
Viral Induced Protein Aggregation: A Mechanism of Immune Evasion



Abstract
:1. Introduction
2. Protein Synthesis and Folding
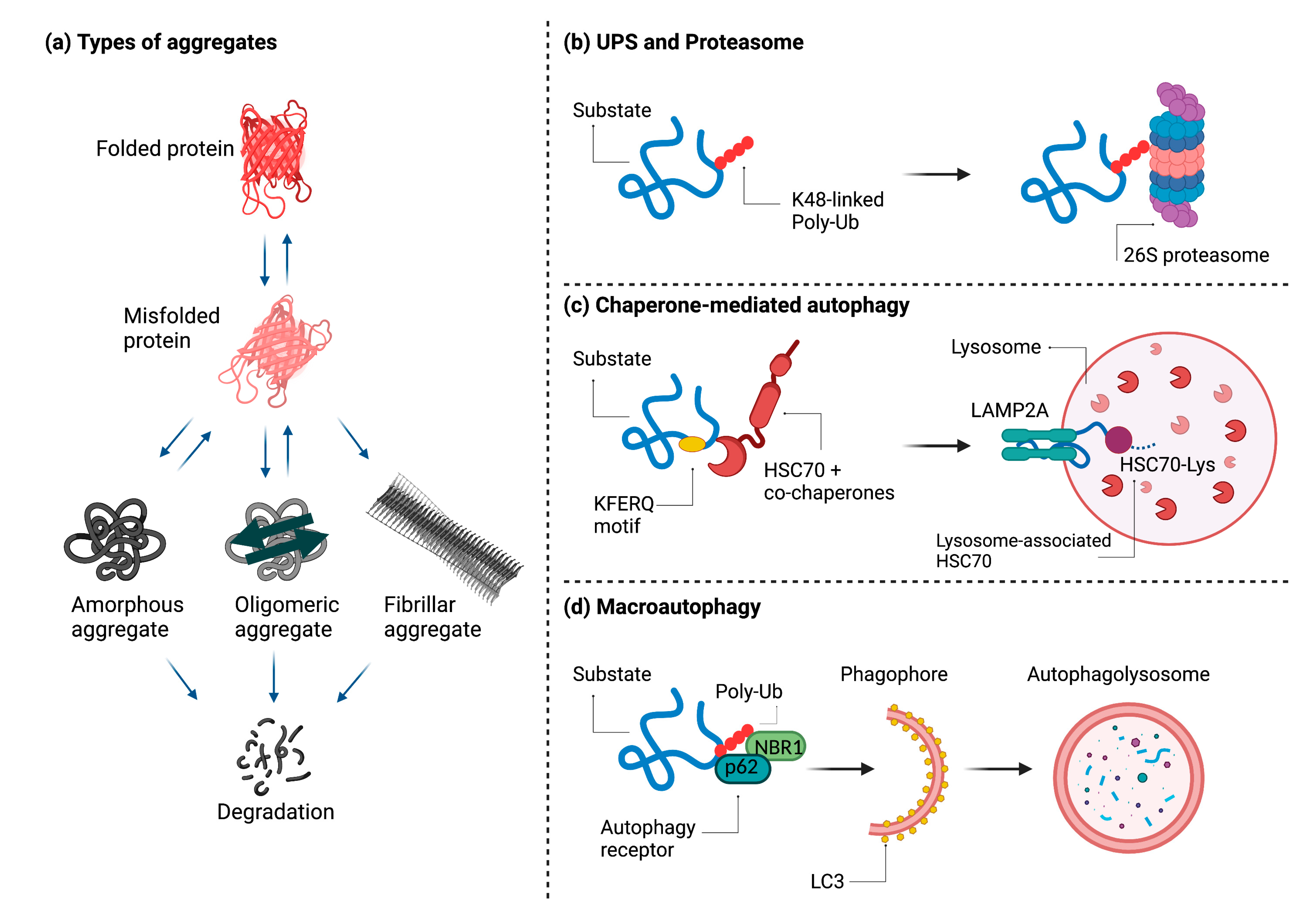
3. Disposal of Protein Aggregates
4. Aggregate Formation during Viral Infection
5. Herpesvirus Proteins and Aggregate Formation
6. Requirements for Viral Induced Protein Aggregation and Disposal: IPAM and Autophagy Adapters
7. Conclusions and Open Question
Author Contributions
Funding
Conflicts of Interest
References
- Lamark, T.; Johansen, T. Aggrephagy: Selective Disposal of Protein Aggregates by Macroautophagy. Int. J. Cell Biol. 2012, 2012, e736905. [Google Scholar] [CrossRef] [Green Version]
- Schmit, J.D.; Ghosh, K.; Dill, K. What Drives Amyloid Molecules to Assemble into Oligomers and Fibrils? Biophys. J. 2011, 100, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Merlini, G.; Bellotti, V. Molecular Mechanisms of Amyloidosis. N. Engl. J. Med. 2003, 349, 583–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, J.B. Molecular Basis of the Neurodegenerative Disorders. N. Engl. J. Med. 1999, 340, 1970–1980. [Google Scholar] [CrossRef]
- Gomes, E.; Shorter, J. The Molecular Language of Membraneless Organelles. J. Biol. Chem. 2019, 294, 7115–7127. [Google Scholar] [CrossRef] [Green Version]
- Kedersha, N.; Anderson, P. Mammalian Stress Granules and Processing Bodies. Methods Enzymol. 2007, 431, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress Granules and Processing Bodies Are Dynamically Linked Sites of MRNP Remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Na, Z.; Slavoff, S.A. P-Bodies: Composition, Properties, and Functions. Biochemistry 2018, 57, 2424–2431. [Google Scholar] [CrossRef]
- Tsai, W.-C.; Lloyd, R.E. Cytoplasmic RNA Granules and Viral Infection. Annu. Rev. Virol. 2014, 1, 147–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.P.; Lloyd, R.E. Regulation of Stress Granules in Virus Systems. Trends Microbiol. 2012, 20, 175–183. [Google Scholar] [CrossRef]
- Kuechler, E.R.; Budzyńska, P.M.; Bernardini, J.P.; Gsponer, J.; Mayor, T. Distinct Features of Stress Granule Proteins Predict Localization in Membraneless Organelles. J. Mol. Biol. 2020, 432, 2349–2368. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Hess, D.; Eglinger, J.; Fritsch, A.W.; Kreysing, M.; Weinert, B.T.; Choudhary, C.; Matthias, P. Acetylation of Intrinsically Disordered Regions Regulates Phase Separation. Nat. Chem. Biol. 2019, 15, 51–61. [Google Scholar] [CrossRef]
- Gorbenko, G.; Trusova, V. Protein Aggregation in a Membrane Environment. Adv. Protein Chem. Struct. Biol. 2011, 84, 113–142. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, R.; Pincus, P. Protein Aggregation in Membranes. Curr. Opin. Solid State Mater. Sci. 1996, 1, 401–406. [Google Scholar] [CrossRef]
- Parton, D.L.; Klingelhoefer, J.W.; Sansom, M.S.P. Aggregation of Model Membrane Proteins, Modulated by Hydrophobic Mismatch, Membrane Curvature, and Protein Class. Biophys. J. 2011, 101, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Naiki, H.; Higuchi, K.; Hosokawa, M.; Takeda, T. Fluorometric Determination of Amyloid Fibrils In Vitro Using the Fluorescent Dye, Thioflavine T. Anal. Biochem. 1989, 177, 244–249. [Google Scholar] [CrossRef]
- Viegas, M.S.; Martins, T.C.; Seco, F.; do Carmo, A. An Improved and Cost-Effective Methodology for the Reduction of Autofluorescence in Direct Immunofluorescence Studies on Formalin-Fixed Paraffin-Embedded Tissues. Eur. J. Histochem. 2007, 51, 59–66. [Google Scholar]
- Shen, D.; Coleman, J.; Chan, E.; Nicholson, T.P.; Dai, L.; Sheppard, P.W.; Patton, W.F. Novel Cell- and Tissue-Based Assays for Detecting Misfolded and Aggregated Protein Accumulation within Aggresomes and Inclusion Bodies. Cell Biochem. Biophys. 2011, 60, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Skovronsky, D.M.; Zhang, B.; Kung, M.P.; Kung, H.F.; Trojanowski, J.Q.; Lee, V.M. In Vivo Detection of Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2000, 97, 7609–7614. [Google Scholar] [CrossRef] [Green Version]
- De Boer, P.; Hoogenboom, J.P.; Giepmans, B.N.G. Correlated Light and Electron Microscopy: Ultrastructure Lights Up! Nat. Methods 2015, 12, 503–513. [Google Scholar] [CrossRef]
- Ishikawa-Ankerhold, H.C.; Ankerhold, R.; Drummen, G.P.C. Advanced Fluorescence Microscopy Techniques—FRAP, FLIP, FLAP, FRET and FLIM. Molecules 2012, 17, 4047–4132. [Google Scholar] [CrossRef] [Green Version]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In Vivo Aspects of Protein Folding and Quality Control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Vembar, S.S.; Brodsky, J.L. One Step at a Time: Endoplasmic Reticulum-Associated Degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef]
- Bascos, N.A.D.; Landry, S.J. A History of Molecular Chaperone Structures in the Protein Data Bank. Int. J. Mol. Sci. 2019, 20, 6195. [Google Scholar] [CrossRef] [Green Version]
- Vabulas, R.M.; Raychaudhuri, S.; Hayer-Hartl, M.; Hartl, F.U. Protein Folding in the Cytoplasm and the Heat Shock Response. Cold Spring Harb. Perspect. Biol. 2010, 2, a004390. [Google Scholar] [CrossRef]
- Ran, R.; Lu, A.; Xu, H.; Tang, Y.; Sharp, F.R. Heat-Shock Protein Regulation of Protein Folding, Protein Degradation, Protein Function, and Apoptosis. In Handbook of Neurochemistry and Molecular Neurobiology: Acute Ischemic Injury and Repair in the Nervous System; Lajtha, A., Chan, P.H., Eds.; Springer: Boston, MA, USA, 2007; pp. 89–107. ISBN 978-0-387-30383-3. [Google Scholar]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Weissman, A.M. The Unfolded Protein Response, Degradation from the Endoplasmic Reticulum, and Cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR Pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Lim, H.-S.; Masliah, E.; Lee, H.-J. Protein Aggregate Spreading in Neurodegenerative Diseases: Problems and Perspectives. Neurosci. Res. 2011, 70, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Wong, E. Chapter Fifteen—Kinetics of Protein Aggregates Disposal by Aggrephagy. In Methods in Enzymology; Molecular Characterization of Autophagic Responses, Part B; Galluzzi, L., Bravo-San Pedro, J.M., Kroemer, G., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 588, pp. 245–281. [Google Scholar]
- Kopito, R.R. Aggresomes, Inclusion Bodies and Protein Aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Hochstrasser, M. Ubiquitin-Dependent Protein Degradation. Annu. Rev. Genet. 1996, 30, 405–439. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A Role for Ubiquitin in Selective Autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fernández, M.R.; Gragera, M.; Ochoa-Ibarrola, L.; Quintana-Gallardo, L.; Valpuesta, J.M. Hsp70—A Master Regulator in Protein Degradation. FEBS Lett. 2017, 591, 2648–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terlecky, S.R.; Chiang, H.L.; Olson, T.S.; Dice, J.F. Protein and Peptide Binding and Stimulation of In Vitro Lysosomal Proteolysis by the 73-KDa Heat Shock Cognate Protein. J. Biol. Chem. 1992, 267, 9202–9209. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. A Receptor for the Selective Uptake and Degradation of Proteins by Lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef]
- Øverbye, A.; Fengsrud, M.; Seglen, P.O. Proteomic Analysis of Membrane-Associated Proteins from Rat Liver Autophagosomes. Autophagy 2007, 3, 300–322. [Google Scholar] [CrossRef] [Green Version]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular Chaperones and Protein Quality Control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A Cellular Response to Misfolded Proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [Green Version]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. P62/SQSTM1 Forms Protein Aggregates Degraded by Autophagy and Has a Protective Effect on Huntingtin-Induced Cell Death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The Deacetylase HDAC6 Regulates Aggresome Formation and Cell Viability in Response to Misfolded Protein Stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and Microtubules Are Required for Autophagic Degradation of Aggregated Huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef] [Green Version]
- Yao, T.-P. The Role of Ubiquitin in Autophagy-Dependent Protein Aggregate Processing. Genes Cancer 2010, 1, 779–786. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective Autophagy Mediated by Autophagic Adapter Proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded Proteins Partition between Two Distinct Quality Control Compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Netherton, C.L.; Wileman, T. Virus Factories, Double Membrane Vesicles and Viroplasm Generated in Animal Cells. Curr. Opin. Virol. 2011, 1, 381–387. [Google Scholar] [CrossRef]
- Netherton, C.; Moffat, K.; Brooks, E.; Wileman, T. A Guide to Viral Inclusions, Membrane Rearrangements, Factories, and Viroplasm Produced during Virus Replication. Adv. Virus Res. 2007, 70, 101–182. [Google Scholar] [CrossRef]
- Heath, C.M.; Windsor, M.; Wileman, T. Aggresomes Resemble Sites Specialized for Virus Assembly. J. Cell Biol. 2001, 153, 449–456. [Google Scholar] [CrossRef] [Green Version]
- Novoa, R.R.; Calderita, G.; Arranz, R.; Fontana, J.; Granzow, H.; Risco, C. Virus Factories: Associations of Cell Organelles for Viral Replication and Morphogenesis. Biol. Cell 2005, 97, 147–172. [Google Scholar] [CrossRef] [PubMed]
- Araujo, F.D.; Stracker, T.H.; Carson, C.T.; Lee, D.V.; Weitzman, M.D. Adenovirus Type 5 E4orf3 Protein Targets the Mre11 Complex to Cytoplasmic Aggresomes. J. Virol. 2005, 79, 11382–11391. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Shevchenko, A.; Shevchenko, A.; Berk, A.J. Adenovirus Exploits the Cellular Aggresome Response To Accelerate Inactivation of the MRN Complex. J. Virol. 2005, 79, 14004–14016. [Google Scholar] [CrossRef] [Green Version]
- Muscolino, E.; Schmitz, R.; Loroch, S.; Caragliano, E.; Schneider, C.; Rizzato, M.; Kim, Y.-H.; Krause, E.; Juranić Lisnić, V.; Sickmann, A.; et al. Herpesviruses Induce Aggregation and Selective Autophagy of Host Signalling Proteins NEMO and RIPK1 as an Immune-Evasion Mechanism. Nat. Microbiol. 2020, 5, 331–342. [Google Scholar] [CrossRef]
- Vidic, J.; Richard, C.-A.; Péchoux, C.; Da Costa, B.; Bertho, N.; Mazerat, S.; Delmas, B.; Chevalier, C. Amyloid Assemblies of Influenza A Virus PB1-F2 Protein Damage Membrane and Induce Cytotoxicity. J. Biol. Chem. 2016, 291, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Rohrmann, G.F. Baculovirus Molecular Biology; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2019. [Google Scholar]
- Guo, Z.-J.; Tao, L.-X.; Dong, X.-Y.; Yu, M.-H.; Tian, T.; Tang, X.-D. Characterization of Aggregate/Aggresome Structures Formed by Polyhedrin of Bombyx Mori Nucleopolyhedrovirus. Sci. Rep. 2015, 5, 14601. [Google Scholar] [CrossRef] [Green Version]
- Zantema, A.; Fransen, J.A.; Davis-Olivier, A.; Ramaekers, F.C.; Vooijs, G.P.; DeLeys, B.; Van der Eb, A.J. Localization of the E1B Proteins of Adenovirus 5 in Transformed Cells, as Revealed by Interaction with Monoclonal Antibodies. Virology 1985, 142, 44–58. [Google Scholar] [CrossRef]
- Greer, A.E.; Hearing, P.; Ketner, G. The Adenovirus E4 11k Protein Binds and Relocalizes the Cytoplasmic P-Body Component Ddx6 to Aggresomes. Virology 2011, 417, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, C.; Al Bazzal, A.; Vidic, J.; Février, V.; Bourdieu, C.; Bouguyon, E.; Le Goffic, R.; Vautherot, J.-F.; Bernard, J.; Moudjou, M.; et al. PB1-F2 Influenza A Virus Protein Adopts a Beta-Sheet Conformation and Forms Amyloid Fibers in Membrane Environments. J. Biol. Chem. 2010, 285, 13233–13243. [Google Scholar] [CrossRef] [Green Version]
- Henkel, M.; Mitzner, D.; Henklein, P.; Meyer-Almes, F.-J.; Moroni, A.; Difrancesco, M.L.; Henkes, L.M.; Kreim, M.; Kast, S.M.; Schubert, U.; et al. The Proapoptotic Influenza A Virus Protein PB1-F2 Forms a Nonselective Ion Channel. PLoS ONE 2010, 5, e11112. [Google Scholar] [CrossRef] [Green Version]
- Miodek, A.; Vidic, J.; Sauriat-Dorizon, H.; Richard, C.-A.; Le Goffic, R.; Korri-Youssoufi, H.; Chevalier, C. Electrochemical Detection of the Oligomerization of PB1-F2 Influenza A Virus Protein in Infected Cells. Anal. Chem. 2014, 86, 9098–9105. [Google Scholar] [CrossRef]
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The Order Herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef]
- Pellet, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1802–18022. [Google Scholar]
- Lembo, D.; Brune, W. Tinkering with a Viral Ribonucleotide Reductase. Trends Biochem. Sci. 2009, 34, 25–32. [Google Scholar] [CrossRef]
- Brune, W.; Ménard, C.; Heesemann, J.; Koszinowski, U.H. A Ribonucleotide Reductase Homolog of Cytomegalovirus and Endothelial Cell Tropism. Science 2001, 291, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Lembo, D.; Donalisio, M.; Hofer, A.; Cornaglia, M.; Brune, W.; Koszinowski, U.; Thelander, L.; Landolfo, S. The Ribonucleotide Reductase R1 Homolog of Murine Cytomegalovirus Is Not a Functional Enzyme Subunit but Is Required for Pathogenesis. J. Virol. 2004, 78, 4278–4288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Cytomegalovirus M45 Cell Death Suppression Requires Receptor-Interacting Protein (RIP) Homotypic Interaction Motif (RHIM)-Dependent Interaction with RIP1. J. Biol. Chem. 2008, 283, 16966–16970. [Google Scholar] [CrossRef] [Green Version]
- Mack, C.; Sickmann, A.; Lembo, D.; Brune, W. Inhibition of Proinflammatory and Innate Immune Signaling Pathways by a Cytomegalovirus RIP1-Interacting Protein. Proc. Natl. Acad. Sci. USA 2008, 105, 3094–3099. [Google Scholar] [CrossRef] [Green Version]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Virus Inhibition of RIP3-Dependent Necrosis. Cell Host Microbe 2010, 7, 302–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, A.; Wang, Z. Necroptosis: MLKL Polymerization. J. Nat. Sci. 2018, 4, e513. [Google Scholar] [PubMed]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.-T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of Mixed Lineage Kinase Domain-like Protein to Plasma Membrane Leads to Necrotic Cell Death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI Complexes with RIP3 to Mediate Virus-Induced Programmed Necrosis That Is Targeted by Murine Cytomegalovirus VIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.-S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 Necrosome Forms a Functional Amyloid Signaling Complex Required for Programmed Necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Pham, C.L.; Shanmugam, N.; Strange, M.; O’Carroll, A.; Brown, J.W.; Sierecki, E.; Gambin, Y.; Steain, M.; Sunde, M. Viral M45 and Necroptosis-Associated Proteins Form Heteromeric Amyloid Assemblies. EMBO Rep. 2019, 20, e46518. [Google Scholar] [CrossRef]
- Guo, H.; Omoto, S.; Harris, P.A.; Finger, J.N.; Bertin, J.; Gough, P.J.; Kaiser, W.J.; Mocarski, E.S. Herpes Simplex Virus Suppresses Necroptosis in Human Cells. Cell Host Microbe 2015, 17, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Wu, S.-Q.; Liang, Y.; Zhou, X.; Chen, W.; Li, L.; Wu, J.; Zhuang, Q.; Chen, C.; Li, J.; et al. RIP1/RIP3 Binding to HSV-1 ICP6 Initiates Necroptosis to Restrict Virus Propagation in Mice. Cell Host Microbe 2015, 17, 229–242. [Google Scholar] [CrossRef] [Green Version]
- Dufour, F.; Sasseville, A.M.-J.; Chabaud, S.; Massie, B.; Siegel, R.M.; Langelier, Y. The Ribonucleotide Reductase R1 Subunits of Herpes Simplex Virus Types 1 and 2 Protect Cells against TNFα- and FasL-Induced Apoptosis by Interacting with Caspase-8. Apoptosis Int. J. Program. Cell Death 2011, 16, 256–271. [Google Scholar] [CrossRef] [Green Version]
- Fliss, P.M.; Jowers, T.P.; Brinkmann, M.M.; Holstermann, B.; Mack, C.; Dickinson, P.; Hohenberg, H.; Ghazal, P.; Brune, W. Viral Mediated Redirection of NEMO/IKKγ to Autophagosomes Curtails the Inflammatory Cascade. PLoS Pathog. 2012, 8, e1002517. [Google Scholar] [CrossRef] [Green Version]
- Krause, E.; de Graaf, M.; Fliss, P.M.; Dölken, L.; Brune, W. Murine Cytomegalovirus Virion-Associated Protein M45 Mediates Rapid NF-ΚB Activation after Infection. J. Virol. 2014, 88, 9963–9975. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.Z.; Moraes, S.N.; Attarian, C.; Yockteng-Melgar, J.; Jarvis, M.C.; Biolatti, M.; Galitska, G.; Dell’Oste, V.; Frappier, L.; Bierle, C.J.; et al. A Conserved Mechanism of APOBEC3 Relocalization by Herpesviral Ribonucleotide Reductase Large Subunits. J. Virol. 2019, 93, e01539-19. [Google Scholar] [CrossRef]
- Cheng, A.Z.; Yockteng-Melgar, J.; Jarvis, M.C.; Malik-Soni, N.; Borozan, I.; Carpenter, M.A.; McCann, J.L.; Ebrahimi, D.; Shaban, N.M.; Marcon, E.; et al. Epstein-Barr Virus BORF2 Inhibits Cellular APOBEC3B to Preserve Viral Genome Integrity. Nat. Microbiol. 2019, 4, 78–88. [Google Scholar] [CrossRef]
- Harris, R.S.; Dudley, J.P. APOBECs and Virus Restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.S.; Petersen-Mahrt, S.K.; Neuberger, M.S. RNA Editing Enzyme APOBEC1 and Some of Its Homologs Can Act as DNA Mutators. Mol. Cell 2002, 10, 1247–1253. [Google Scholar] [CrossRef]
- Hultquist, J.F.; Lengyel, J.A.; Refsland, E.W.; LaRue, R.S.; Lackey, L.; Brown, W.L.; Harris, R.S. Human and Rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H Demonstrate a Conserved Capacity to Restrict Vif-Deficient HIV-1. J. Virol. 2011, 85, 11220–11234. [Google Scholar] [CrossRef] [Green Version]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad Antiretroviral Defence by Human APOBEC3G through Lethal Editing of Nascent Reverse Transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a Human Gene That Inhibits HIV-1 Infection and Is Suppressed by the Viral Vif Protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The Cytidine Deaminase CEM15 Induces Hypermutation in Newly Synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Ylä-Anttila, P.; Sandalova, T.; Sun, R.; Achour, A.; Masucci, M.G. 14-3-3 Scaffold Proteins Mediate the Inactivation of Trim25 and Inhibition of the Type I Interferon Response by Herpesvirus Deconjugases. PLoS Pathog. 2019, 15, e1008146. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Ylä-Anttila, P.; Sandalova, T.; Achour, A.; Masucci, M.G. Interaction with 14-3-3 Correlates with Inactivation of the RIG-I Signalosome by Herpesvirus Ubiquitin Deconjugases. Front. Immunol. 2020, 11, 437. [Google Scholar] [CrossRef] [Green Version]
- Saito, S.; Murata, T.; Kanda, T.; Isomura, H.; Narita, Y.; Sugimoto, A.; Kawashima, D.; Tsurumi, T. Epstein-Barr Virus Deubiquitinase Downregulates TRAF6-Mediated NF-ΚB Signaling during Productive Replication. J. Virol. 2013, 87, 4060–4070. [Google Scholar] [CrossRef] [Green Version]
- Van Gent, M.; Braem, S.G.E.; de Jong, A.; Delagic, N.; Peeters, J.G.C.; Boer, I.G.J.; Moynagh, P.N.; Kremmer, E.; Wiertz, E.J.; Ovaa, H.; et al. Epstein-Barr Virus Large Tegument Protein BPLF1 Contributes to Innate Immune Evasion through Interference with Toll-like Receptor Signaling. PLoS Pathog. 2014, 10, e1003960. [Google Scholar] [CrossRef]
- Ye, R.; Su, C.; Xu, H.; Zheng, C. Herpes Simplex Virus 1 Ubiquitin-Specific Protease UL36 Abrogates NF-ΚB Activation in DNA Sensing Signal Pathway. J. Virol. 2017, 91, e02417-16. [Google Scholar] [CrossRef] [Green Version]
- Inn, K.-S.; Lee, S.-H.; Rathbun, J.Y.; Wong, L.-Y.; Toth, Z.; Machida, K.; Ou, J.-H.J.; Jung, J.U. Inhibition of RIG-I-Mediated Signaling by Kaposi’s Sarcoma-Associated Herpesvirus-Encoded Deubiquitinase ORF64. J. Virol. 2011, 85, 10899–10904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Ylä-Anttila, P.; Callegari, S.; Tsai, M.-H.; Delecluse, H.-J.; Masucci, M.G. Herpesvirus Deconjugases Inhibit the IFN Response by Promoting TRIM25 Autoubiquitination and Functional Inactivation of the RIG-I Signalosome. PLoS Pathog. 2018, 14, e1006852. [Google Scholar] [CrossRef] [Green Version]
- Kumari, P.; Saha, I.; Narayanan, A.; Narayanan, S.; Takaoka, A.; Kumar, N.S.; Tailor, P.; Kumar, H. Essential Role of HCMV Deubiquitinase in Promoting Oncogenesis by Targeting Anti-Viral Innate Immune Signaling Pathways. Cell Death Dis. 2017, 8, e3078. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, K.; Li, J.; Zheng, C. Herpes Simplex Virus 1 Ubiquitin-Specific Protease UL36 Inhibits Beta Interferon Production by Deubiquitinating TRAF3. J. Virol. 2013, 87, 11851–11860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, K.M.; Oh, S.E.; Kim, Y.E.; Han, T.-H.; Ahn, J.-H. Cooperative Inhibition of RIP1-Mediated NF-ΚB Signaling by Cytomegalovirus-Encoded Deubiquitinase and Inactive Homolog of Cellular Ribonucleotide Reductase Large Subunit. PLoS Pathog. 2017, 13, e1006423. [Google Scholar] [CrossRef] [Green Version]
- Meriin, A.B.; Narayanan, A.; Meng, L.; Alexandrov, I.; Varelas, X.; Cissé, I.I.; Sherman, M.Y. Hsp70–Bag3 Complex Is a Hub for Proteotoxicity-Induced Signaling That Controls Protein Aggregation. Proc. Natl. Acad. Sci. USA 2018, 115, E7043–E7052. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Detection Method | Principle | Additional Remarks |
|---|---|---|
| Thioflavin-T (ThT) [16] | Fluorescent rotor dye Free rotation inhibited upon intercalation into the β-sheet structure of amyloid fibrils leading to (or significantly increasing existing) fluorescence emission. | Emission maximum at ~485 nm overlaps with the intrinsic fluorescence of some cellular constituents [17] |
| ProteoStat® [18] | High signal intensity Emission maximum at 600 nm enables the use of additional fluorescent markers Suitable for high-throughput screening by flow cytometry | |
| BSB (trans, trans)-1- bromo-2, 5-bis-(3-hydroxycarbonyl-4-hydroxy) styrylbenzene [19] | Fluorescent probe for staining amyloids in tissue sections or live mice. | Emission maximum at 520 nm Crosses the blood-brain barrier enabling in vivo staining of characteristic amyloid plaques in neurodegenerative disorders Requires a UV laser source for optimal excitation |
| Separation of cell lysate into insoluble and soluble fractions | Soluble proteins are dissolved with a mild detergent (e.g., Nonidet P-40, Triton X-100). Insoluble fraction is pelleted by centrifugation and lysed using strong reducing agents (e.g., 2-mercaptoethanol, SDS). Both fractions are visualized by immunoblotting. | Laborious, not suitable for large sample volumes |
| Correlative light and electron microscopy (CLEM) [20] | Area of interest containing a fluorescently labeled target is selected using fluorescence microscopy. Target can be analyzed at high resolution by EM. | Combines ultrastructural analysis with dynamics and improved target identification. Laborious and time-consuming Only a limited amount of cells can be examined. |
| Fluorescence recovery after photobleaching (FRAP) [21] | Region of a fluorescently labeled target protein is bleached with a laser. Recovery of fluorescence is recorded over time. In liquid compartments, fluorophores can diffuse, and fluorescence recovers rapidly. In solid aggregates, diffusion is not possible and fluorescence recovery is prevented. | Investigates protein mobility in living cells Laborious and time-consuming |
| Virus | Protein | Involvement in Aggregate Formation | Involvement of Autophagy |
|---|---|---|---|
| African swine fever virus (ASFV) | - | Concentrates vDNA in aggresome-like viral factories | Not known |
| Baculoviruses | BmNPV polyhedrin | Co-localizes with aggregate markers | Co-localizes with LC3 |
| Adenovirus type 5 (Ad5) | E4-11k and E1B-55k | Sequester the MRN DNA repair complex to the insoluble fraction | Re-localize components of RNA processing bodies to aggresomes |
| Influenza A virus (IAV) | PB1-F2 | Capable of oligomerizing into amyloid fibers | Not known |
| Herpesviruses | M45 (MCMV) | Purified N-terminal fragment (90 aa) forms amyloid fibrils in vitro Expression in cells causes accumulation of RIPK1 and NEMO as insoluble aggregates | Recruits autophagy adapters VPS26B and TBC1D5 and degrades aggregates by autophagy |
| ICP6 (HSV-1) | Induces the formation of RIPK1 aggregates | Degrades aggregates by autophagy | |
| ICP6 (HSV-1) | Re-localize nuclear APOBEC3 proteins to distinct structures in the cytoplasm | Not known | |
| BORF2 (EBV) | |||
| ORF61 (KSHV) | |||
| UL48 (HCMV) | Sequester TRIM25 in aggregate structures | Bind to 14-3-3 scaffold proteins and co-localize with p62 | |
| BPLF1 (EBV) | |||
| ORF64 (KSHV) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muscolino, E.; Luoto, L.-M.; Brune, W. Viral Induced Protein Aggregation: A Mechanism of Immune Evasion. Int. J. Mol. Sci. 2021, 22, 9624. https://doi.org/10.3390/ijms22179624
Muscolino E, Luoto L-M, Brune W. Viral Induced Protein Aggregation: A Mechanism of Immune Evasion. International Journal of Molecular Sciences. 2021; 22(17):9624. https://doi.org/10.3390/ijms22179624
Chicago/Turabian StyleMuscolino, Elena, Laura-Marie Luoto, and Wolfram Brune. 2021. "Viral Induced Protein Aggregation: A Mechanism of Immune Evasion" International Journal of Molecular Sciences 22, no. 17: 9624. https://doi.org/10.3390/ijms22179624
APA StyleMuscolino, E., Luoto, L.-M., & Brune, W. (2021). Viral Induced Protein Aggregation: A Mechanism of Immune Evasion. International Journal of Molecular Sciences, 22(17), 9624. https://doi.org/10.3390/ijms22179624