
Slow Off-Rate Modified Aptamer (SOMAmer) Proteomic Analysis of Patient-Derived Malignant Glioma Identifies Distinct Cellular Proteomes


 , , , , and
, , , , and 
Abstract
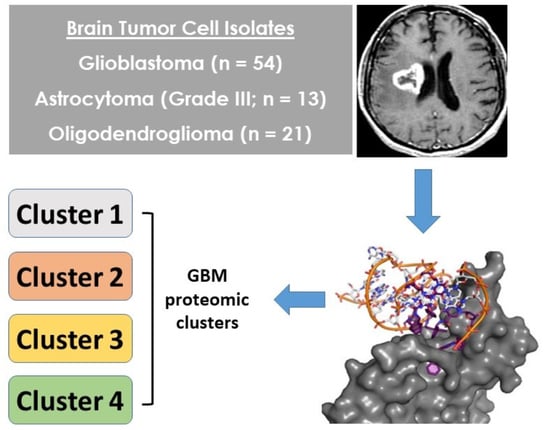
1. Introduction
2. Results
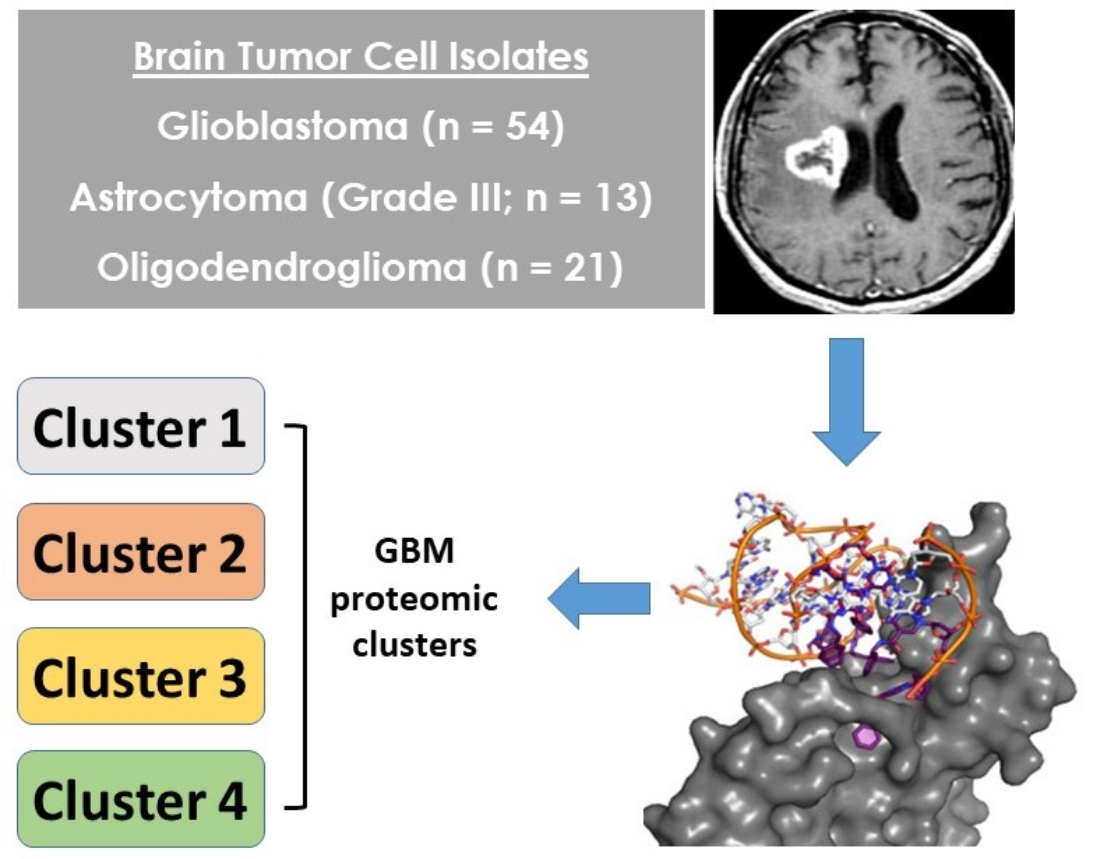
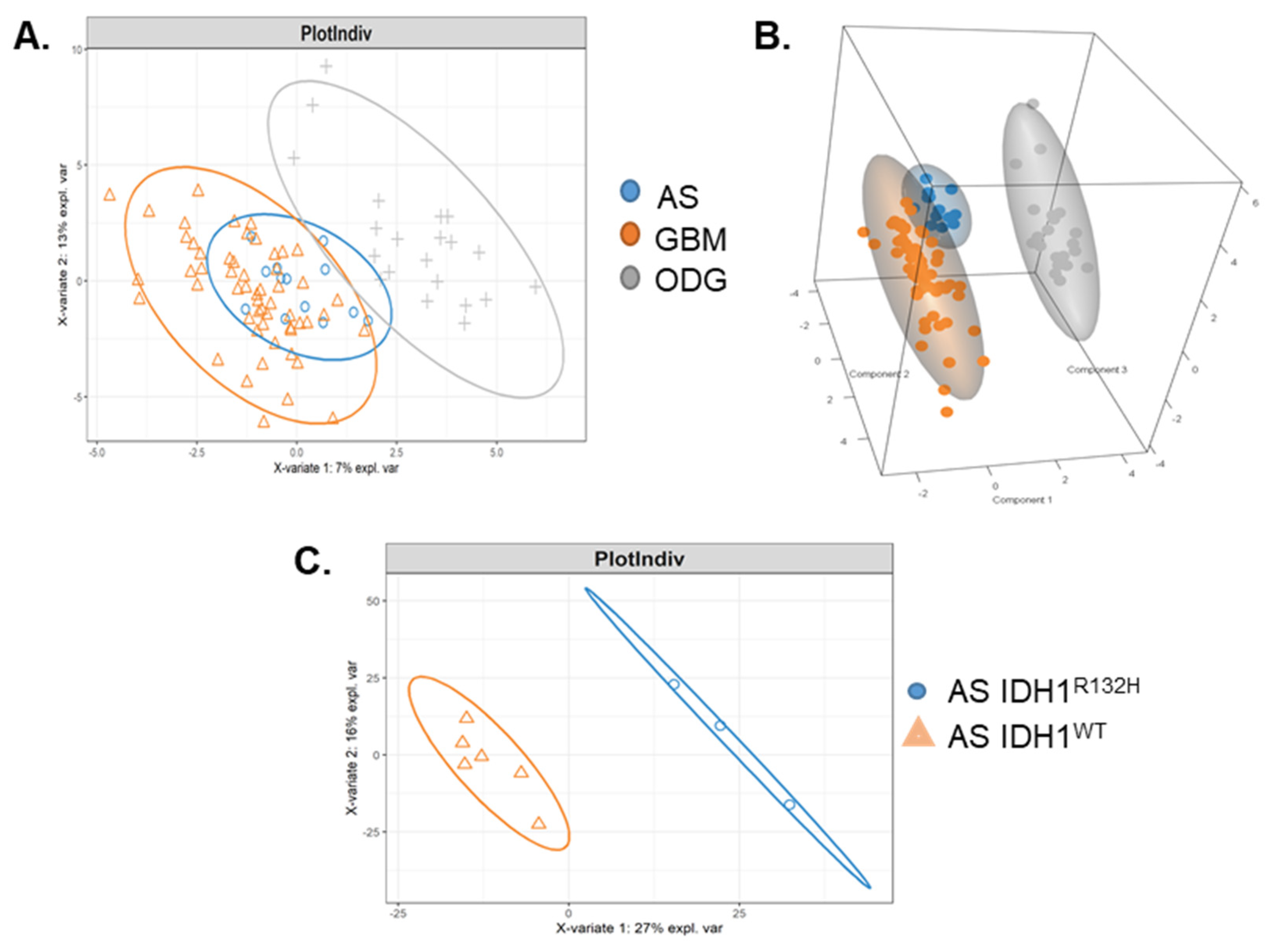
2.1. Malignant Glioma Pathologies Have Distinct SOMAscan® Cellular Proteomes
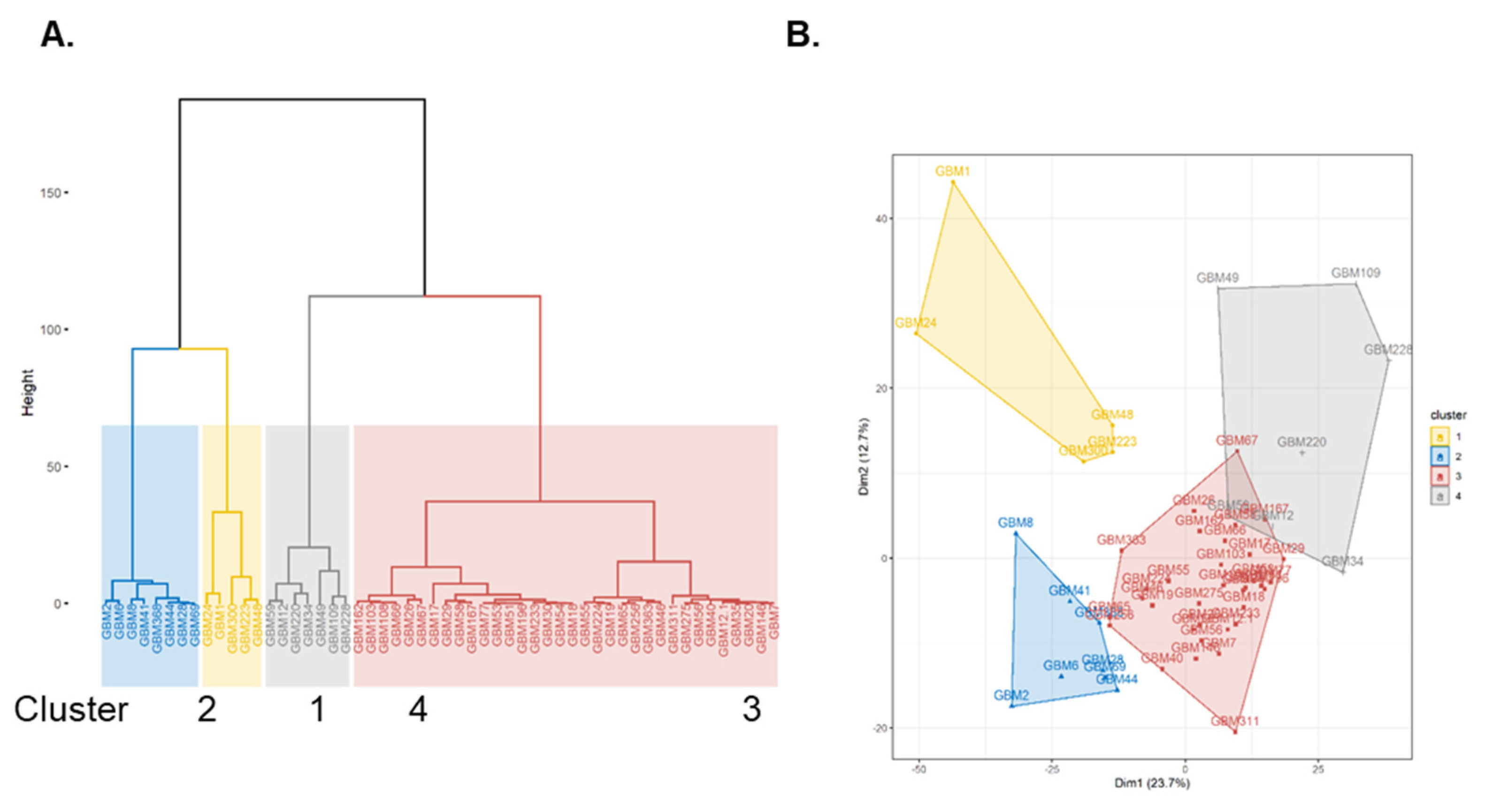
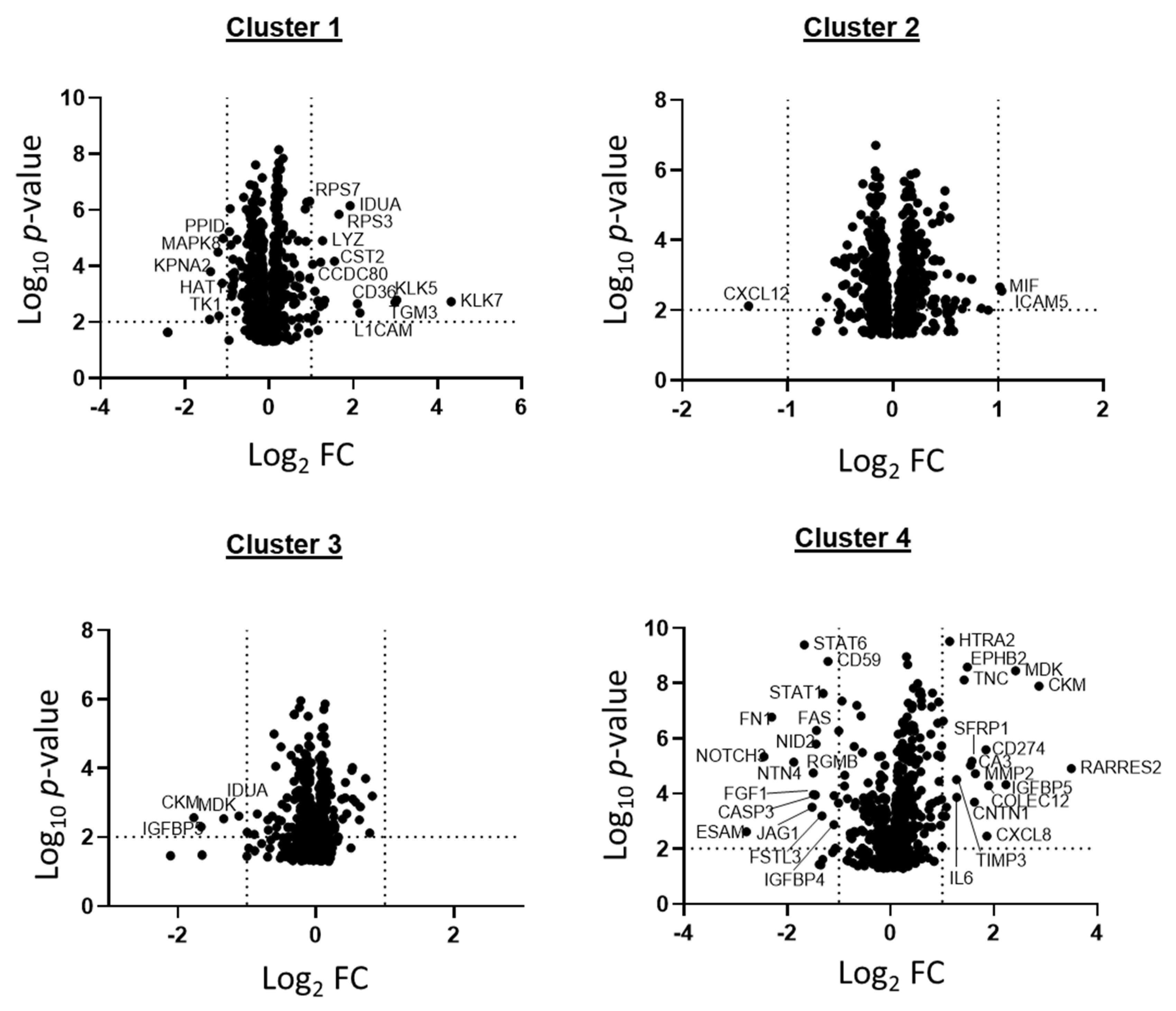
2.2. Patient GBM Cell Isolates Segregate into Four SOMAscan® Proteomic Clusters
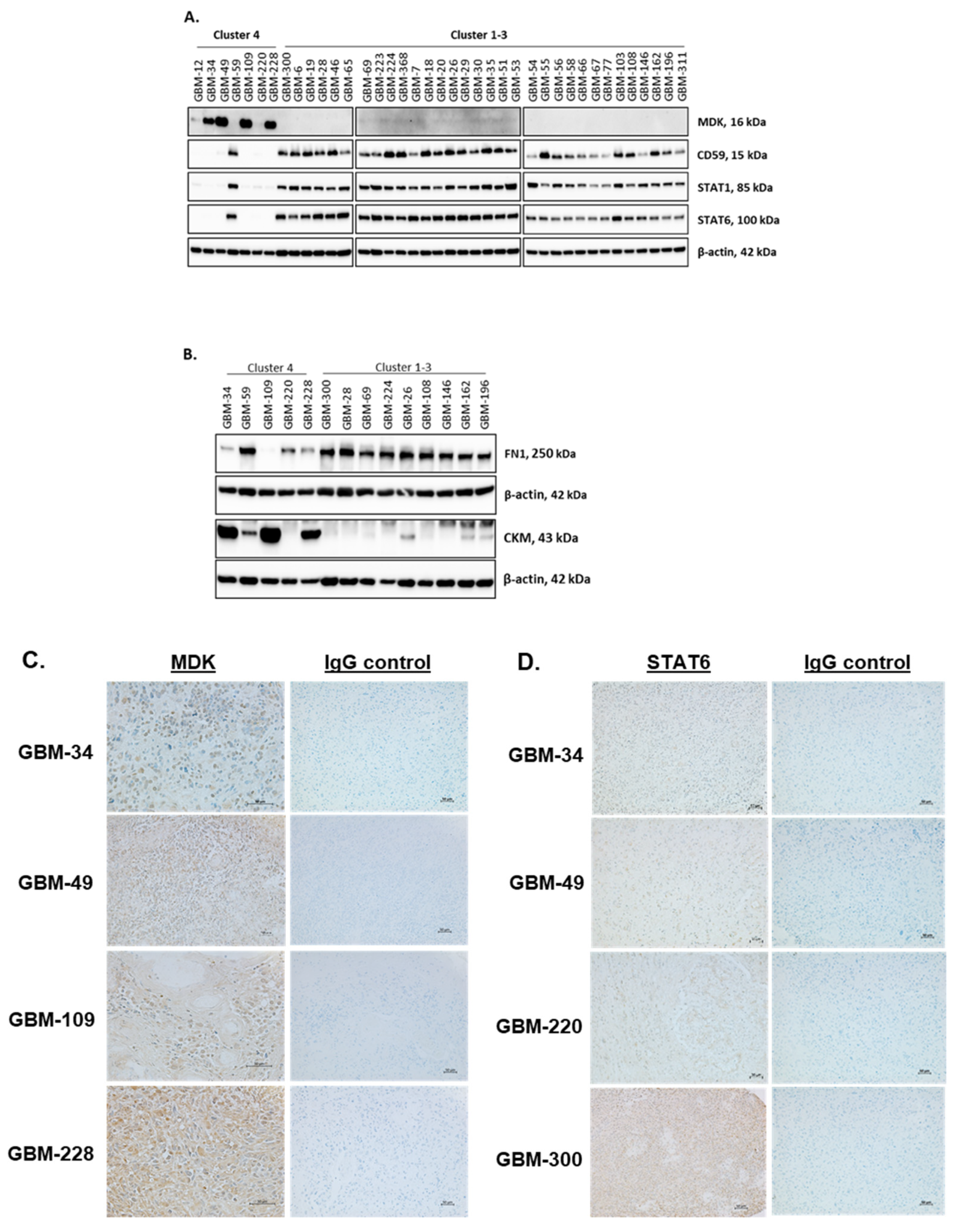
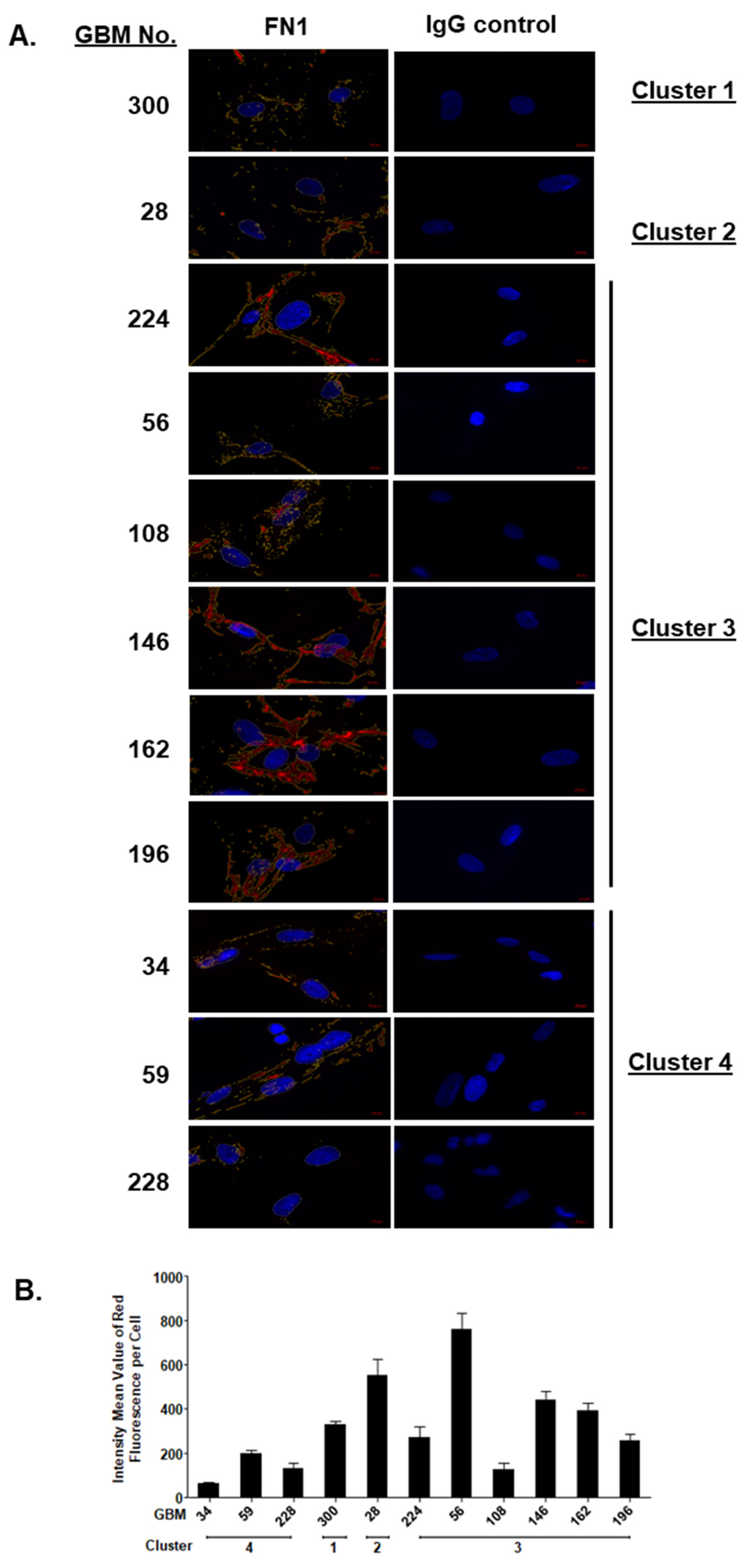
2.3. SOMAscan® Proteomic Clusters Are Validated in GBM Cells and Corresponding Tissues
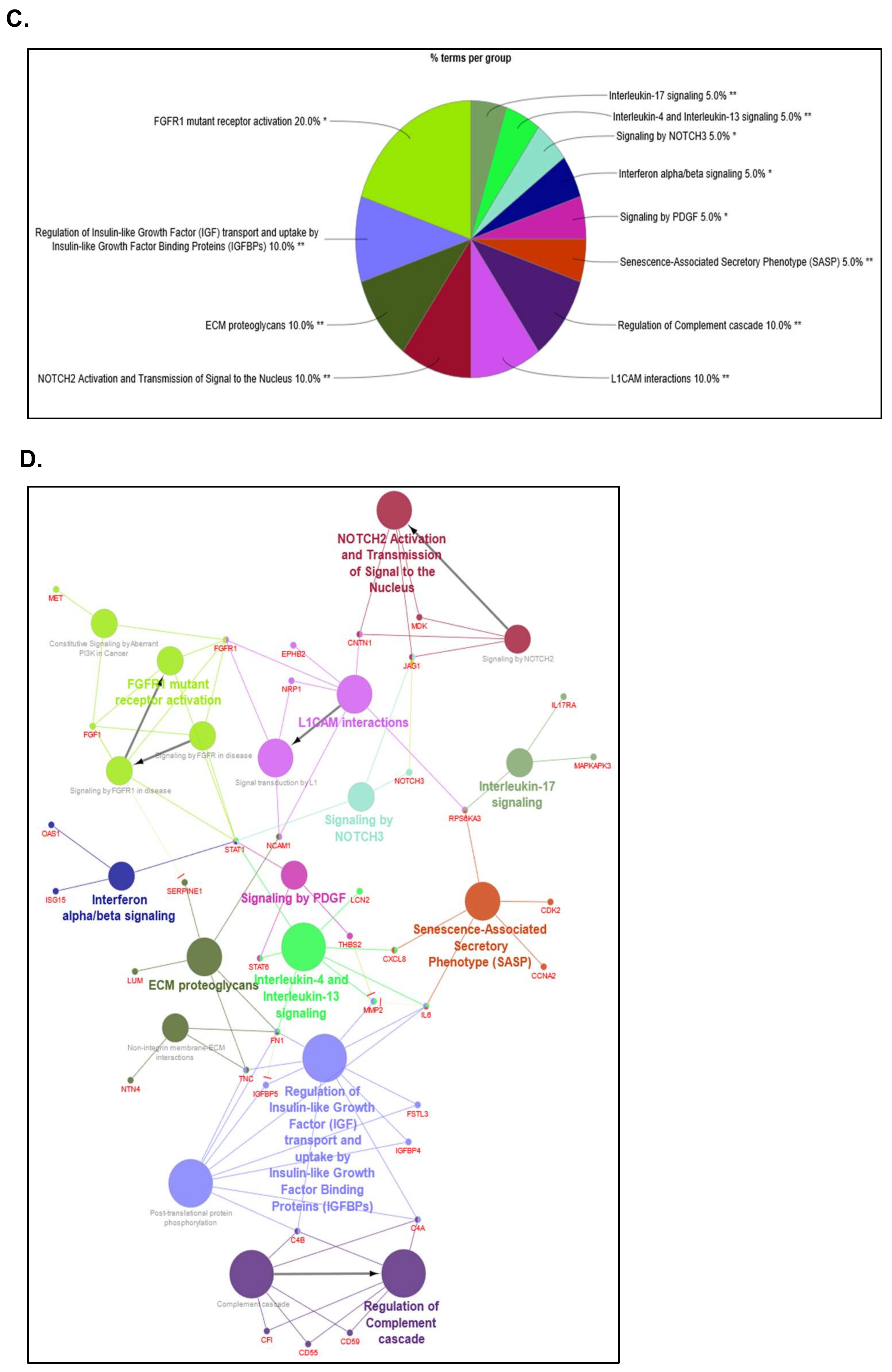
2.4. Different Signaling Networks among SOMAscan® GBM Proteomic Clusters
3. Discussion
4. Materials and Methods
4.1. GB Patient Tissue Samples and Cell Culture
4.2. Sample Preparation and SOMAscan® Analysis
4.3. Sparse PLS-DA
4.4. Hierarchical Clustering
4.5. Principal Component Analysis and Partial Least Squares Discriminate Analysis
4.6. Western Blot Analysis
4.7. Immunodetection of Proteomic Targets in Patient GBM Cells and Tissues
4.8. RNA Isolation and Quantitative Reverse Transcriptase Polymerase Chain Reaction (qPCR)
4.9. Bioinformatics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar]
- Krex, D.; Klink, B.; Hartmann, C.; von Deimling, A.; Pietsch, T.; Simon, M.; Sabel, M.; Steinbach, J.P.; Heese, O.; Reifenberger, G.; et al. Long-term survival with glioblastoma multiforme. Brain 2007, 130, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Tonn, J.C.; Brada, M.; Pentheroudakis, G. High-grade malignant glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2010, 21 (Suppl. 5), v190–v193. [Google Scholar] [CrossRef] [PubMed]
- Gurney, J.G.; Kadan-Lottick, N. Brain and other central nervous system tumors: Rates, trends, and epidemiology. Curr. Opin. Oncol. 2001, 13, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef]
- Stupp, R.; Bent, M.V.D.; Hegi, M. Optimal role of temozolomide in the treatment of malignant gliomas. Curr. Neurol. Neurosci. Rep. 2005, 5, 198–206. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Mrugala, M.M.; Chamberlain, M.C. Mechanisms of Disease: Temozolomide and glioblastoma—look to the future. Nat. Clin. Pract. Oncol. 2008, 5, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Shen, Y.; Grisdale, C.; Islam, S.A.; Bose, P.; Lever, J.; Zhao, E.Y.; Grinshtein, N.; Ma, Y.; Mungall, A.J.; Moore, R.A.; et al. Comprehensive genomic profiling of glioblastoma tumors, BTICs, and xenografts reveals stability and adaptation to growth environments. Proc. Natl. Acad. Sci. USA 2019, 116, 19098–19108. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Dekker, L.J.M.; Kannegieter, N.M.; Haerkens, F.; Toth, E.; Kros, J.M.; Hov, D.A.S.; Fillebeen, J.; Verschuren, L.; Leenstra, S.; Ressa, A.; et al. Multiomics profiling of paired primary and recurrent glioblastoma patient tissues. Neuro-Oncol. Adv. 2020, 2, vdaa083. [Google Scholar] [CrossRef] [PubMed]
- Daubon, T.; Guyon, J.; Raymond, A.-A.; Dartigues, B.; Rudewicz, J.; Ezzoukhry, Z.; Dupuy, J.-W.; Herbert, J.M.J.; Saltel, F.; Bjerkvig, R.; et al. The invasive proteome of glioblastoma revealed by laser-capture microdissection. Neuro-Oncol. Adv. 2019, 1, vdz029. [Google Scholar] [CrossRef] [PubMed]
- De Aquino, P.F.; Carvalho, P.C.; Nogueira, F.C.; da Fonseca, C.O.; de Souza Silva, J.C.; da Costa Carvalho, M.d.G.; Domont, G.B.; Zanchin, N.I.; da Gama Fischer, S.F. A Time-Based and Intratumoral Proteomic Assessment of a Recurrent Glioblastoma Multiforme. Front. Oncol. 2016, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Polisetty, R.V.; Gupta, M.K.; Nair, S.C.; Ramamoorthy, K.; Tiwary, S.; Shiras, A.; Chandak, G.R.; Sirdeshmukh, R. Glioblastoma cell secretome: Analysis of three glioblastoma cell lines reveal 148 non-redundant proteins. J. Proteom. 2011, 74, 1918–1925. [Google Scholar] [CrossRef]
- Okawa, S.; Gagrica, S.; Blin, C.; Ender, C.; Pollard, S.M.; Krijgsveld, J. Proteome and Secretome Characterization of Glioblastoma-Derived Neural Stem Cells. Stem Cells 2017, 35, 967–980. [Google Scholar] [CrossRef]
- Tian, Q.; Sangar, V.; Price, N.D. Emerging Proteomic Technologies Provide Enormous and Underutilized Potential for Brain Cancer Research. Mol. Cell. Proteom. 2016, 15, 362–367. [Google Scholar] [CrossRef]
- Wang, L.-B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528.e20. [Google Scholar] [CrossRef]
- Karsy, M.; Neil, J.A.; Guan, J.; Mahan, M.A.; Colman, H.; Jensen, R.L. A practical review of prognostic correlations of molecular biomarkers in glioblastoma. Neurosurg. Focus 2015, 38, E4. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Weller, M.; Bent, M.V.D.; Sanson, M.; Weiler, M.; Von Deimling, A.; Plass, C.; Hegi, M.; Platten, M.; Reifenberger, G. MGMT testing-the challenges for biomarker-based glioma treatment. Nat. Rev. Neurol. 2014, 10, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Aldape, K.D. The Evolving Role of Molecular Markers in the Diagnosis and Management of Diffuse Glioma. Clin. Cancer Res. 2014, 20, 5601–5611. [Google Scholar] [CrossRef]
- Sreekanthreddy, P.; Srinivasan, H.; Kumar, D.M.; Nijaguna, M.B.; Sridevi, S.; Vrinda, M.; Arivazhagan, A.; Balasubramaniam, A.; Hegde, A.S.; Chandramouli, B.A.; et al. Identification of Potential Serum Biomarkers of Glioblastoma: Serum Osteopontin Levels Correlate with Poor Prognosis. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.; Ayers, D.; Bertino, J.; Bock, C.; Bock, A.; Brody, E.N.; Carter, J.; Dalby, A.B.; Eaton, B.E.; Fitzwater, T.; et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 2010, 5, e15004. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.; Walker, J.J.; Wilcox, S.K.; Williams, S. Advances in human proteomics at high scale with the SOMAscan proteomics platform. New Biotechnol. 2012, 29, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Mehan, M.R.; Ayers, D.; Thirstrup, D.; Xiong, W.; Ostroff, R.M.; Brody, E.N.; Walker, J.J.; Gold, L.; Jarvis, T.C.; Janjic, N.; et al. Protein Signature of Lung Cancer Tissues. PLoS ONE 2012, 7, e35157. [Google Scholar] [CrossRef] [PubMed]
- Graumann, J.; Finkernagel, F.; Reinartz, S.; Stief, T.; Brödje, D.; Renz, H.; Jansen, J.M.; Wagner, U.; Worzfeld, T.; Von Strandmann, E.P.; et al. Multi-platform Affinity Proteomics Identify Proteins Linked to Metastasis and Immune Suppression in Ovarian Cancer Plasma. Front. Oncol. 2019, 9, 1150. [Google Scholar] [CrossRef] [PubMed]
- Mysona, D.; Pyrzak, A.; Purohit, S.; Zhi, W.; Sharma, A.; Tran, L.; Tran, P.; Bai, S.; Rungruang, B.; Ghamande, S.; et al. A combined score of clinical factors and serum proteins can predict time to recurrence in high grade serous ovarian cancer. Gynecol. Oncol. 2019, 152, 574–580. [Google Scholar] [CrossRef]
- Tsim, S.; Kelly, C.; Alexander, L.; McCormick, C.; Thomson, F.; Woodward, R.; Foster, J.E.; Stobo, D.B.; Paul, J.; Maskell, N.A.; et al. Diagnostic and Prognostic Biomarkers in the Rational Assessment of Mesothelioma (DIAPHRAGM) study: Protocol of a prospective, multicentre, observational study. BMJ Open 2016, 6, e013324. [Google Scholar] [CrossRef]
- Ostroff, R.M.; Mehan, M.R.; Stewart, A.; Ayers, D.; Brody, E.N.; Williams, S.A.; Levin, S.; Black, B.; Harbut, M.; Carbone, M.; et al. Early Detection of Malignant Pleural Mesothelioma in Asbestos-Exposed Individuals with a Noninvasive Proteomics-Based Surveillance Tool. PLoS ONE 2012, 7, e46091. [Google Scholar] [CrossRef]
- Qiao, Z.; Pan, X.; Parlayan, C.; Ojima, H.; Kondo, T. Proteomic study of hepatocellular carcinoma using a novel modified aptamer-based array (SOMAscan) platform. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 434–443. [Google Scholar] [CrossRef]
- Brody, E.; Gold, L.; Mehan, M.; Ostroff, R.; Rohloff, J.; Walker, J.; Zichi, D. Life’s simple measures: Unlocking the proteome. J. Mol. Biol. 2012, 422, 595–606. [Google Scholar] [CrossRef]
- Gold, L.; Janjic, N.; Jarvis, T.; Schneider, D.; Walker, J.J.; Wilcox, S.K.; Zichi, D. Aptamers and the RNA World, Past and Present. Cold Spring Harb. Perspect. Biol. 2010, 4, a003582. [Google Scholar] [CrossRef]
- Cao, K.-A.L.; Boitard, S.; Besse, P. Sparse PLS discriminant analysis: Biologically relevant feature selection and graphical displays for multiclass problems. BMC Bioinform. 2011, 12, 253. [Google Scholar]
- Minniti, G.; Scaringi, C.; Arcella, A.; Lanzetta, G.; Di Stefano, D.; Scarpino, S.; Bozzao, A.; Pace, A.; Villani, V.; Salvati, M.; et al. IDH1 mutation and MGMT methylation status predict survival in patients with anaplastic astrocytoma treated with temozolomide-based chemoradiotherapy. J. Neuro-Oncol. 2014, 118, 377–383. [Google Scholar] [CrossRef]
- Christians, A.; Adel-Horowski, A.; Banan, R.; Lehmann, U.; Bartels, S.; Behling, F.; Barrantes-Freer, A.; Stadelmann, C.; Rohde, V.; Stockhammer, F.; et al. The prognostic role of IDH mutations in homogeneously treated patients with anaplastic astrocytomas and glioblastomas. Acta Neuropathol. Commun. 2019, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Wu, B.; Fu, Z.; Feng, F.; Qiao, E.; Li, Q.; Sun, C.; Ge, M. Prognostic role of IDH mutations in gliomas: A meta-analysis of 55 observational studies. Oncotarget 2015, 6, 17354–17365. [Google Scholar] [CrossRef] [PubMed]
- Gadji, M.; Fortin, D.; Tsanaclis, A.-M.; Drouin, R. Is the 1p/19q deletion a diagnostic marker of oligodendrogliomas? Cancer Genet. Cytogenet. 2009, 194, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Civita, P.; Franceschi, S.; Aretini, P.; Ortenzi, V.; Menicagli, M.; Lessi, F.; Pasqualetti, F.; Naccarato, A.G.; Mazzanti, C.M. Laser Capture Microdissection and RNA-Seq Analysis: High Sensitivity Approaches to Explain Histopathological Heterogeneity in Human Glioblastoma FFPE Archived Tissues. Front. Oncol. 2019, 9, 482. [Google Scholar] [CrossRef] [PubMed]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.K.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef]
- Schäfer, N.; Gielen, G.H.; Rauschenbach, L.; Kebir, S.; Till, A.; Reinartz, R.; Simon, M.; Niehusmann, P.; Kleinschnitz, C.; Herrlinger, U.; et al. Longitudinal heterogeneity in glioblastoma: Moving targets in recurrent versus primary tumors. J. Transl. Med. 2019, 17, 96. [Google Scholar] [CrossRef]
- Shi, L.; Westwood, S.; Baird, A.L.; Winchester, L.; Dobricic, V.; Kilpert, F.; Hong, S.; Franke, A.; Hye, A.; Ashton, N.J.; et al. Discovery and validation of plasma proteomic biomarkers relating to brain amyloid burden by SOMAscan assay. Alzheimer’s Dement. 2019, 15, 1478–1488. [Google Scholar] [CrossRef]
- Muramatsu, T. Structure and function of midkine as the basis of its pharmacological effects. Br. J. Pharmacol. 2014, 171, 814–826. [Google Scholar] [CrossRef]
- Jono, H.; Ando, Y. Midkine: A Novel Prognostic Biomarker for Cancer. Cancers 2010, 2, 624–641. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Ishihara, K.; Shinozawa, T.; Yamaguchi, H.; Asano, Y.; Saito, M.; Kato, M.; Terada, T.; Awaya, A.; Hirano, A.; et al. Monoclonal antibody to human midkine reveals increased midkine expression in human brain tumors. J. Neuropathol. Exp. Neurol. 1999, 58, 430–441. [Google Scholar] [CrossRef][Green Version]
- Mishima, K.; Asai, A.; Kadomatsu, K.; Ino, Y.; Nomura, K.; Narita, Y.; Muramatsu, T.; Kirino, T. Increased expression of midkine during the progression of human astrocytomas. Neurosci. Lett. 1997, 233, 29–32. [Google Scholar] [CrossRef]
- Luo, J.; Wang, X.; Xia, Z.; Yang, L.; Ding, Z.; Chen, S.; Lai, B.; Zhang, N. Transcriptional factor specificity protein 1 (SP1) promotes the proliferation of glioma cells by up-regulating midkine (MDK). Mol. Biol. Cell 2015, 26, 430–439. [Google Scholar] [CrossRef]
- Kishida, S.; Mu, P.; Miyakawa, S.; Fujiwara, M.; Abe, T.; Sakamoto, K.; Onishi, A.; Nakamura, Y.; Kadomatsu, K. Midkine Promotes Neuroblastoma through Notch2 Signaling. Cancer Res. 2013, 73, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Herradon, G.; Ramos-Alvarez, M.P.; Gramage, E. Connecting Metainflammation and Neuroinflammation Through the PTN-MK-RPTPbeta/zeta Axis: Relevance in Therapeutic Development. Front. Pharm. 2019, 10, 377. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Takeuchi, H.; Sonobe, Y.; Jin, S.; Mizuno, T.; Miyakawa, S.; Fujiwara, M.; Nakamura, Y.; Kato, T.; Muramatsu, H.; et al. Inhibition of midkine alleviates experimental autoimmune encephalomyelitis through the expansion of regulatory T cell population. Proc. Natl. Acad. Sci. USA 2008, 105, 3915–3920. [Google Scholar] [CrossRef]
- Hao, H.; Maeda, Y.; Fukazawa, T.; Yamatsuji, T.; Takaoka, M.; Bao, X.H.; Matsuoka, J.; Okui, T.; Shimo, T.; Takigawa, N.; et al. Inhibition of the growth factor MDK/midkine by a novel small molecule compound to treat non-small cell lung cancer. PLoS ONE 2013, 8, e71093. [Google Scholar] [CrossRef]
- Huang, H.-L.; Shen, J.-F.; Min, L.-S.; Ping, J.-L.; Lu, Y.-L.; Dai, L.-C. Inhibitory effect of midkine-binding peptide on tumor proliferation and migration. Int. J. Clin. Exp. Pathol. 2015, 8, 5387–5394. [Google Scholar]
- Ma, J.; Lang, B.; Wang, X.; Wang, L.; Dong, Y.; Hu, H. Co-expression of midkine and pleiotrophin predicts poor survival in human glioma. J. Clin. Neurosci. 2014, 21, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Toledano-Katchalski, H.; Nir, R.; Volohonsky, G.; Volk, T. Post-transcriptional repression of the Drosophila midkine and pleiotrophin homolog miple by HOW is essential for correct mesoderm spreading. Development 2007, 134, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Shibata, Y.; Urano, T.; Murohara, T.; Muramatsu, T.; Kadomatsu, K. Proteasomal Degradation of the Nuclear Targeting Growth Factor Midkine. J. Biol. Chem. 2004, 279, 17785–17791. [Google Scholar] [CrossRef]
- Zhang, D.; Ding, L.; Li, Y.; Ren, J.; Shi, G.; Wang, Y.; Zhao, S.; Ni, Y.; Hou, Y. Midkine derived from cancer-associated fibroblasts promotes cisplatin-resistance via up-regulation of the expression of lncRNA ANRIL in tumour cells. Sci. Rep. 2017, 7, 16231. [Google Scholar] [CrossRef]
- Rawnaq, T.; Kunkel, M.; Simon, R.; Zander, H.; Bachmann, K.; Sauter, G.; Izbicki, J.R.; Kaifi, J.T. Serum midkine correlates with tumor progression and imatinib response in gastrointestinal stromal tumors. Ann. Surg. Oncol. 2011, 18, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Hu, C.; Yang, Z.; Zhang, X.; Zhao, L.; Xiong, J.; Ma, J.; Chen, L.; Qian, H.; Luo, X.; et al. Midkine promotes hepatocellular carcinoma metastasis by elevating anoikis resistance of circulating tumor cells. Oncotarget 2017, 8, 32523–32535. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-Y.; Mao, X.-Y.; Song, Y.-X.; Zhao, F.; Wang, Z.-N.; Zhang, W.-X.; Xu, H.-M.; Jin, F. Midkine confers Adriamycin resistance in human gastric cancer cells. Tumor Biol. 2012, 33, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Zeller, C.; Dai, W.; Steele, N.L.; Siddiq, A.; Walley, A.; Wilhelm-Benartzi, C.; Rizzo, S.; Van Der Zee, A.; Plumb, J.A.; Brown, R. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene 2012, 31, 4567–4576. [Google Scholar] [CrossRef]
- Kadomatsu, K.; Muramats, T. Midkine and pleiotrophin in neural development and cancer. Cancer Lett. 2004, 204, 127–143. [Google Scholar] [CrossRef]
- Muramatsu, H.; Zou, P.; Suzuki, H.; Oda, Y.; Chen, G.Y.; Sakaguchi, N.; Sakuma, S.; Maeda, N.; Noda, M.; Takada, Y.; et al. alpha4beta1- and alpha6beta1-integrins are functional receptors for midkine, a heparin-binding growth factor. J. Cell Sci. 2004, 117, 5405–5415. [Google Scholar] [CrossRef]
- Mitsiadis, T.A.; Salmivirta, M.; Muramatsu, T.; Muramatsu, H.; Rauvala, H.; Lehtonen, E.; Jalkanen, M.; Thesleff, I. Expression of the heparin-binding cytokines, midkine (MK) and HB-GAM (pleiotrophin) is associated with epithelial-mesenchymal interactions during fetal development and organogenesis. Development 1995, 121, 37–51. [Google Scholar] [CrossRef]
- Kojima, T.; Katsumi, A.; Yamazaki, T.; Muramatsu, T.; Nagasaka, T.; Ohsumi, K.; Saito, H. Human ryudocan from endothelium-like cells binds basic fibroblast growth factor, midkine, and tissue factor pathway inhibitor. J. Biol. Chem. 1996, 271, 5914–5920. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Kadomatsu, K.; Okamoto, T.; Ichihara-Tanaka, K.; Kojima, T.; Saito, H.; Tomoda, Y.; Muramatsu, T. Expression of syndecan-1 and -3 during embryogenesis of the central nervous system in relation to binding with midkine. J. Biochem. 1997, 121, 197–205. [Google Scholar] [PubMed]
- XGuo, X.; Pan, Y.; Xiong, M.; Sanapala, S.; Anastasaki, C.; Cobb, O.; Dahiya, S.; Gutmann, D.H. Midkine activation of CD8+ T cells establishes a neuron–immune–cancer axis responsible for low-grade glioma growth. Nat. Commun. 2020, 11, 1–15. [Google Scholar]
- Hemmer, W.; Wallimann, T. Functional Aspects of Creatine Kinase in Brain. Dev. Neuroscience 1993, 15, 249–260. [Google Scholar]
- Róna, E.; Nagy, A.; Wollemann, M.; Slowik, F. Localization of various isoenzymes in different cell fractions of brain tumours. Neuropatol. Pol. 1972, 10, 207–220. [Google Scholar]
- Tsung, S.H. Creatine kinase activity and isoenzyme pattern in various normal tissues and neoplasms. Clin. Chem. 1983, 29, 2040–2043. [Google Scholar] [CrossRef] [PubMed]
- Wallimann, T.; Tokarska-Schlattner, M.; Schlattner, U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 2011, 40, 1271–1296. [Google Scholar] [CrossRef]
- Andres, R.H.; Wallimann, T.; Widmer, H.R. Creatine supplementation improves neural progenitor cell survival in Huntington’s disease. Brain Circ. 2016, 2, 133–137. [Google Scholar]
- Gramage, E.; Herradon, G.; Martin, Y.B.; Vicente-Rodriguez, M.; Rojo, L.; Gnekow, H.; Barbero, A.; Perez-Garcia, C. Differential phosphoproteome of the striatum from pleiotrophin knockout and midkine knockout mice treated with amphetamine: Correlations with amphetamine-induced neurotoxicity. Toxicology 2013, 306, 147–156. [Google Scholar] [CrossRef]
- Mäenpää, A.; Junnikkala, S.; Hakulinen, J.; Timonen, T.; Meri, S. Expression of complement membrane regulators membrane cofactor protein (CD46), decay accelerating factor (CD55), and protectin (CD59) in human malignant gliomas. Am. J. Pathol. 1996, 148, 1139–1152. [Google Scholar] [PubMed]
- Marx, S.; Wilken, F.; Wagner, I.; Marx, M.; Troschke-Meurer, S.; Zumpe, M.; Bien-Moeller, S.; Weidemeier, M.; Baldauf, J.; Fleck, S.K.; et al. GD2 targeting by dinutuximab beta is a promising immunotherapeutic approach against malignant glioma. J. Neuro Oncol. 2020, 147, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Sonmez, H.A.; Kökoǧlu, E.; Süer, S.; Özyurt, E. Fibronectin and sialic acid levels in human meningiomas and gliomas. Cancer Lett. 1995, 90, 119–122. [Google Scholar] [CrossRef]
- Kochi, N.; Tani, E.; Morimura, T.; Itagaki, T. Immunohistochemical study of fibronectin in human glioma and meningioma. Acta Neuropathol. 1983, 59, 119–126. [Google Scholar] [CrossRef]
- Paetau, A. Glial fibrillary acidic protein, vimentin and fibronectin in primary cultures of human glioma and fetal brain. Acta Neuropathol. 1988, 75, 448–455. [Google Scholar] [CrossRef]
- Yang, C.H.; Wang, Y.; Sims, M.; Cai, C.; Pfeffer, L.M. MicroRNA-1 suppresses glioblastoma in preclinical models by targeting fibronectin. Cancer Lett. 2019, 465, 59–67. [Google Scholar] [CrossRef]
- Sengupta, S.; Nandi, S.; Hindi, E.S.; Wainwright, D.A.; Han, Y.; Lesniak, M.S. Short hairpin RNA-mediated fibronectin knockdown delays tumor growth in a mouse glioma model. Neoplasia 2010, 12, 837–847. [Google Scholar] [CrossRef]
- Wang, J.P.; Hielscher, A. Fibronectin: How Its Aberrant Expression in Tumors May Improve Therapeutic Targeting. J. Cancer 2017, 8, 674–682. [Google Scholar] [CrossRef]
- Daniels, D.A.; Chen, H.; Hicke, B.J.; Swiderek, K.M.; Gold, L. A tenascin-C aptamer identified by tumor cell SELEX: Systematic evolution of ligands by exponential enrichment. Proc. Natl. Acad. Sci. USA 2003, 100, 15416–15421. [Google Scholar] [CrossRef]
- Chung, D.; Keles, S. Sparse Partial Least Squares Classification for High Dimensional Data. Stat. Appl. Genet. Mol. Biol. 2010, 9, 1–30. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Le Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. | ||||||
| No. | Sex | Age at Diagnosis | IDH1 Status | Survival (Months) | Proteomic Cluster | |
| 1 | f | 57 | ND | 24.1 | 1 | |
| 24 | f | 45 | ND | 8.9 | ||
| 48 | f | 76 | ND | 20.7 | ||
| 223 | f | 83 | negative for IDH1 (R132H) | 9.1 | ||
| 300 | f | 34 | negative for IDH1 (R132H) | 18.6 | ||
| Median survival | 16.3 | |||||
| 2 | f | 72 | ND | 18.5 | 2 | |
| 6 | f | 63 | ND | 0.4 | ||
| 8 | m | 78 | ND | 2.2 | ||
| 28 | f | 45 | ND | 29.1 | ||
| 41 | m | 72 | ND | 29.5 | ||
| 44 | m | 63 | ND | 58.4 | ||
| 69 | m | 49 | mutant IDH1 (R132H) | 67.6 | ||
| 368 | m | 51 | negative for IDH1 (R132H) | 11.5 | ||
| Median survival | 27.2 | |||||
| 7 | f | 34 | ND | 72.8 | 3 | |
| 12.1 | m | 59 | ND | 86.9 | ||
| 17 | m | 63 | ND | 2.8 | ||
| 18 | f | 55 | ND | 6.9 | ||
| 19 | f | 49 | ND | 19.3 | ||
| 20 | m | 65 | ND | 3.1 | ||
| 26 | m | 76 | ND | 7.9 | ||
| 29 | m | 59 | ND | 10.7 | ||
| 30 | m | 35 | ND | 9.2 | ||
| 35 | f | 51 | ND | 20.8 | ||
| 40 | m | 52 | ND | 30.9 | ||
| 46 | m | 36 | ND | 54.5 | ||
| 51 | f | 45 | ND | 9.7 | ||
| 53 | m | 63 | ND | 1 | ||
| 54 | f | 40 | ND | 26.1 | ||
| 55 | m | 25 | ND | 10.7 | ||
| 56 | m | 66 | ND | 7.9 | ||
| 58 | m | 68 | ND | 7.5 | ||
| 65 | f | 59 | ND | 19.4 | ||
| 66 | m | 53 | ND | 6.2 | ||
| 67 | f | 67 | ND | 3.7 | ||
| 77 | m | 75 | ND | 0.6 | ||
| 103 | m | 64 | ND | 36.2 | ||
| 108 | m | 55 | ND | 6.7 | ||
| 146 | f | 38 | negative for IDH1 (R132H) | 11.8 | ||
| 162 | m | 58 | negative for IDH1 (R132H) | 19.9 | ||
| 167 | f | 63 | negative for IDH1 (R132H) | 5 | ||
| 196 | m | 50 | negative for IDH1 (R132H) | 3.4 | ||
| 224 | f | 43 | negative for IDH1 (R132H) | 10.9 | ||
| 233 | m | 66 | negative for IDH1 (R132H) | 39.1 | ||
| 256 | m | 52 | mutated IDH1 (R132H) | 34.8 | ||
| 275 | m | 60 | negative for IDH1 (R132H) | 17.7 | ||
| 311 | m | 28 | mutated IDH1 (R132H) | 26.6 | ||
| 363 | m | 40 | negative for IDH1 (R132H) | 7 | ||
| Median survival | 18.8 | |||||
| 12 | m | 59 | ND | 86.9 | 4 | |
| 34 | m | 62 | ND | 1.8 | ||
| 49 | m | 75 | ND | 1.8 | ||
| 59 | m | 65 | ND | 8.5 | ||
| 109 recurrence of GBM54 | f | 41 | 26.1 | |||
| 220 | m | 58 | negative for IDH1 (R132H) | 14.5 | ||
| 228 | m | 83 | negative for IDH1 (R132H) | 0.3 | ||
| Median survival | 20 | |||||
| B. | ||||||
| No. | Sex | Age at Diagnosis | IDH1 Status | Survival (months) | Recurrence | |
| 13 | f | 47 | ND | 20.9 | ||
| 42 | f | 27 | ND | 17.4 | ||
| 60 | m | 51 | ND | 42 | ||
| 115 | f | 27 | negative for IDH1 (R132H) | 57.5 | ||
| 173 | m | 46 | negative for IDH1 (R132H) | 7.8 | ||
| 236 | m | 17 | negative for IDH1 (R132H) | 24.6 | ||
| 287 | f | 52 | mutated IDH1 (R132H) | 30.5 | ||
| 295 | m | 33 | negative for IDH1 (R132H) | 29.8 | ||
| 302 | m | 36 | mutated IDH1 (R132H) | 28.4 | ||
| 310 | m | 18 | ND | 24.6 | recurrence of AS-236 | |
| 337 | m | 65 | negative for IDH1 (R132H) | 17.5 | ||
| 355 | f | 31 | negative for IDH1 (R132H) | 57.5 | recurrence of AS-115 | |
| 382 | m | 32 | mutated IDH1 (R132H) | 8.1 | ||
| Median survival | 28.2 | |||||
| C. | ||||||
| No. | Sex | Age at Diagnosis | IDH1 Status | Survival (Months) | WHO Grade | Recurrence |
| 22 | f | 36 | ND | 83 | 2 | |
| 32 | m | 35 | ND | 80.7 | 3 | |
| 37 | f | 63 | ND | 2.4 | 2 | |
| 62 | m | 27 | ND | 69.7 | 2 | |
| 83 | m | 39 | ND | 64.5 | 2 | |
| 134 | m | 32 | mutated IDH1 (R132H) | 102.3 | 3 | Yes |
| 152 | m | 28 | negative for IDH1 (R132H) | 69.7 | 3 | Yes |
| 158 | f | 41 | negative for IDH1 (R132H) | 51.2 | 2 | |
| 160 | m | 33 | mutated IDH1 (R132H) | 53.3 | 3 | |
| 172 | m | 55 | mutated IDH1 (R132H) | 18.9 | 3 | |
| 188 | f | 26 | negative for mutated IDH1 (R132H) | 46.4 | 2 | |
| 193 | m | 35 | negative for mutated IDH1 (R132H) | 45.2 | 3 | |
| 197 | f | 29 | negative for mutated IDH1 (R132H) | 45 | 2 | |
| 211 | m | 43 | mutated IDH1 (R132H) | 42.1 | 2 | |
| 218 | m | 31 | mutated IDH1 (R132H) | 41.4 | 2 | |
| 225 | m | 33 | mutated IDH1 (R132H) | 102.3 | 3 | Yes |
| 238 | m | 66 | mutated IDH1 (R132H) | 38.5 | 3 | |
| 242 | m | 23 | mutated IDH1 (R132H) | 67.3 | 2 | Yes |
| 325 | m | 30 | mutated IDH1 (R132H) | 23.7 | 2 | |
| 341 | m | 45 | mutated IDH1 (R132H) | 22.8 | 2 | |
| 372 | m | 28 | mutated IDH1 (R132H) | 10.6 | 3 | |
| Median survival | 51.5 | |||||
| Target Protein | Company and Catalog Number | Species | Dilution |
|---|---|---|---|
| CD59 | CST, #65055 | Rabbit monoclonal | 1:1000 |
| STAT1 | CST, #9176 | Mouse monoclonal | 1:1000 |
| STAT6 | ABCAM, ab32520 | Rabbit monoclonal | 1:1000 Western blot; 1:50 IHC |
| Creatinine Kinase, M-Type (CKM) | ABCAM, ab151465 | Rabbit polyclonal | 1:1000 |
| Fibronectin 1 (FN1) | ABCAM, ab2413 | Rabbit polyclonal | 1:1000 Western blot; 1:200 IF |
| Midkine (MDK) | ABCAM, ab52637 | Rabbit monoclonal | 1:1000 Western blot |
| Midkine (MDK) | ABCAM, ab170820 | Rabbit polyclonal | 1:50 IHC |
| β-actin | SCBT, sc47778 | Mouse monoclonal | 1:10,000 |
| Biotinylated Goat anti Rabbit | Vector Laboratories, BA-1000 | 1:200 | |
| HRP conjugated Goat anti-Mouse | CST, 7076 | 1:2000 | |
| Alexa Fluor 594 conjugated Goat anti Rabbit | Thermo Fisher, A11012 | 1:1000 | |
| HRP conjugated Goat anti-Rabbit | CST, 7074 | 1:2000 |
| Target | Forward | Reverse |
|---|---|---|
| MDK | 5′-CGCGGTCGCCAAAAAGAAAG-3′ | 5′-ACTTGCAGTCGGCTCCAAAC-3′ |
| PTP | 5′-GTGGAGACTGTGG GCTGGG-3′ | 5′-GCCTTCCTTTTTCTTCTTCTTAG-3′ |
| PTPRZ1 | 5′-GTGTCAGCGGAGGAGTTTCAG-3′ | 5′-CTGCTTCCCAAAACGACTAACAC-3′ |
| ALK1 | 5′-ACCGACTACAACCCCAACTAC-3′ | 5′-ACCCCAATGCAGCGAACAATG-3′ |
| ALK2 | 5′-CTTCATCCACCGAGACATTGCT-3′ | 5′-GGGCAGTTCTTGGGTGGGTC-3′ |
| NOTCH2 | 5′- CAGAAGATGTGGATGAATGTGC-3′ | 5′- GACTTTATCCACACACTGCCC -3′ |
| Nucleolin | 5′-GAAGGCACAGAACCGACTACG-3′ | 5′-CCTTTACTTTTCCCATCCTTGC-3′ |
| SDC3 | 5′-CGATGATGAACTGGATGACCTC-3′ | 5′-ATGGTAGTGGAGACGGTGGTG-3′ |
| SDC4 | 5′-CCAGACGATGAGGATGTAGTG-3′ | 5′-ACACATCCTCACTCTCTTCAAC-3′ |
| LRP6 | 5′-GAGAAGTGCCAAAGATAGAACG-3′ | 5′-TTCACGCAGACCCTCACCAG-3′ |
| LRP8 | 5′-CTACCCTGGCTACGAGATGG-3′ | 5′-CTCCTGCTCTTTCGGGTCAC-3′ |
| CSPG5 | 5′-TCAGTGTGCGACCTCTTCCC-3′ | 5′-GGGAGAAGTTATCATTGTGGAG-3′ |
| GAPDH | 5′-GTCTCCTCTGACTTCAACAGCG-3′ | 5′-ACCACCCTGTTGCTGTAGCCAA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thanasupawat, T.; Glogowska, A.; Pascoe, C.; Krishnan, S.N.; Munir, M.; Begum, F.; Beiko, J.; Krcek, J.; Del Bigio, M.R.; Pitz, M.; et al. Slow Off-Rate Modified Aptamer (SOMAmer) Proteomic Analysis of Patient-Derived Malignant Glioma Identifies Distinct Cellular Proteomes. Int. J. Mol. Sci. 2021, 22, 9566. https://doi.org/10.3390/ijms22179566
Thanasupawat T, Glogowska A, Pascoe C, Krishnan SN, Munir M, Begum F, Beiko J, Krcek J, Del Bigio MR, Pitz M, et al. Slow Off-Rate Modified Aptamer (SOMAmer) Proteomic Analysis of Patient-Derived Malignant Glioma Identifies Distinct Cellular Proteomes. International Journal of Molecular Sciences. 2021; 22(17):9566. https://doi.org/10.3390/ijms22179566
Chicago/Turabian StyleThanasupawat, Thatchawan, Aleksandra Glogowska, Christopher Pascoe, Sai Nivedita Krishnan, Maliha Munir, Farhana Begum, Jason Beiko, Jerry Krcek, Marc R. Del Bigio, Marshall Pitz, and et al. 2021. "Slow Off-Rate Modified Aptamer (SOMAmer) Proteomic Analysis of Patient-Derived Malignant Glioma Identifies Distinct Cellular Proteomes" International Journal of Molecular Sciences 22, no. 17: 9566. https://doi.org/10.3390/ijms22179566
APA StyleThanasupawat, T., Glogowska, A., Pascoe, C., Krishnan, S. N., Munir, M., Begum, F., Beiko, J., Krcek, J., Del Bigio, M. R., Pitz, M., Shen, Y., Spicer, V., Coombs, K. M., Wilkins, J., Hombach-Klonisch, S., & Klonisch, T. (2021). Slow Off-Rate Modified Aptamer (SOMAmer) Proteomic Analysis of Patient-Derived Malignant Glioma Identifies Distinct Cellular Proteomes. International Journal of Molecular Sciences, 22(17), 9566. https://doi.org/10.3390/ijms22179566