
Transcriptome and Resequencing Analyses Provide Insight into Differences in Organic Acid Accumulation in Two Pear Varieties

Abstract
:1. Introduction
2. Results
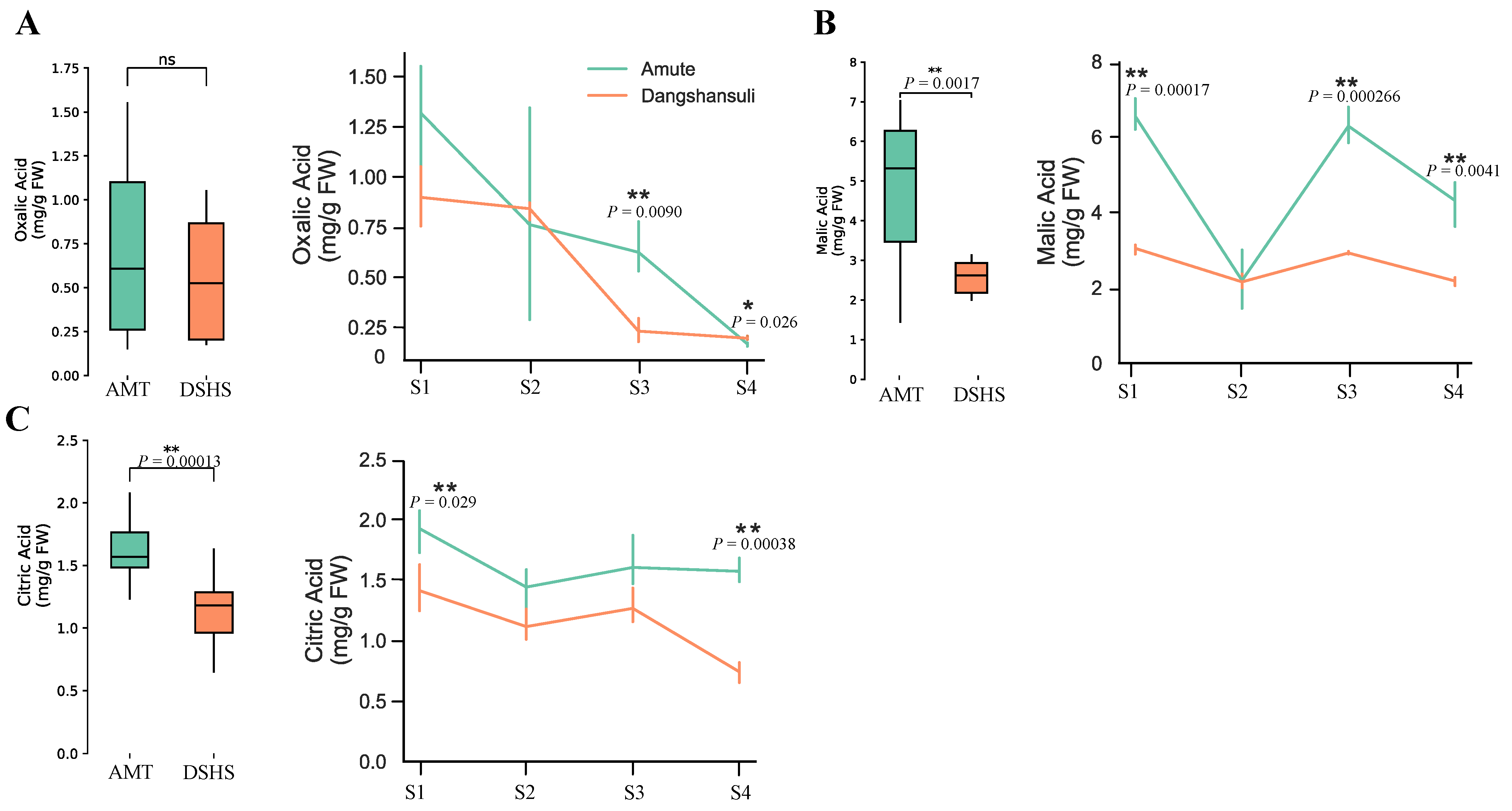
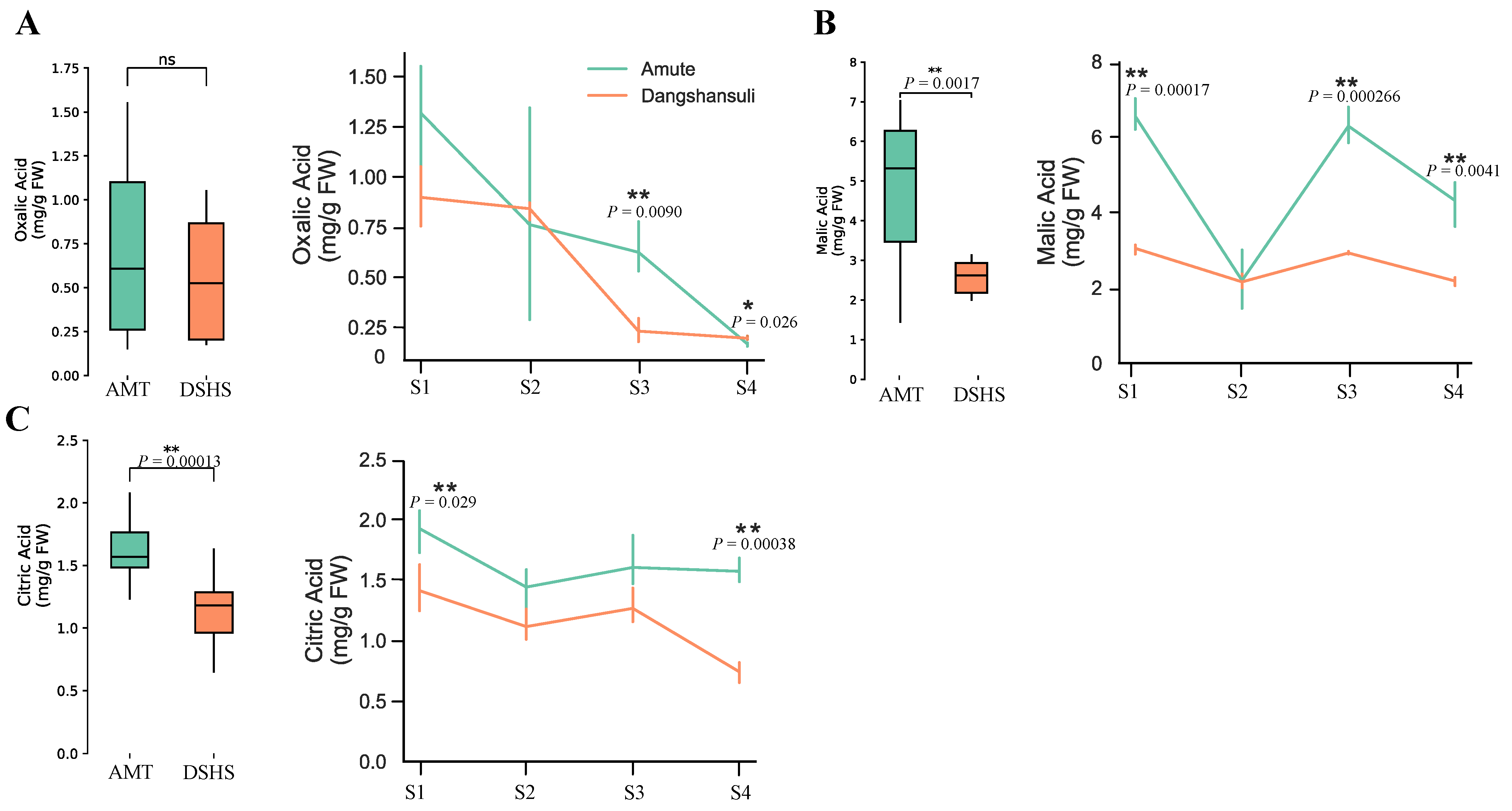
2.1. Quantitation of Organic Acid Contents in ‘Dangshansuli’ and ‘Amute’ Pear
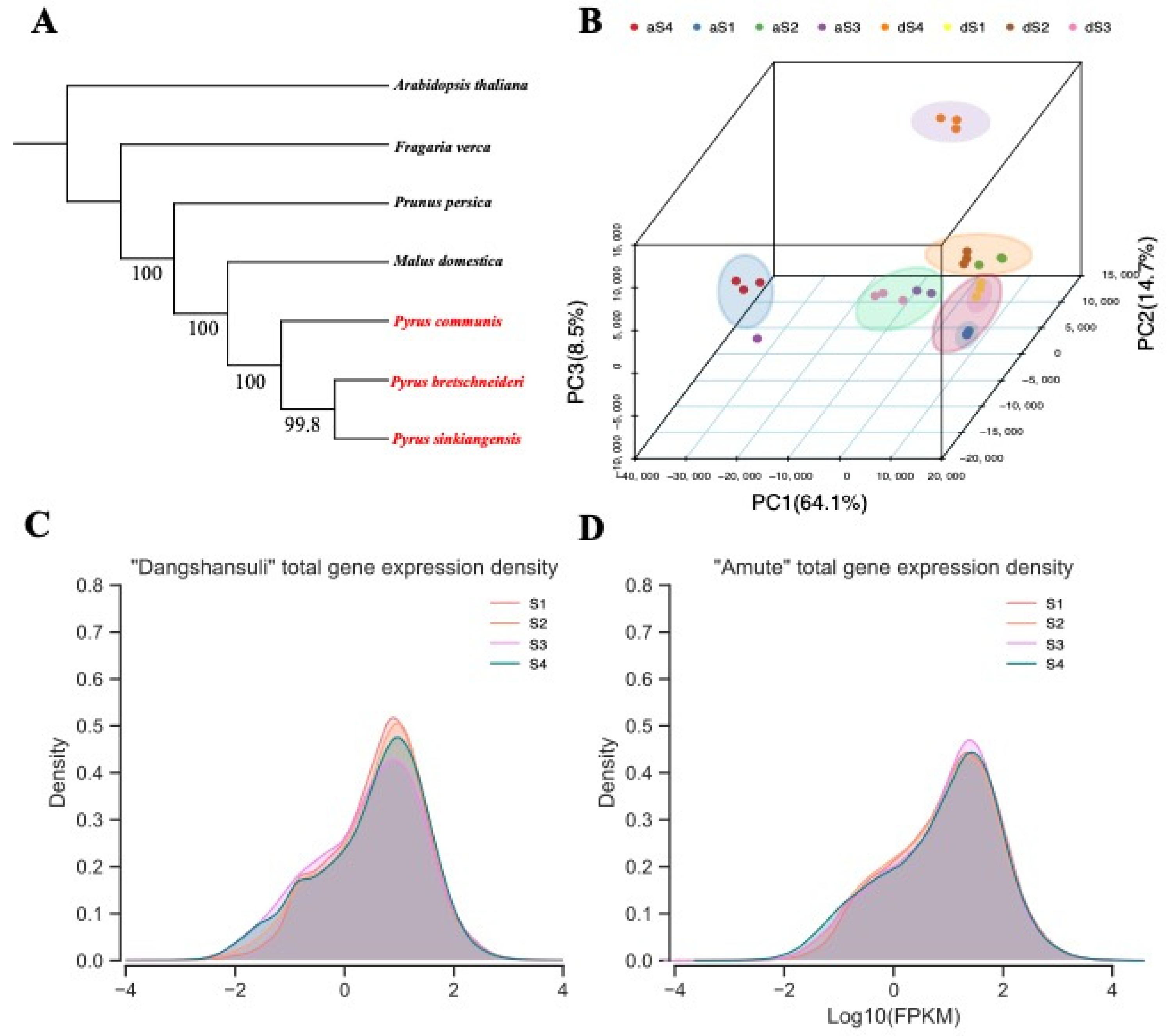
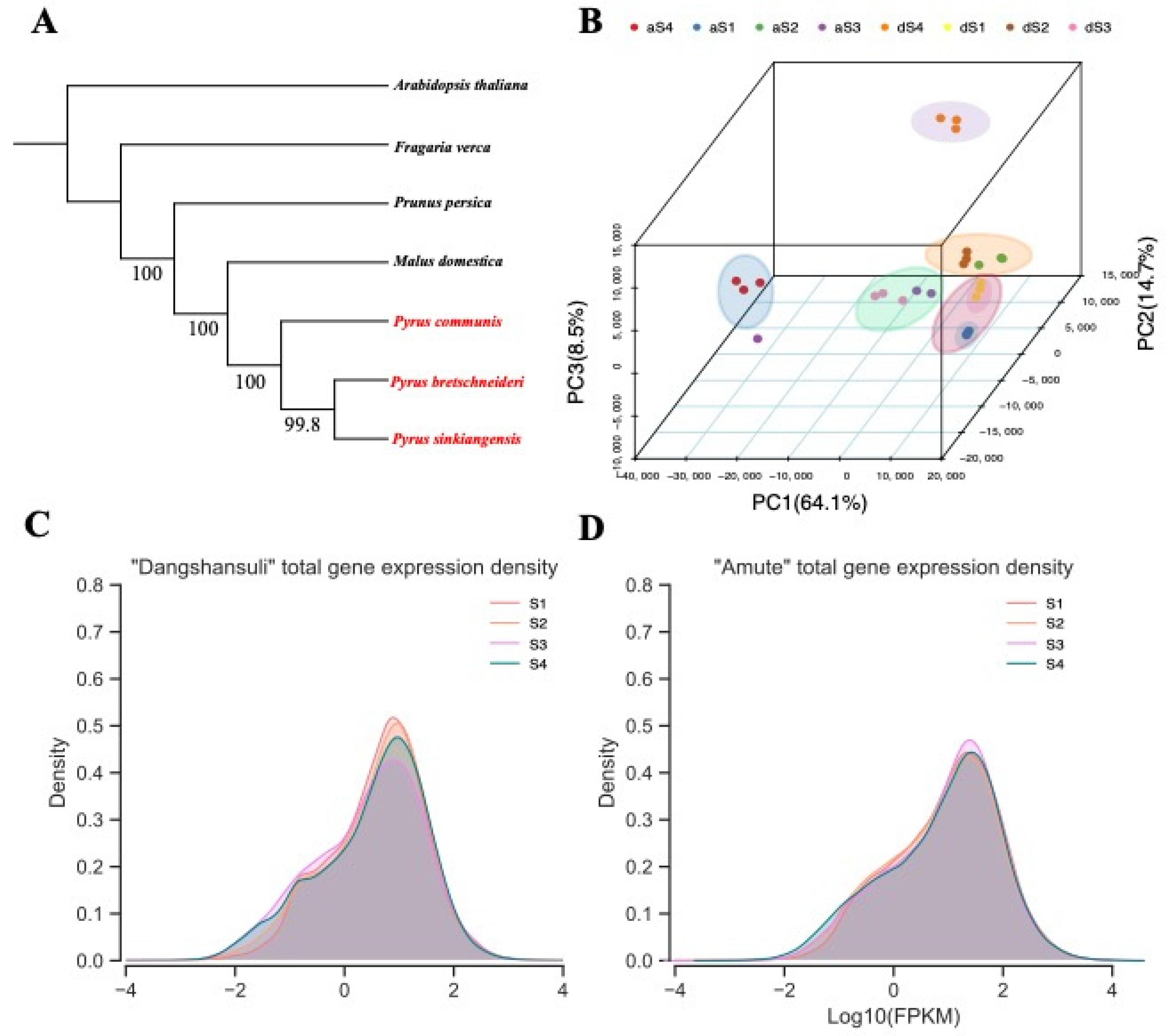
2.2. Phylogenetic Analysis and Expression Landscapes between Two Varieties
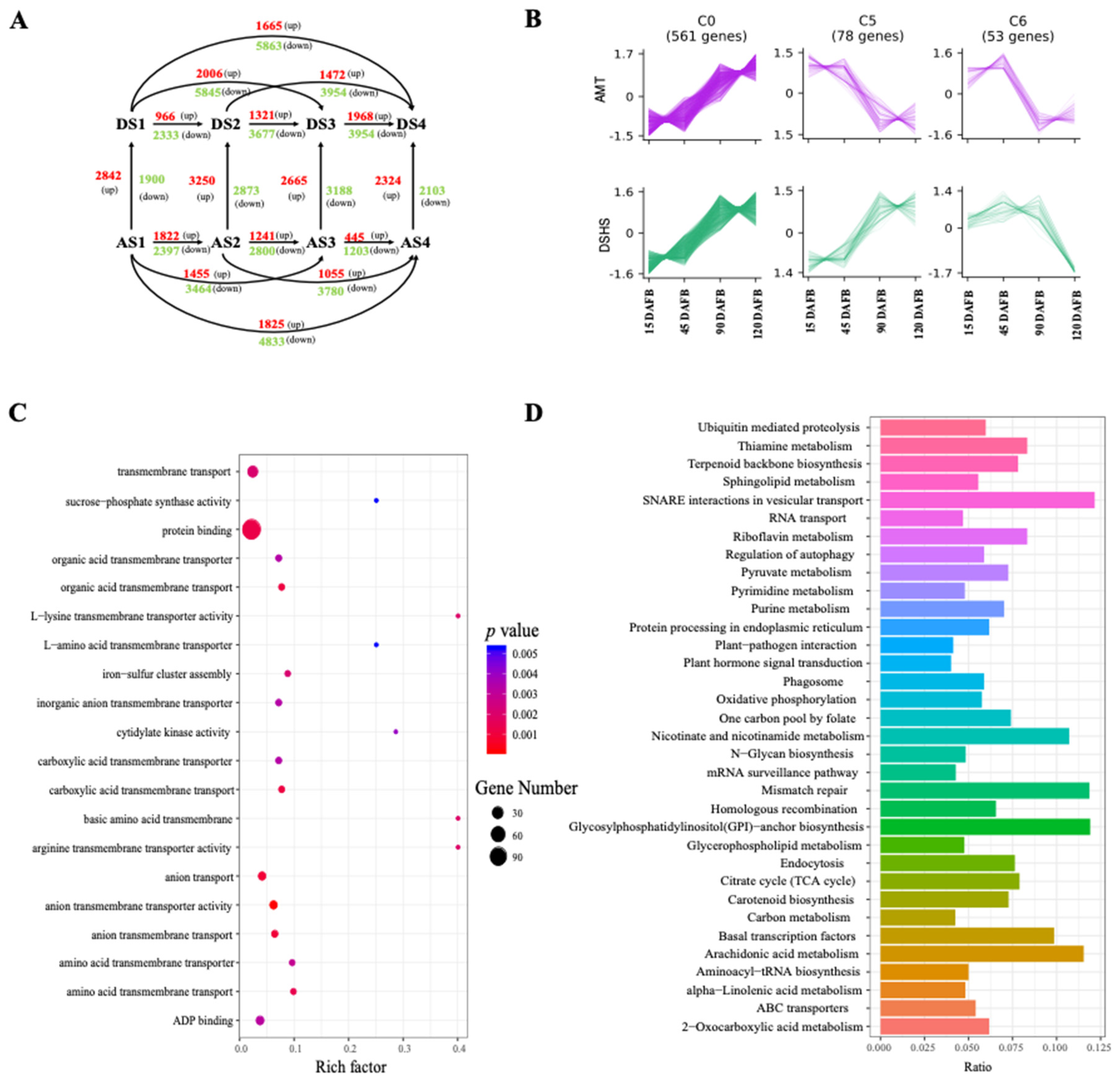
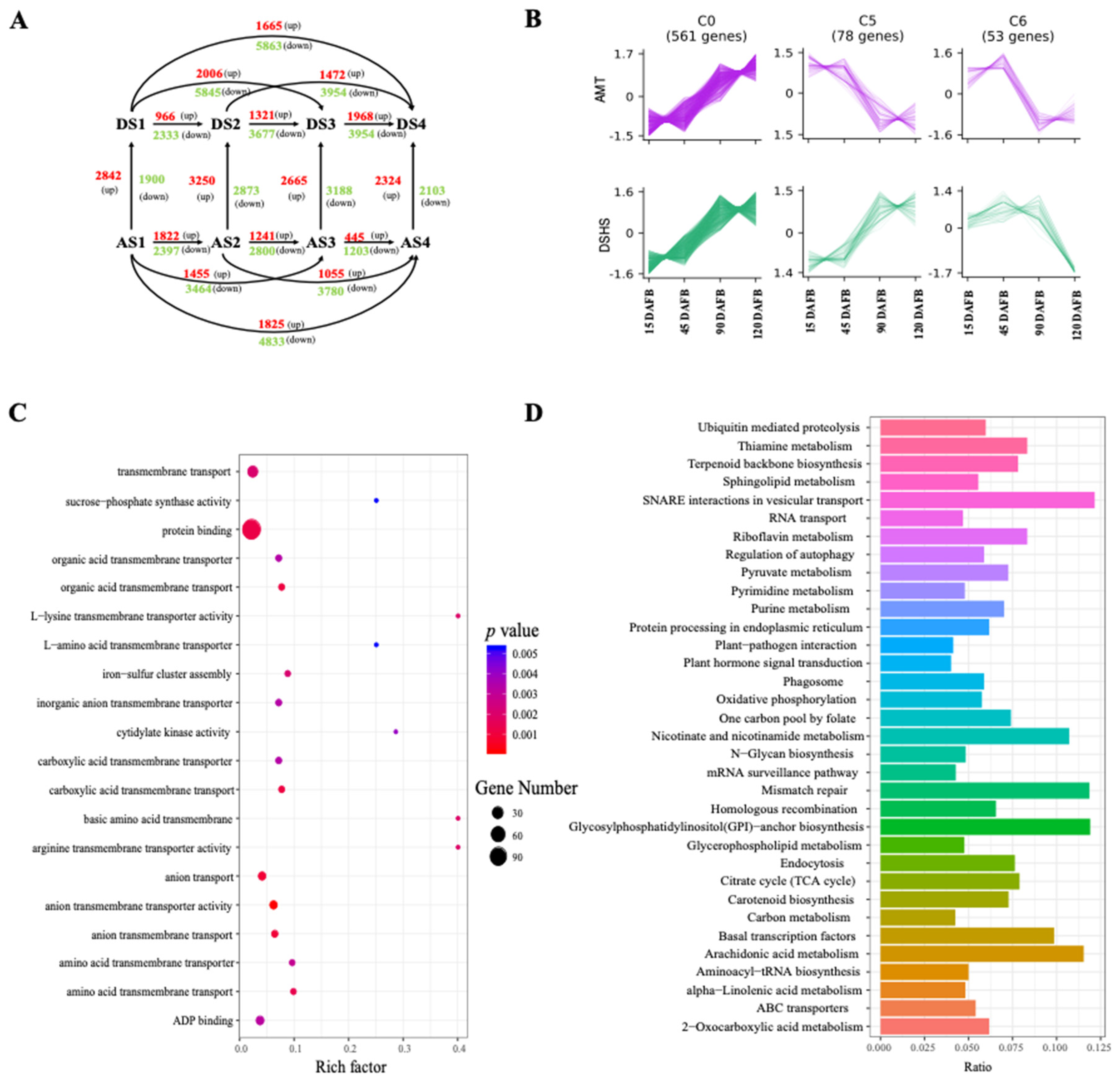
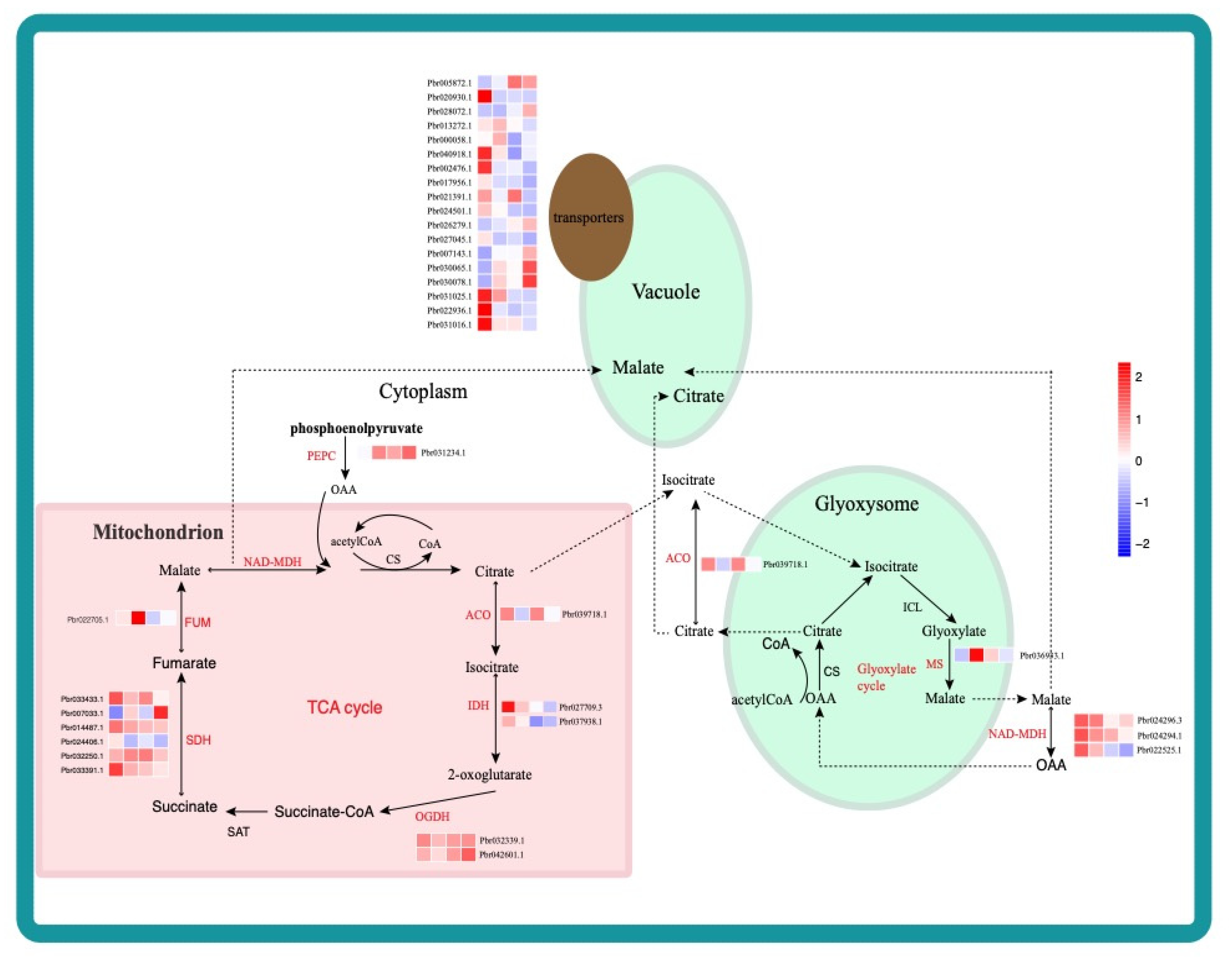
2.3. Identification and Functional Analysis of DEGs during Different Fruit Developmental Stages
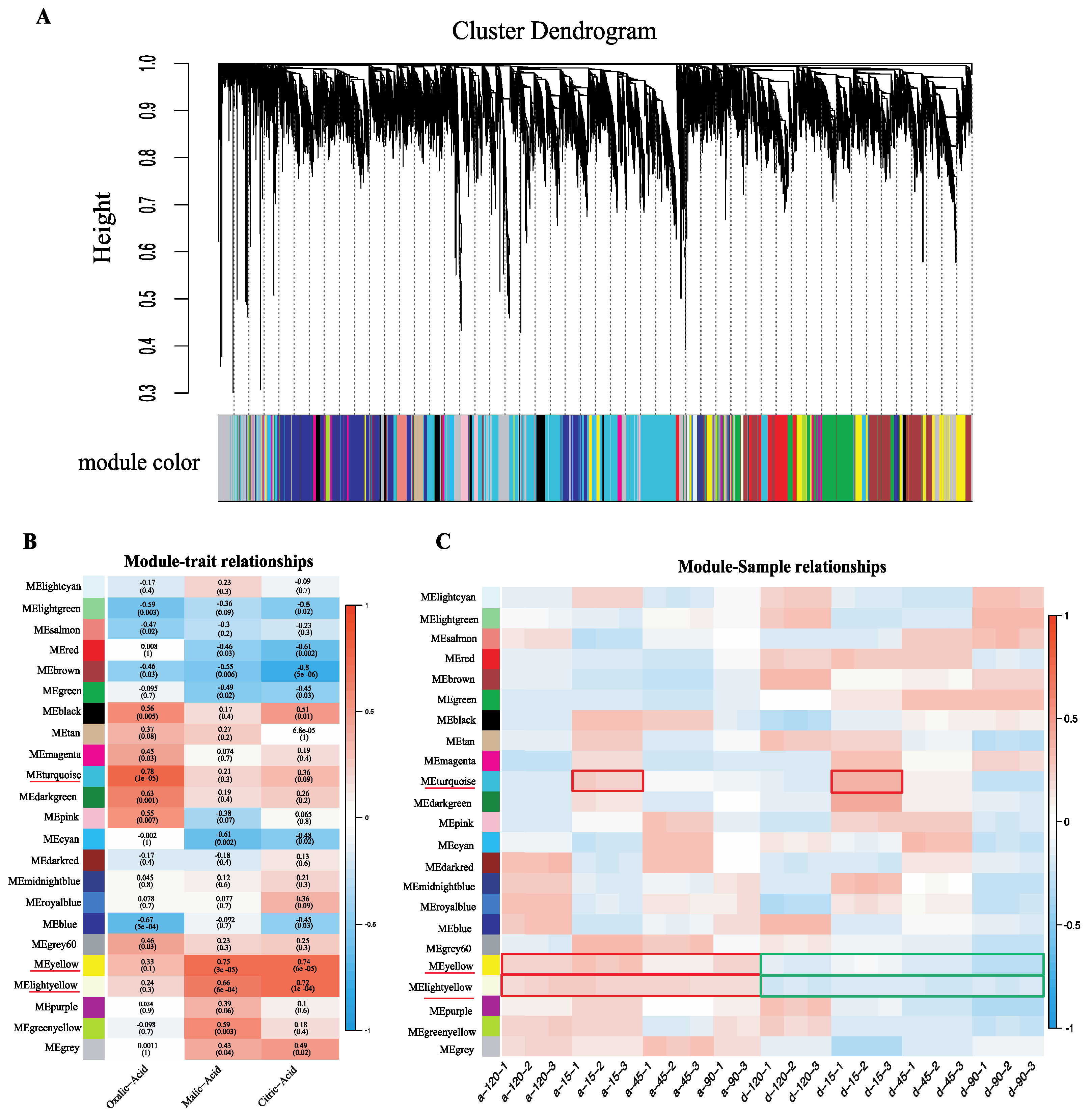
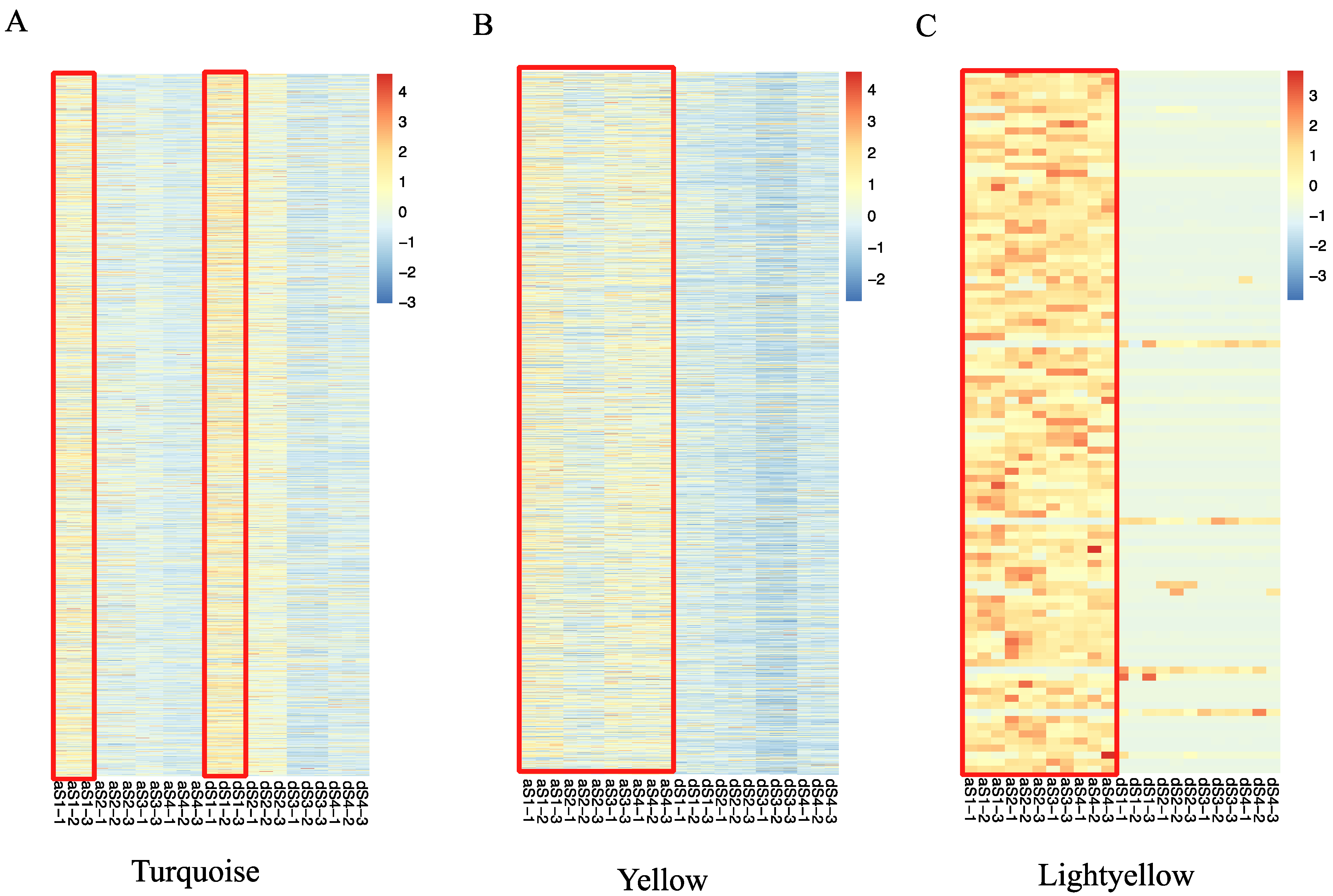
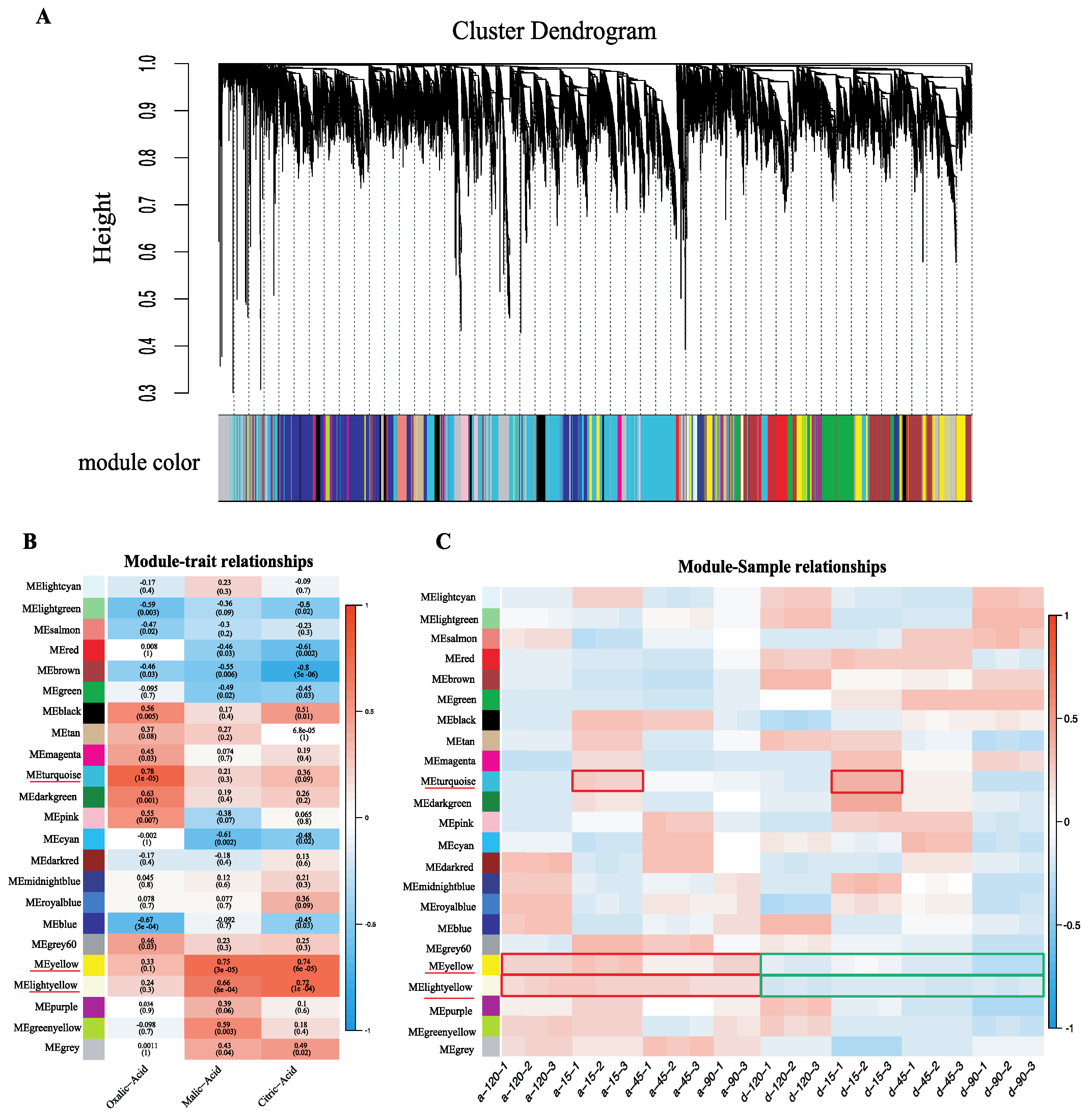
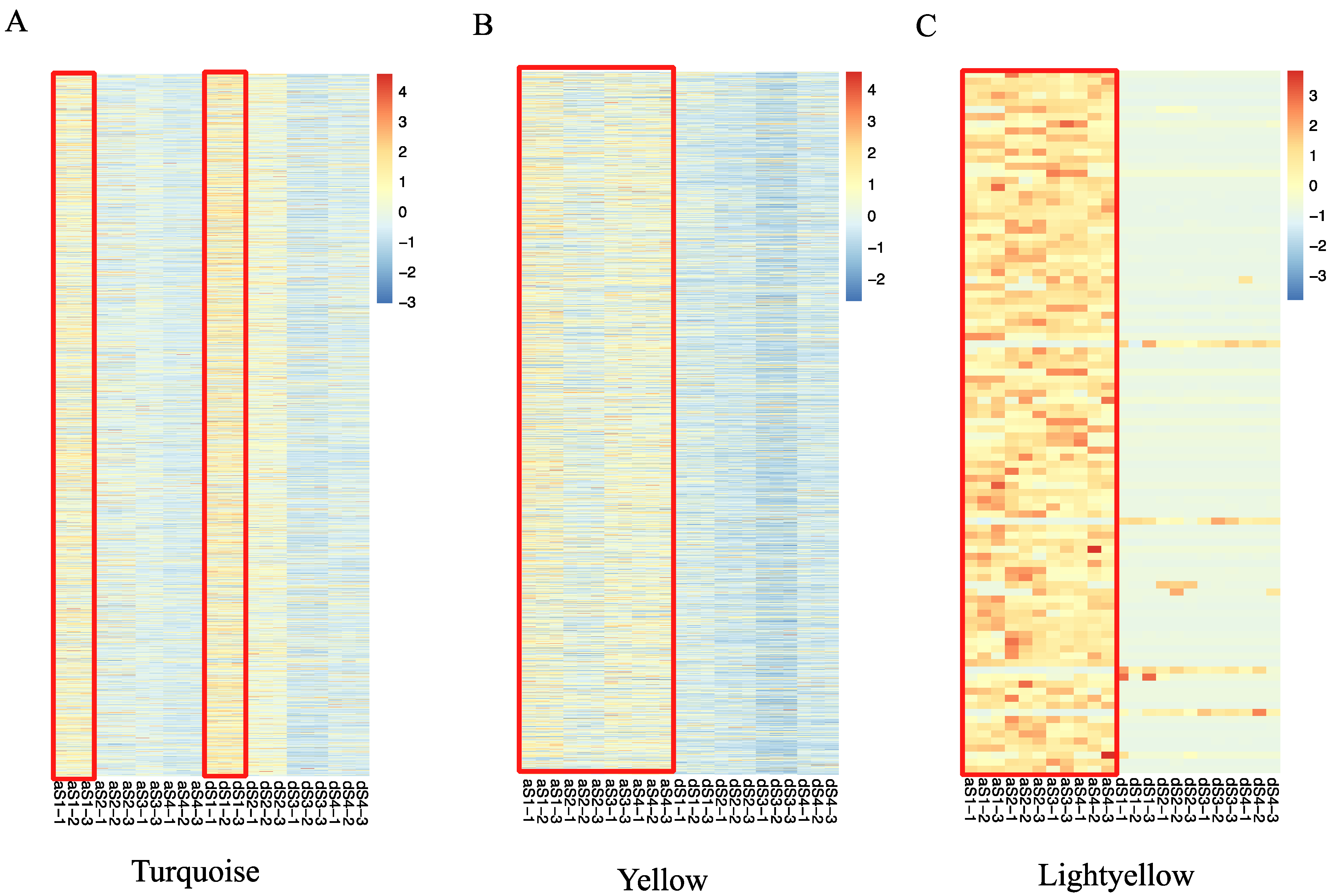
2.4. Weighted Gene Co-Expression Network Construction and Analysis
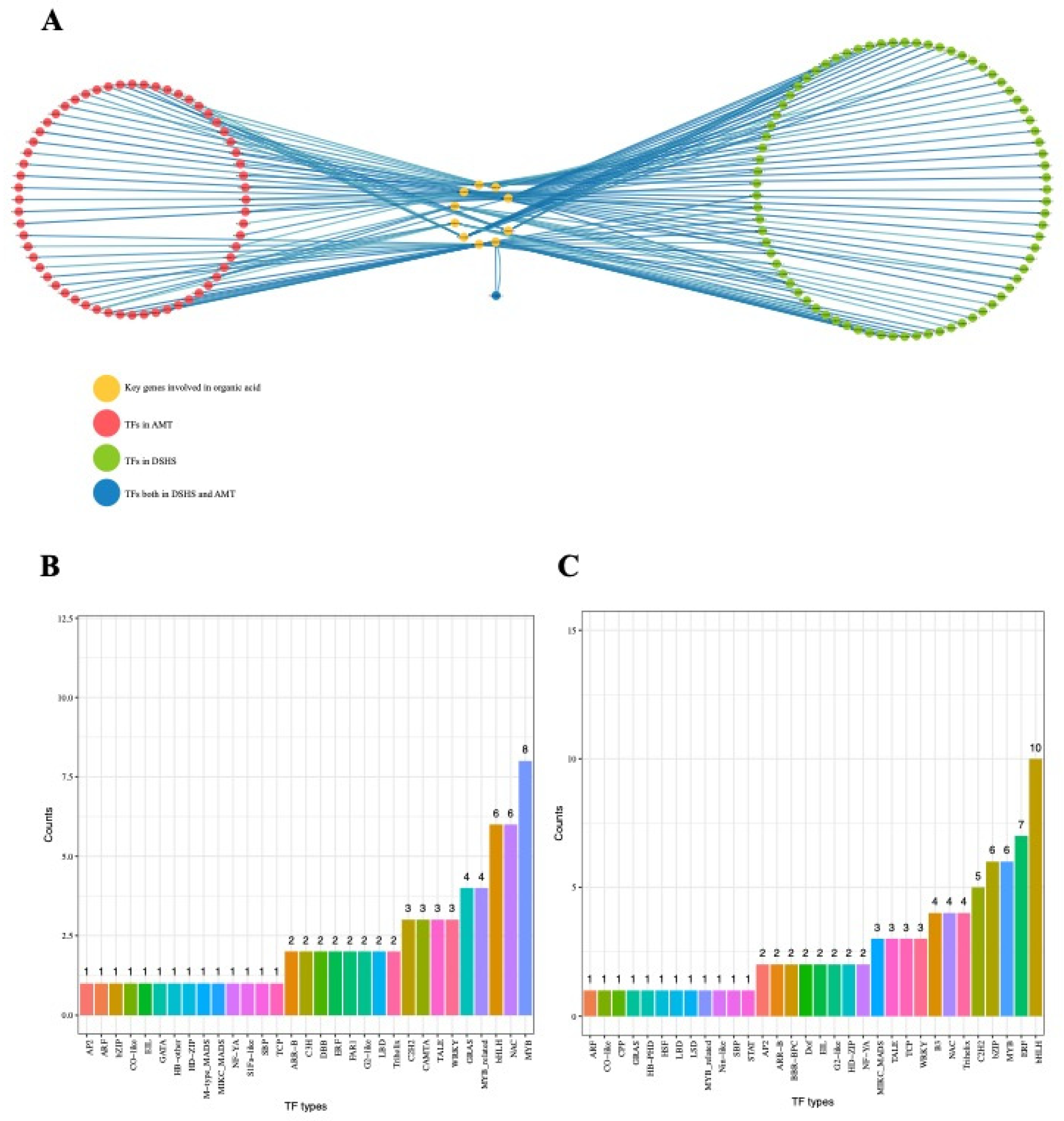
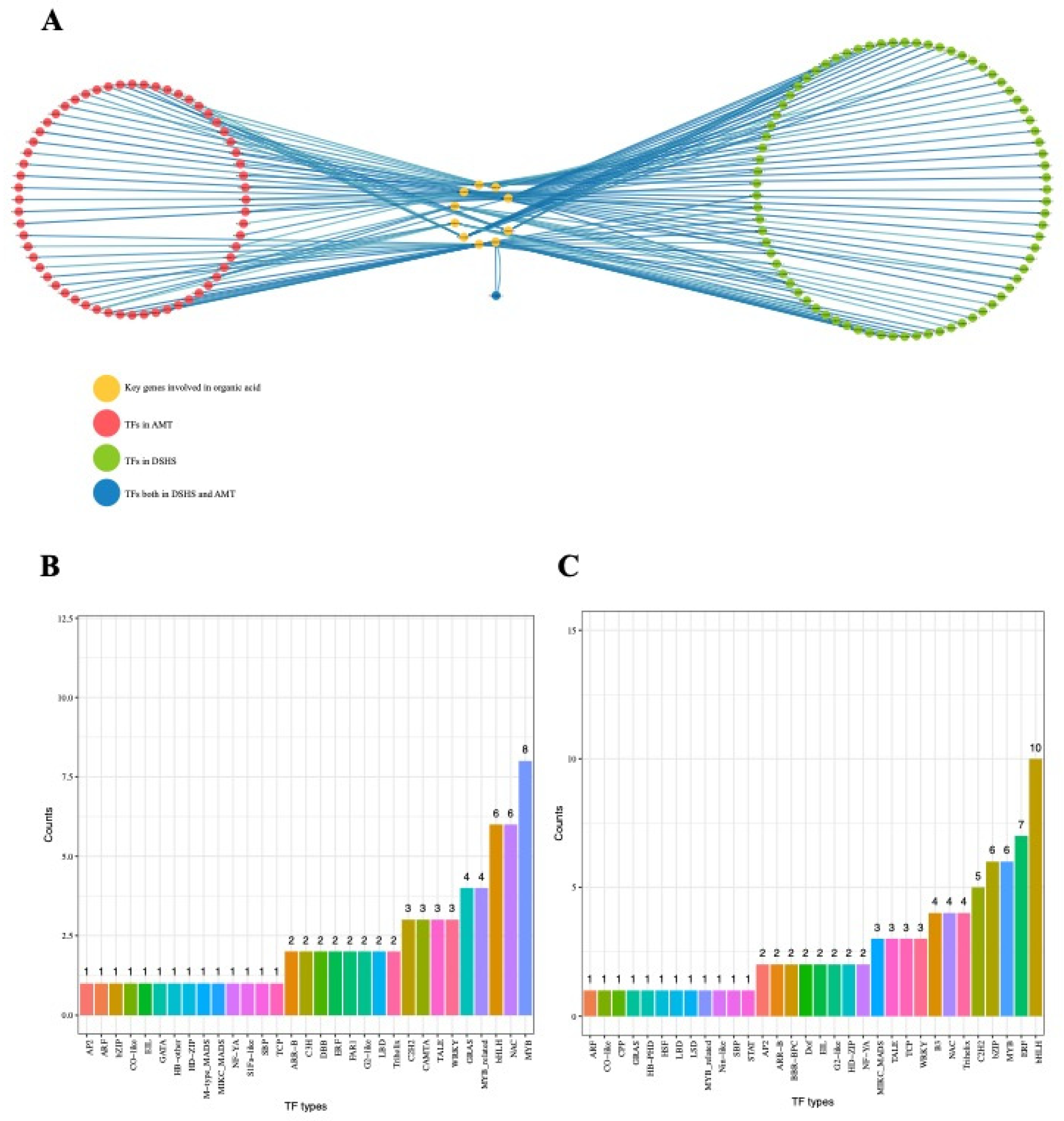
2.5. Identification of Transcriptional Regulators Networks
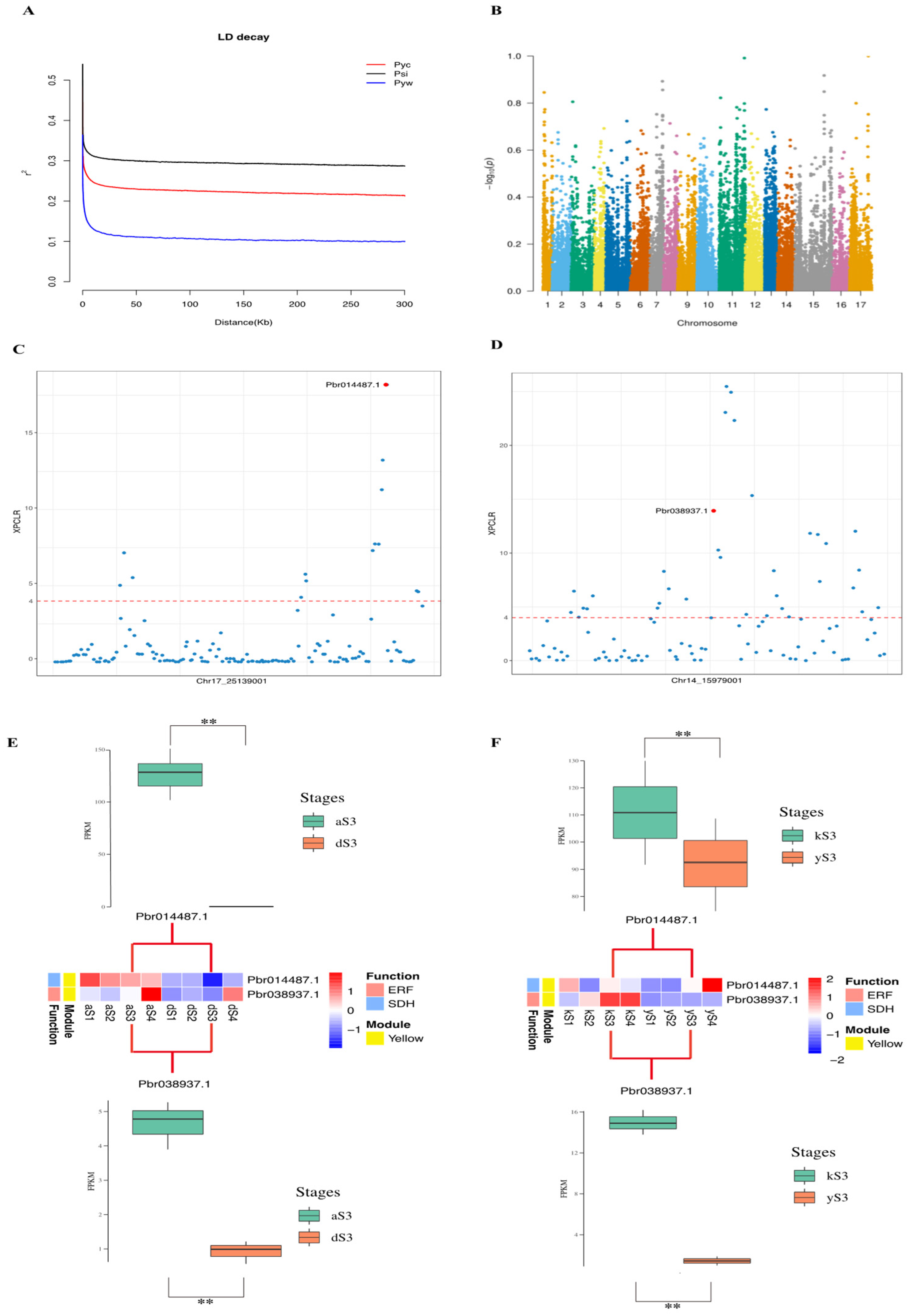
2.6. Selective Sweeps Reveal Domestication Genes in Transcriptional Regulators Networks
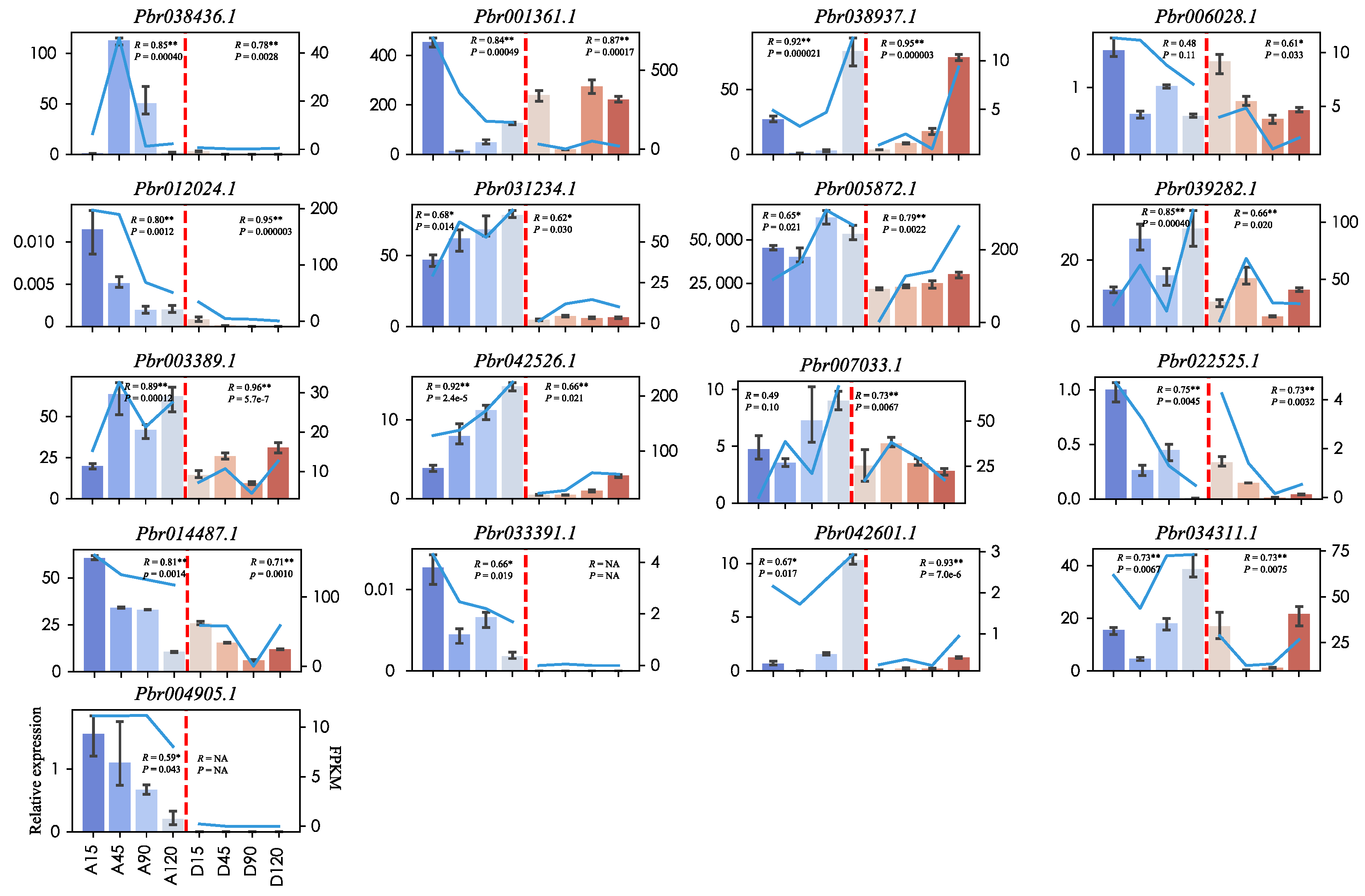
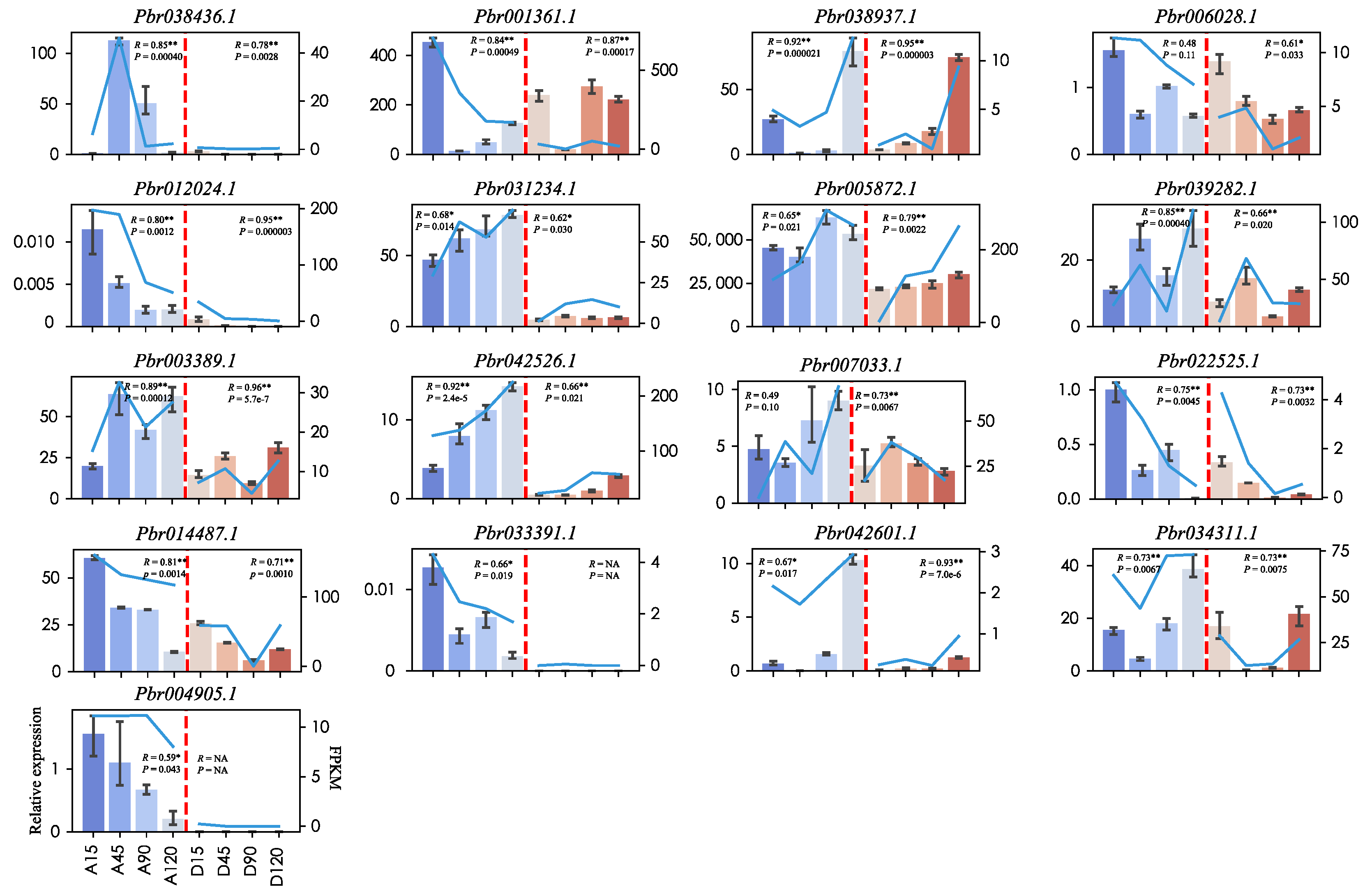
2.7. qRT-PCR Validation
3. Discussion
3.1. Divergent Gene Expression Contributes to Phenotypic Differences
3.2. Divergent Gene Expression Influenced by Differential Selection Process
4. Materials and Methods
4.1. Plant Sample Collection
4.2. Determination of Organic Acid Contents
4.3. RNA Isolation, Library Construction and Sequencing
4.4. RNA-seq Data Processing
4.5. RNA-seq Data De Novo Assembly and Phylogenetic Analysis
4.6. Resequencing Data Processing and Detecting Selective Signals
4.7. Weighted Correlation Network Analysis (WGCNA) and Co-Expression Network Construction
4.8. qRT-PCR Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Ma, M.; Zhang, Y.; Wu, Z.; Guo, L.; Luo, W.; Wang, L.; Zhang, Z.; Zhang, S. Characterization of the Genes Involved in Malic Acid Metabolism from Pear Fruit and Their Expression Profile after Postharvest 1-MCP/Ethrel Treatment. J. Agric. Food Chem. 2018, 66, 8772–8782. [Google Scholar] [CrossRef]
- Seymour, G.B.; Taylor, J.E.; Tucker, G.A. Biochemistry of Fruit Ripening; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Katz, E.; Fon, M.; Lee, Y.J.; Phinney, B.S.; Sadka, A.; Blumwald, E. The citrus fruit proteome: Insights into citrus fruit metabolism. Planta 2007, 226, 989–1005. [Google Scholar] [CrossRef]
- Yao, Y.-X.; Li, M.; Zhai, H.; You, C.-X.; Hao, Y.-J. Isolation and characterization of an apple cytosolic malate dehydrogenase gene reveal its function in malate synthesis. J. Plant Physiol. 2011, 168, 474–480. [Google Scholar] [CrossRef]
- Yao, Y.-X.; Li, M.; Liu, Z.; You, C.-X.; Wang, D.-M.; Zhai, H.; Hao, Y.-J. Molecular cloning of three malic acid related genes MdPEPC, MdVHA-A, MdcyME and their expression analysis in apple fruits. Sci. Hortic. 2009, 122, 404–408. [Google Scholar] [CrossRef]
- Sadka, A.; Dahan, E.; Or, E.; Cohen, L. NADP+-isocitrate dehydrogenase gene expression and isozyme activity during citrus fruit development. Plant Sci. 2000, 158, 173–181. [Google Scholar] [CrossRef]
- Chen, J.; Hao, Z.; Guang, X.; Zhao, C.; Wang, P.; Xue, L.; Zhu, Q.; Yang, L.; Sheng, Y.; Zhou, Y.; et al. Liriodendron genome sheds light on angiosperm phylogeny and species–pair differentiation. Nat. Plants 2018, 5, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Emmerlich, V.; Linka, N.; Reinhold, T.; Hurth, M.A.; Traub, M.; Martinoia, E.; Neuhaus, H.E. The plant homolog to the human sodium/dicarboxylic cotransporter is the vacuolar malate carrier. Proc. Natl. Acad. Sci. USA 2003, 100, 11122–11126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovermann, P.; Meyer, S.; Hörtensteiner, S.; Picco, C.; Scholz-Starke, J.; Ravera, S.; Lee, Y.; Martinoia, E. The Arabidopsis vacuolar malate channel is a member of the ALMT family. Plant J. 2007, 52, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; Scholz-Starke, J.; De Angeli, A.; Kovermann, P.; Burla, B.; Gambale, F.; Martinoia, E. Malate transport by the vacuolar AtALMT6 channel in guard cells is subject to multiple regulation. Plant J. 2011, 67, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Dougherty, L.; Cheng, L.; Xu, K. A co-expression gene network associated with developmental regulation of apple fruit acidity. Mol. Genet. Genom. 2015, 290, 1247–1263. [Google Scholar] [CrossRef]
- Guo, L.-X.; Shi, C.-Y.; Liu, X.; Ning, D.-Y.; Jing, L.-F.; Yang, H.; Liu, Y.-Z. Citrate Accumulation-Related Gene Expression and/or Enzyme Activity Analysis Combined With Metabolomics Provide a Novel Insight for an Orange Mutant. Sci. Rep. 2016, 6, 29343. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.L.; Qiao, X.; Zhang, M.Y.; Zhang, S.L. Genome-Wide analysis of aluminum-activated malate transporter family genes in six rosaceae species, and expression analysis and functional characterization on malate accumulation in Chinese white pear. Plant Sci. 2018, 274, 451–465. [Google Scholar]
- Jia, D.; Shen, F.; Wang, Y.; Wu, T.; Xu, X.; Zhang, X.; Han, Z. Apple fruit acidity is genetically diversified by natural variations in three hierarchical epistatic genes: MdSAUR37, MdPP2CH and MdALMTII. Plant J 2018, 95, 427–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.; Tzuri, G.; Harel-Beja, R.; Itkin, M.; Portnoy, V.; Sa’Ar, U.; Lev, S.; Yeselson, L.; Petrikov, M.; Rogachev, I.; et al. Co-mapping studies of QTLs for fruit acidity and candidate genes of organic acid metabolism and proton transport in sweet melon (Cucumis melo L.). Theor. Appl. Genet. 2012, 125, 343–353. [Google Scholar] [CrossRef]
- Verma, S.; Evans, K.; Guan, Y.; Luby, J.J.; Rosyara, U.R.; Howard, N.P.; Bassil, N.; Bink, M.C.A.M.; Van De Weg, W.E.; Peace, C.P. Two large-effect QTLs, Ma and Ma3, determine genetic potential for acidity in apple fruit: Breeding insights from a multi-family study. Tree Genet. Genomes 2019, 15, 18. [Google Scholar] [CrossRef] [Green Version]
- You, Q.; Zhang, L.; Yi, X.; Zhang, K.; Yao, D.; Zhang, X.; Wang, Q.; Zhao, X.; Ling, Y.; Xu, W.; et al. Co-expression network analyses identify functional modules associated with development and stress response in Gossypium arboreum. Sci. Rep. 2016, 6, 38436. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; He, C.; Qin, Y.; Lin, G.; Park, W.D.; Sun, M.; Li, J.; Lu, X.; Zhang, C.; Yeh, C.; et al. Co-expression analysis aids in the identification of genes in the cuticular wax pathway in maize. Plant J. 2018, 97, 530–542. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Li, K.; Hao, S.; Zhang, J.; Song, T.; Tian, J.; Yao, Y. The Use of RNA Sequencing and Correlation Network Analysis to Study Potential Regulators of Crabapple Leaf Color Transformation. Plant Cell Physiol. 2018, 59, 1027–1042. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Ming, M.; Hu, H.; Zhang, M.; Fan, J.; Song, B.; Zhang, S.; Wu, J. Comparative Transcriptomic Analysis Provides Insight into the Domestication and Improvement of Pear (P. pyrifolia) Fruit. Plant Physiol. 2019, 180, 435–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nham, N.T.; De Freitas, S.T.; Macnish, A.J.; Carr, K.M.; Kietikul, T.; Guilatco, A.J.; Jiang, C.-Z.; Zakharov, F.; Mitcham, E.J. A transcriptome approach towards understanding the development of ripening capacity in ‘Bartlett’ pears (Pyrus communis L.). BMC Genom. 2015, 16, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Busatto, N.; Farneti, B.; Tadiello, A.; Oberkofler, V.; Cellini, A.; Biasioli, F.; Delledonne, M.; Cestaro, A.; Noutsos, C.; Costa, F. Wide transcriptional investigation unravel novel insights of the on-tree maturation and postharvest ripening of ‘Abate Fetel’ pear fruit. Hortic. Res. 2019, 6, 32. [Google Scholar] [CrossRef] [Green Version]
- Yi, X.; Zhao, B.; Tang, Y.; Xu, Z. Transcriptome analysis reveals the regulation of metabolic processes during the post-harvest cold storage of pear. Genomics 2020, 112, 3933–3942. [Google Scholar] [CrossRef]
- Zhou, H.; Tian, M.; Huang, W.; Luo, S.; Hu, H.; Zhang, Y.; Zhang, L.; Li, P. Physiological and transcriptomic analysis of ‘Whangkeumbae’ pear core browning during low-temperature storage. Patterns 2020, 36, 119113. [Google Scholar] [CrossRef]
- Chen, G.; Li, X.; Chen, Q.; Wang, L.; Qi, K.; Yin, H.; Qiao, X.; Wang, P.; Zhang, S.; Wu, J.; et al. Dynamic transcriptome analysis of root nitrate starvation and re-supply provides insights into nitrogen metabolism in pear (Pyrus bretschneideri). Plant Sci. 2018, 277, 322–333. [Google Scholar] [CrossRef]
- Zhang, M.-Y.; Xue, C.; Xu, L.; Sun, H.; Qin, M.-F.; Zhang, S.; Wu, J. Distinct transcriptome profiles reveal gene expression patterns during fruit development and maturation in five main cultivated species of pear (Pyrus L.). Sci. Rep. 2016, 6, 28130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauvage, C.; Rau, A.; Aichholz, C.; Chadoeuf, J.; Sarah, G.; Ruiz, M.; Santoni, S.; Causse, M.; David, J.; Glemin, S. Domesti-cation rewired gene expression and nucleotide diversity patterns in tomato. Plant J. 2017, 91, 631–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Fernie, A.R.; Yan, J. The Past, Present, and Future of Maize Improvement: Domestication, Genomics, and Functional Genomic Routes toward Crop Enhancement. Plant Commun. 2019, 1. [Google Scholar] [CrossRef]
- Koenig, D.; Jiménez-Gómez, J.M.; Kimura, S.; Fulop, D.; Chitwood, D.H.; Headland, L.; Kumar, R.; Covington, M.F.; Devisetty, U.K.; Tat, A.V.; et al. Comparative transcriptomics reveals patterns of selection in domesticated and wild tomato. Proc. Natl. Acad. Sci. 2013, 110, E2655–E2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Li, Q.; Xiong, Q.; Li, W.; Bi, Y.; Lai, Y.; Liu, X.; Man, W.; Zhang, W.; Ma, B.; et al. The transcriptomic signature of developing soybean seeds reveals the genetic basis of seed trait adaptation during domestication. Plant J. 2016, 86, 530–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Wang, Y.; Xu, J.; Korban, S.S.; Fei, Z.; Tao, S.; Ming, R.; Tai, S.; Khan, A.M.; Postman, J.D.; et al. Diversification and independent domestication of Asian and European pears. Genome Biol. 2018, 19, 1–16. [Google Scholar] [CrossRef]
- Sha, S.F.; Li, J.C.; Wu, J.; Zhang, S.L. Characteristics of organic acids in the fruit of different pear species. Afr. J. Agr. Res. 2011, 6, 2403–2410. [Google Scholar]
- Zheng, X.; Cai, D.; Potter, D.; Postman, J.; Liu, J.; Teng, Y. Phylogeny and evolutionary histories of Pyrus L. revealed by phylogenetic trees and networks based on data from multiple DNA sequences. Mol. Phylogenetics Evol. 2014, 80, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.-G.; Li, Y.-Y.; Zhang, Q.-Y.; Li, M.; Sun, C.-H.; Yu, J.-Q.; Hao, Y.-J. The R2R3-MYB transcription factor MdMYB73 is involved in malate accumulation and vacuolar acidification in apple. Plant J. 2017, 91, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Dougherty, L.; Li, M.J.; Fazio, G.; Cheng, L.L.; Xu, K.N. A natural mutation-led truncation in one of the two alu-minum-activated malate transporter-like genes at the Ma locus is associated with low fruit acidity in apple. Mol Genet Genomics 2012, 287, 663–678. [Google Scholar] [CrossRef] [Green Version]
- Moenga, S.M.; Gai, Y.; Carrasquilla-Garcia, N.; Perilla-Henao, L.M.; Cook, D.R. Gene co-expression analysis reveals tran-scriptome divergence between wild and cultivated chickpea under drought stress. Plant J. 2020, 104, 1195–1214. [Google Scholar] [CrossRef]
- Studer, A.; Zhao, Q.; Ross-Ibarra, J.; Doebley, J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 2011, 43, 1160–1163. [Google Scholar] [CrossRef] [Green Version]
- Wills, D.M.; Whipple, C.; Takuno, S.; Kursel, L.; Shannon, L.M.; Ross-Ibarra, J.; Doebley, J.F. From Many, One: Genetic Control of Prolificacy during Maize Domestication. PLoS Genet. 2013, 9, e1003604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.B.; Li, W.; Unger-Wallace, E.; Yang, J.L.; Vollbrecht, E.; Chuck, G. Ideal crop plant architecture is mediated by tassels replace upper ears1, a BTB/POZ ankyrin repeat gene directly targeted by TEOSINTE BRANCHED1. Proc. Natl. Acad. Sci. USA 2017, 114, E8656–E8664. [Google Scholar] [CrossRef] [Green Version]
- Sigmon, B.; Vollbrecht, E. Evidence of selection at the ramosa1 locus during maize domestication. Molecular Ecology 2010, 19, 1296–1311. [Google Scholar] [CrossRef]
- Cong, B.; Barrero, L.S.; Tanksley, S.D. Regulatory change in YABBY-like transcription factor led to evolution of extreme fruit size during tomato domestication. Nat. Genet. 2008, 40, 800–804. [Google Scholar] [CrossRef]
- Lin, Z.; Li, X.; Shannon, L.M.; Yeh, C.-T.; Wang, M.L.; Bai, G.; Peng, Z.; Li, J.; Trick, H.; Clemente, T.E.; et al. Parallel domestication of the Shattering1 genes in cereals. Nat. Genet. 2012, 44, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Wang, Y.; Li, W.; Gu, Y.; Lai, Y.; Bi, Y.; He, C. Transcriptomic comparison reveals genetic variation potentially un-derlying seed developmental evolution of soybeans. J. Exp. Bot. 2018, 69, 5089–5104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doebley, J.F.; Gaut, B.S.; Smith, B.D. The Molecular Genetics of Crop Domestication. Cell 2006, 127, 1309–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Li, Z.; Li, W.Q.; Ku, L.X.; Wang, C.; Ye, J.R.; Li, K.; Yang, N.; Li, Y.P.; Zhong, T.; et al. CACTA-like transposable element in ZmCCT attenuated photoperiod sensitivity and accelerated the post-domestication spread of maize. Proc. Natl. Acad. Sci. USA 2013, 110, 16969–16974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.K.; Garg, R.; Jain, M. A global view of transcriptome dynamics during flower development in chickpea by deep sequencing. Plant Biotechnol. J. 2013, 11, 691–701. [Google Scholar] [CrossRef]
- Chen, Q.; Li, W.; Tan, L.; Tian, F. Harnessing Knowledge from Maize and Rice Domestication for New Crop Breeding. Mol. Plant 2020, 14, 9–26. [Google Scholar] [CrossRef]
- Or, E.; Baybik, J.; Sadka, A.; Saks, Y. Isolation of mitochondrial malate dehydrogenase and phosphoenolpyruvate carboxylase cDNA clones from grape berries and analysis of their expression pattern throughout berry development. J. Plant Physiol. 2000, 157, 527–534. [Google Scholar] [CrossRef]
- Terrier, N.; Glissant, D.; Grimplet, J.; Barrieu, F.; Abbal, P.; Couture, C.; Ageorges, A.; Atanassova, R.; Leon, C.; Renaudin, J.P.; et al. Isogene specific oligo arrays reveal multifaceted changes in gene ex-pression during grape berry (Vitis vinifera L.) development. Planta 2005, 222, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Diakou, P.; Svanella, L.; Raymond, P.; Gaudillere, J.P.; Moing, A. Phosphoenolpyruvate carboxylase during grape berry de-velopment: Protein level, enzyme activity and regulation. Aust J Plant Physiol 2000, 27, 221–229. [Google Scholar]
- Araujo, W.L.; Nunes-Nesi, A.; Osorio, S.; Usadel, B.; Fuentes, D.; Nagy, R.; Balbo, I.; Lehmann, M.; Studart-Witkowski, C.; Tohge, T.; et al. Antisense Inhibition of the Iron-Sulphur Subunit of Suc-cinate Dehydrogenase Enhances Photosynthesis and Growth in Tomato via an Organic Acid-Mediated Effect on Stomatal Aperture. Plant Cell 2011, 23, 600–627. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-J.; Yin, X.-R.; Wang, W.-L.; Liu, X.-F.; Zhang, B.; Chen, K.-S. Citrus CitNAC62 cooperates with CitWRKY1 to participate in citric acid degradation via up-regulation of CitAco3. J. Exp. Bot. 2017, 68, 3419–3426. [Google Scholar] [CrossRef]
- Li, S.-J.; Yin, X.-R.; Xie, X.-L.; Allan, A.C.; Ge, H.; Shen, S.-L.; Chen, K.-S. The Citrus transcription factor, CitERF13, regulates citric acid accumulation via a protein-protein interaction with the vacuolar proton pump, CitVHA-c4. Sci. Rep. 2016, 6, 20151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.-J.; Liu, X.-J.; Xie, X.-L.; Sun, C.-D.; Grierson, D.; Yin, X.-R.; Chen, K.-S. CrMYB73, a PH -like gene, contributes to citric acid accumulation in citrus fruit. Sci. Hortic. 2015, 197, 212–217. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Ge-nome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Abu-Jamous, B.; Kelly, S. Clust: Automatic extraction of optimal co-expressed gene clusters from gene expression data. Genome Biol. 2018, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenführer, J. Gene set enrichment analysis with topGO. Bioconductor Improv 2009, 27, 1–26. [Google Scholar]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Mirarab, S.; Warnow, T.J. ASTRAL-II: Coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 2015, 31, i44–i52. [Google Scholar] [CrossRef] [Green Version]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 10–11. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.B.; Daly, M.J.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obayashi, T.; Kinoshita, K. Rank of Correlation Coefficient as a Comparable Measure for Biological Significance of Gene Co-expression. DNA Research 2009, 16, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T) (-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organic Acid Type | Varieties | S1 | S2 | S3 | S4 |
|---|---|---|---|---|---|
| OA | AMT | 1.3182 ± 0.22 | 0.7615 ± 0.44 | 0.6233 ± 0.11 | 0.1669 ± 0.0085 |
| DSHS | 0.8982 ± 0.12 | 0.8406 ± 0.040 | 0.2305 ± 0.048 | 0.1959 ± 0.0084 | |
| MA | AMT | 6.543 ± 0.35 | 2.195 ± 0.64 | 6.232 ± 0.39 | 4.310 ± 0.50 |
| DSHS | 3.045 ± 0.11 | 2.153 ± 0.16 | 2.920 ± 0.034 | 2.178 ± 0.093 | |
| CA | AMT | 1.927 ± 0.15 | 1.443 ± 0.15 | 1.606 ± 0.19 | 1.574 ± 0.082 |
| DSHS | 1.412 ± 0.16 | 1.116 ± 0.11 | 1.265 ± 0.12 | 0.7402 ± 0.068 |
| Genes | Function | DS1 vs. AS1 | DS2 vs. AS2 | DS3 vs. AS3 | DS4 vs. AS4 |
|---|---|---|---|---|---|
| Pbr039718.1 | ACO | + | |||
| Pbr008056.1 | ACO | − | |||
| Pbr022705.1 | FUM | + | + | + | |
| Pbr037938.1 | IDH | - | + | ||
| Pbr027709.3 | IDH | + | |||
| Pbr024296.3 | MDH | + | + | + | |
| Pbr024294.1 | MDH | + | + | + | + |
| Pbr002553.1 | MDH | − | |||
| Pbr024269.1 | MDH | − | |||
| Pbr022525.1 | MDH | + | |||
| Pbr011434.1 | MDH | − | |||
| Pbr027217.2 | MS | − | − | ||
| Pbr036933.1 | MS | + | |||
| Pbr032339.1 | OGDH | + | + | + | + |
| Pbr042601.1 | OGDH | + | + | + | |
| Pbr015662.1 | OGDH | − | − | − | − |
| Pbr031234.1 | PEPC | + | + | + | + |
| Pbr017951.1 | SDH | − | |||
| Pbr033433.1 | SDH | + | + | + | + |
| Pbr022657.1 | SDH | − | |||
| Pbr007033.1 | SDH | + | |||
| Pbr014487.1 | SDH | + | |||
| Pbr024406.1 | SDH | − | + | ||
| Pbr032250.1 | SDH | + | |||
| Pbr033391.1 | SDH | + | + | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Qiao, X.; Jia, L.; Zhang, Y.; Zhang, S. Transcriptome and Resequencing Analyses Provide Insight into Differences in Organic Acid Accumulation in Two Pear Varieties. Int. J. Mol. Sci. 2021, 22, 9622. https://doi.org/10.3390/ijms22179622
Li Q, Qiao X, Jia L, Zhang Y, Zhang S. Transcriptome and Resequencing Analyses Provide Insight into Differences in Organic Acid Accumulation in Two Pear Varieties. International Journal of Molecular Sciences. 2021; 22(17):9622. https://doi.org/10.3390/ijms22179622
Chicago/Turabian StyleLi, Qionghou, Xin Qiao, Luting Jia, Yuxin Zhang, and Shaoling Zhang. 2021. "Transcriptome and Resequencing Analyses Provide Insight into Differences in Organic Acid Accumulation in Two Pear Varieties" International Journal of Molecular Sciences 22, no. 17: 9622. https://doi.org/10.3390/ijms22179622
APA StyleLi, Q., Qiao, X., Jia, L., Zhang, Y., & Zhang, S. (2021). Transcriptome and Resequencing Analyses Provide Insight into Differences in Organic Acid Accumulation in Two Pear Varieties. International Journal of Molecular Sciences, 22(17), 9622. https://doi.org/10.3390/ijms22179622