramolecular complexes. J. Mol. Liq. 2018, 259, 55–64. [Google Scholar] [CrossRef]
- Figueiras, A.; Sarraguça, J.M.G.; Pais, A.A.C.C.; Carvalho, R.A.; Veiga, J.F. The role of L-arginine in inclusion complexes of omeprazole with cyclodextrins. AAPS PharmSciTech 2010, 11, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Varghese, B.; Al-Busafi, S.N.; Suliman, F.O.; Al-Kindy, S.M.Z. Tuning the constrained photophysics of a pyrazoline dye 3-naphthyl-1-phenyl-5-(4-carboxyphenyl)-2-pyrazoline inside the cyclodextrin nanocavities: A detailed insight via experimental and theoretical approach. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 173, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Carmona, T.; Marcelo, G.; Rinaldi, L.; Martina, K.; Cravotto, G.; Mendicuti, F. Soluble cyanine dye/β-cyclodextrin derivatives: Potential carriers for drug delivery and optical imaging. Dye. Pigment. 2015, 114, 204–214. [Google Scholar] [CrossRef]
- Lao, W.; Song, C.; You, J.; Ou, Q. Fluorescence and β-cyclodextrin inclusion properties of three carbazole-based dyes. Dye. Pigment. 2012, 95, 619–626. [Google Scholar] [CrossRef]
- Zhao, Q.; Miriyala, N.; Su, Y.; Chen, W.; Gao, X.; Shao, L.; Yan, R.; Li, H.; Yao, X.; Cao, D.; et al. Computer-aided formulation design for a highly soluble lutein–cyclodextrin multiple-component delivery system. Mol. Pharm. 2018, 15, 1664–1673. [Google Scholar] [CrossRef]
- Manas, N.H.A.; Bakar, F.D.A.; Illias, R.M. Computational docking, molecular dynamics simulation and subsite structure analysis of a maltogenic amylase from Bacillus lehensis G1 provide insights into substrate and product specificity. J. Mol. Graph. Model. 2016, 67, 1–13. [Google Scholar] [CrossRef]
- Melani, F.; Pasquini, B.; Caprini, C.; Gotti, R.; Orlandini, S.; Furlanetto, S. Combination of capillary electrophoresis, molecular modeling and NMR to study the enantioselective complexation of sulpiride with double cyclodextrin systems. J. Pharm. Biomed. Anal. 2015, 114, 265–271. [Google Scholar] [CrossRef]
- Li, S.; Wang, L.; Jiang, J.; Tang, P.; Wang, Q.; Wu, D.; Li, H. Investigations of bisacodyl with modified β-cyclodextrins: Characterization, molecular modeling, and effect of PEG. Carbohydr. Polym. 2015, 134, 82–91. [Google Scholar] [CrossRef]
- Wang, Z.; Landy, D.; Sizun, C.; Cézard, C.; Solgadi, A.; Przybylski, C.; de Chaisemartin, L.; Herfindal, L.; Barratt, G.; Legrand, F.-X. Cyclodextrin complexation studies as the first step for repurposing of chlorpromazine. Int. J. Pharm. 2020, 584, 119391. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Bomzan, P.; Nath Roy, M. Probing Host-Guest inclusion complexes of Ambroxol Hydrochloride with α- & β-Cyclodextrins by physicochemical contrivance subsequently optimized by molecular modeling simulations. Chem. Phys. Lett. 2020, 748, 137372. [Google Scholar] [CrossRef]
- Adeoye, O.; Conceição, J.; Serra, P.A.; Bento da Silva, A.; Duarte, N.; Guedes, R.C.; Corvo, M.C.; Aguiar-Ricardo, A.; Jicsinszky, L.; Casimiro, T.; et al. Cyclodextrin solubilization and complexation of antiretroviral drug lopinavir: In silico prediction; Effects of derivatization, molar ratio and preparation method. Carbohydr. Polym. 2020, 227, 115287. [Google Scholar] [CrossRef]
- Floresta, G.; Rescifina, A. Metyrapone-β-cyclodextrin supramolecular interactions inferred by complementary spectroscopic/spectrometric and computational studies. J. Mol. Struct. 2019, 1176, 815–824. [Google Scholar] [CrossRef]
- Garnier, L.; Sarraute, S.; Israëli, Y.; Bonal, C.; Malfreyt, P. Associations of water-soluble macrocyclic hosts with 4-aminoazobenzene: Impact of pH. J. Phys. Chem. B 2018, 122, 11953–11961. [Google Scholar] [CrossRef]
- Yakavets, I.; Lassalle, H.P.; Yankovsky, I.; Ingrosso, F.; Monari, A.; Bezdetnaya, L.; Zorin, V. Evaluation of temoporfin affinity to β-cyclodextrins assuming self-aggregation. J. Photochem. Photobiol. A Chem. 2018, 367, 13–21. [Google Scholar] [CrossRef]
- Das, S.; Maharana, J.; Mohanty, S.; Subuddhi, U. Spectroscopic and computational insights into theophylline/β-cyclodextrin complexation: Inclusion accomplished by diverse methods. J. Microencapsul. 2018, 35, 667–679. [Google Scholar] [CrossRef]
- Bethanis, K.; Christoforides, E.; Tsorteki, F.; Fourtaka, K.; Mentzafos, D. Structural studies of the inclusion compounds of α-naphthaleneacetic acid in heptakis(2,6-di-O-methyl)-β-Cyclodextrin and heptakis(2,3,6-tri-O-methyl)-β-Cyclodextrin by X-ray crystallography and molecular dynamics. J. Incl. Phenom. Macrocycl. Chem. 2018, 92, 157–171. [Google Scholar] [CrossRef]
- Krait, S.; Salgado, A.; Chankvetadze, B.; Gago, F.; Scriba, G.K.E. Investigation of the complexation between cyclodextrins and medetomidine enantiomers by capillary electrophoresis, NMR spectroscopy and molecular modeling. J. Chromatogr. A 2018, 1567, 198–210. [Google Scholar] [CrossRef]
- Dang, P.; Ye, R.; Meng, F.; Han, Y.; Zhou, Y.; Gong, X.; Zhou, B. Microencapsulation thermodynamics of methylated β-cyclodextrins with bile salt: Enthalpy, entropy, and solvent effect. J. Incl. Phenom. Macrocycl. Chem. 2017, 88, 181–189. [Google Scholar] [CrossRef]
- Salgado, A.; Tatunashvili, E.; Gogolashvili, A.; Chankvetadze, B.; Gago, F. Structural rationale for the chiral separation and migration order reversal of clenpenterol enantiomers in capillary electrophoresis using two different β-cyclodextrins. Phys. Chem. Chem. Phys. 2017, 19, 27935–27939. [Google Scholar] [CrossRef] [PubMed]
- Al-Burtomani, S.K.S.; Suliman, F.O. Inclusion complexes of norepinephrine with β-cyclodextrin, 18-crown-6 and cucurbit[7]uril: Experimental and molecular dynamics study. RSC Adv. 2017, 7, 9888–9901. [Google Scholar] [CrossRef] [Green Version]
- Martini, M.F.; Glisoni, R.J.; Sosnik, A.; Moglioni, A.; Pickholz, M. Insights on self-aggregation phenomena of 1-indanone thiosemicarbazones and the formation of inclusion complexes with hydroxypropyl-β-cyclodextrin by molecular dynamics simulations. J. Mol. Liq. 2016, 222, 963–971. [Google Scholar] [CrossRef]
- Mayer, B.P.; Kennedy, D.J.; Lau, E.Y.; Valdez, C.A. Solution-state structure and affinities of cyclodextrin: Fentanyl complexes by nuclear magnetic resonance spectroscopy and molecular dynamics simulation. J. Phys. Chem. B 2016, 120, 2423–2433. [Google Scholar] [CrossRef] [PubMed]
- Braga, M.A.; Martini, M.F.; Pickholz, M.; Yokaichiya, F.; Franco, M.K.D.; Cabeça, L.F.; Guilherme, V.A.; Silva, C.M.G.; Limia, C.E.G.; de Paula, E. Clonidine complexation with hydroxypropyl-beta-cyclodextrin: From physico-chemical characterization to in vivo adjuvant effect in local anesthesia. J. Pharm. Biomed. Anal. 2016, 119, 27–36. [Google Scholar] [CrossRef]
- Minea, B.; Marangoci, N.; Peptanariu, D.; Rosca, I.; Nastasa, V.; Corciova, A.; Varganici, C.; Nicolescu, A.; Fifere, A.; Neamtu, A.; et al. Inclusion complexes of propiconazole nitrate with substituted β-cyclodextrins: The synthesis and in silico and in vitro assessment of their antifungal properties. New J. Chem. 2016, 40, 1765–1776. [Google Scholar] [CrossRef]
- Ateba, B.A.; Lissouck, D.; Azébazé, A.; Ebelle, C.T.; Nassi, A.; Ngameni, E.; Duportail, G.; Mbazé, L.; Kenfack, C.A. Characterization of Mammea A/AA in solution and in interaction with β-cyclodextrin: UV–visible spectroscopy, cyclic voltammetry and DFT-TDDFT/MD study. J. Mol. Liq. 2016, 213, 294–303. [Google Scholar] [CrossRef]
- Medarević, D.; Kachrimanis, K.; Djurić, Z.; Ibrić, S. Influence of hydrophilic polymers on the complexation of carbamazepine with hydroxypropyl-β-cyclodextrin. Eur. J. Pharm. Sci. 2015, 78, 273–285. [Google Scholar] [CrossRef]
- Barbosa, J.A.A.; Zoppi, A.; Quevedo, M.A.; De Melo, P.N.; De Medeiros, A.S.A.; Streck, L.; De Oliveira, A.R.; Fernandes-Pedrosa, M.F.; Longhi, M.R.; Da Silva-Júnior, A.A. Triethanolamine stabilization of methotrexate-β-cyclodextrin interactions in ternary complexes. Int. J. Mol. Sci. 2014, 15, 17077–17099. [Google Scholar] [CrossRef] [Green Version]
- Jiao, A.; Zhou, X.; Xu, X.; Jin, Z. Molecular dynamics simulations of cyclodextrin–cumene hydroperoxide complexes in water. Comput. Theor. Chem. 2013, 1013, 1–6. [Google Scholar] [CrossRef]
- Pacioni, N.L.; Pierini, A.B.; Veglia, A.V. Structural characterization of N-methylcarbamate: β-Cyclodextrin complexes by experimental methods and molecular dynamics simulations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 103, 319–324. [Google Scholar] [CrossRef]
- Mouria-Bellabdelli, F.; Potier, J.; Bouterfas, M.; Cavrot, J.-P.; Sayede, A.; Menuel, S.; Monflier, E.; Machut-Binkowski, C. Characterization of β-cyclodextrins and isosorbide diesters self-assemblies: Towards new renewable surfactants. Colloids Surf. A Physicochem. Eng. Asp. 2012, 415, 380–387. [Google Scholar] [CrossRef]
- Schönbeck, C. Charge determines guest orientation: A combined NMR and molecular dynamics study of β-cyclodextrins and adamantane derivatives. J. Phys. Chem. B 2018, 122, 4821–4827. [Google Scholar] [CrossRef] [Green Version]
- Akhondi, M.; Jamalizadeh, E.; Mohebbi, A. MD and DFT calculations on the structural variations of amino-cyclodextrin as a pH-sensitive carrier for smart carriage and release of Doxorubicin. J. Mol. Struct. 2021, 1230, 129855. [Google Scholar] [CrossRef]
- Shao, M.; Li, S.; Tan, C.P.; Kraithong, S.; Gao, Q.; Fu, X.; Zhang, B.; Huang, Q. Encapsulation of caffeine into starch matrices: Bitterness evaluation and suppression mechanism. Int. J. Biol. Macromol. 2021, 173, 118–127. [Google Scholar] [CrossRef]
- Jana, M.; Bandyopadhyay, S. Molecular dynamics study of β-cyclodextrin–phenylalanine (1:1) inclusion complex in aqueous medium. J. Phys. Chem. B 2013, 117, 9280–9287. [Google Scholar] [CrossRef] [PubMed]
- Tomeček, J.; Čablová, A.; Hromádková, A.; Novotný, J.; Marek, R.; Durník, I.; Kulhánek, P.; Prucková, Z.; Rouchal, M.; Dastychová, L.; et al. Modes of micromolar host–guest binding of β-cyclodextrin complexes revealed by NMR spectroscopy in salt water. J. Org. Chem. 2021, 86, 4483–4496. [Google Scholar] [CrossRef] [PubMed]
- González-Méndez, I.; Aguayo-Ortiz, R.; Sorroza-Martínez, K.; Solano, J.D.; Porcu, P.; Rivera, E.; Dominguez, L. Conformational analysis by NMR and molecular dynamics of adamantane-doxorubicin prodrugs and their assemblies with β-cyclodextrin: A focus on the design of platforms for controlled drug delivery. Bioorg. Med. Chem. 2020, 28, 115510. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Liu, C.; Abroshan, H.; Li, Z.; Qiu, R.; Li, G. Surface modification of adamantane-terminated gold nanoclusters using cyclodextrins. Phys. Chem. Chem. Phys. 2016, 18, 23358–23364. [Google Scholar] [CrossRef] [PubMed]
- Kiriakidi, S.; Mavromoustakos, T. Study of CandesartanCandesartan (CAN)Cilexetil: 2-hydroxypropyl-β-cyclodextrin interactionsinteractions: A computational approach using steered molecular dynamics-molecular dynamics simulations. In Supramolecules in Drug Discovery and Drug Delivery: Methods and Protocols; Mavromoustakos, T., Tzakos, A.G., Durdagi, S., Eds.; Springer: New York, NY, USA, 2021; pp. 45–70. [Google Scholar] [CrossRef]
- Kicuntod, J.; Sangpheak, K.; Mueller, M.; Wolschann, P.; Viernstein, H.; Yanaka, S.; Kato, K.; Chavasiri, W.; Pongsawasdi, P.; Kungwan, N.; et al. Theoretical and experimental studies on inclusion complexes of pinostrobin and β-cyclodextrins. Sci. Pharm. 2018, 86, 5. [Google Scholar] [CrossRef] [Green Version]
- Herrera, A.; Rodríguez, F.J.; Bruna, J.E.; Abarca, R.L.; Galotto, M.J.; Guarda, A.; Mascayano, C.; Sandoval-Yáñez, C.; Padula, M.; Felipe, F.R.S. Antifungal and physicochemical properties of inclusion complexes based on β-cyclodextrin and essential oil derivatives. Food Res. Int. 2019, 121, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tan, T.; Feng, W.; van der Spoel, D. Molecular recognition in different environments: β-cyclodextrin dimer Formation in organic solvents. J. Phys. Chem. B 2012, 116, 12684–12693. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Zhang, D.; Zhan, J. Theoretical investigation on the inclusion of TCDD with β-cyclodextrin by performing QM calculations and MD simulations. J. Hazard. Mater. 2011, 192, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhang, D.; Zhan, J. Investigation on the inclusions of PCB52 with cyclodextrins by performing DFT calculations and molecular dynamics simulations. J. Phys. Chem. A 2010, 114, 13122–13128. [Google Scholar] [CrossRef]
- Ren, B.; Zhang, M.; Gao, H.; Zheng, J.; Jia, L. Atomic elucidation of the cyclodextrin effects on DDT solubility and biodegradation. Phys. Chem. Chem. Phys. 2016, 18, 17380–17388. [Google Scholar] [CrossRef]
- Poór, M.; Kunsági-Máté, S.; Szente, L.; Matisz, G.; Secenji, G.; Czibulya, Z.; Kőszegi, T. Interaction of ochratoxin A with quaternary ammonium beta-cyclodextrin. Food Chem. 2015, 172, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Xu, H.; Zhang, D.; Zhan, J. Molecular inclusion of PCB126 by beta-cyclodextrin: A combined molecular dynamics simulation and quantum chemical study. J. Incl. Phenom. Macrocycl. Chem. 2013, 76, 301–309. [Google Scholar] [CrossRef]
- Škvára, J.; Nezbeda, I.; Izák, P. Molecular dynamics study of racemic mixtures. II. Temperature dependence of the separation of ibuprofen racemic mixture with β-cyclodextrin in methanol solvent. J. Mol. Liq. 2020, 302, 112575. [Google Scholar] [CrossRef]
- Cai, C.; Liu, M.; Yan, H.; Zhao, Y.; Shi, Y.; Guo, Q.; Pei, W.; Han, J.; Wang, Z. A combined calorimetric, spectroscopic and molecular dynamic simulation study on the inclusion complexation of (E)-piceatannol with hydroxypropyl-β-cyclodextrin in various alcohol + water cosolvents. J. Chem. Thermodyn. 2019, 132, 341–351. [Google Scholar] [CrossRef]
- Boonyarattanakalin, K.; Wolschann, P.; Toochinda, P.; Lawtrakul, L. Molecular dynamics simulations of UC781-cyclodextrins inclusion complexes in aqueous solution. Eur. J. Pharm. Sci. 2012, 47, 752–758. [Google Scholar] [CrossRef]
- Slavgorodska, M.V.; Gurova, Y.O.; Kyrychenko, A. γ-Cyclodextrin as a capping agent for gold nanoparticles. Comput. Theor. Chem. 2021, 1194, 113060. [Google Scholar] [CrossRef]
- Dou, R.; Chen, K.; Chi, G.; Luo, J.; Wong, C.F.; Zhou, B. Why heptakis(2,3-di-O-acetyl)-β-cyclodextrin can separate terbutaline enantiomers better than β-cyclodextrin: Nonbonding and hydrophobic interactions. J. Incl. Phenom. Macrocycl. Chem. 2021, 100, 189–195. [Google Scholar] [CrossRef]
- Ikeda, A.; Ishikawa, M.; Aono, R.; Kikuchi, J.-i.; Akiyama, M.; Shinoda, W. Regioselective recognition of a [60]fullerene-bisadduct by cyclodextrin. J. Org. Chem. 2013, 78, 2534–2541. [Google Scholar] [CrossRef] [PubMed]
- Bikádi, Z.; Fodor, G.; Hazai, I.; Hári, P.; Szemán, J.; Szente, L.; Fülöp, F.; Péter, A.; Hazai, E. Molecular modeling of enantioseparation of phenylazetidin derivatives by cyclodextrins. Chromatographia 2010, 71, 21–28. [Google Scholar] [CrossRef]
- Alvira, E. Molecular simulation of the separation of isoleucine enantiomers by β-cyclodextrin. Molecules 2019, 24, 1021. [Google Scholar] [CrossRef] [Green Version]
- Altarsha, M.; Yeguas, V.; Ingrosso, F.; López, R.; Ruiz-López, M.F. Taste for chiral guests: Investigating the stereoselective binding of peptides to β-cyclodextrins. J. Phys. Chem. B 2013, 117, 3091–3097. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef]
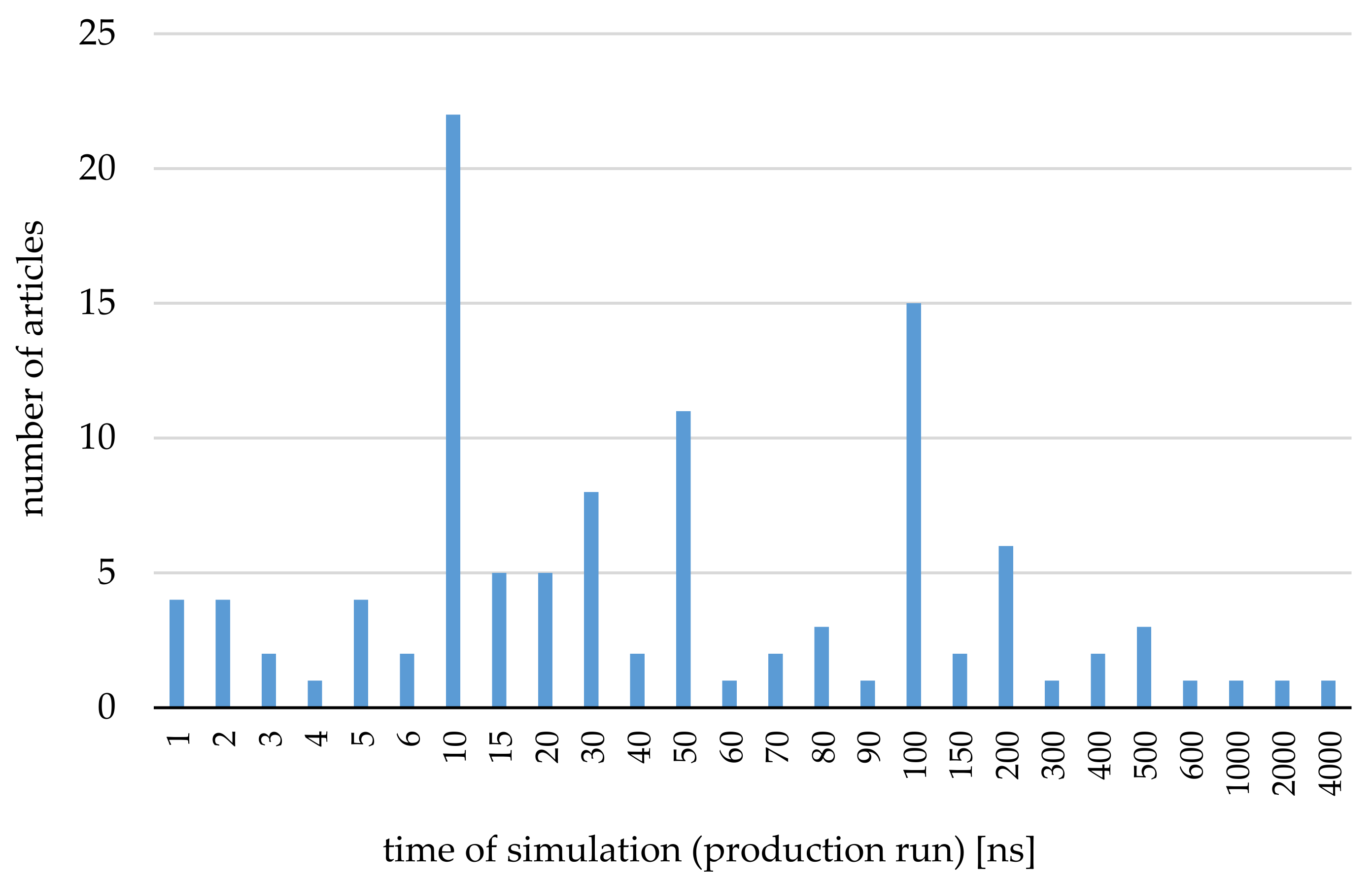

| N° | Software/Code | Force Field | License | Ref. |
|---|---|---|---|---|
| 1 | GROMACS | GROMOS CHARMM Carbohydrate Solution force field, CSFF (for CD) | General Public License (GPL) | [32] |
| 2 | AMBER | GAFF, Glycam06 (for CD), q4md-CD (for CD) | Commercial | [33] |
| 3 | CHARMM | CHARMM FF | Commercial | [34] |
| 4 | Desmond (Schrödinger, Inc.) | OPLS | Commercial | [35] |
| 5 | NAMD | CHARMM | Commercial, academic | [36] |
| 6 | Forcite (BIOVIA Materials Studio, Accelrys) | COMPASS | Commercial | [37] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazurek, A.H.; Szeleszczuk, Ł.; Gubica, T. Application of Molecular Dynamics Simulations in the Analysis of Cyclodextrin Complexes. Int. J. Mol. Sci. 2021, 22, 9422. https://doi.org/10.3390/ijms22179422
Mazurek AH, Szeleszczuk Ł, Gubica T. Application of Molecular Dynamics Simulations in the Analysis of Cyclodextrin Complexes. International Journal of Molecular Sciences. 2021; 22(17):9422. https://doi.org/10.3390/ijms22179422
Chicago/Turabian StyleMazurek, Anna Helena, Łukasz Szeleszczuk, and Tomasz Gubica. 2021. "Application of Molecular Dynamics Simulations in the Analysis of Cyclodextrin Complexes" International Journal of Molecular Sciences 22, no. 17: 9422. https://doi.org/10.3390/ijms22179422
APA StyleMazurek, A. H., Szeleszczuk, Ł., & Gubica, T. (2021). Application of Molecular Dynamics Simulations in the Analysis of Cyclodextrin Complexes. International Journal of Molecular Sciences, 22(17), 9422. https://doi.org/10.3390/ijms22179422