
Increase in Akkermansiaceae in Gut Microbiota of Prostate Cancer-Bearing Mice

 ,
,  and
and
Abstract
:1. Introduction
2. Results
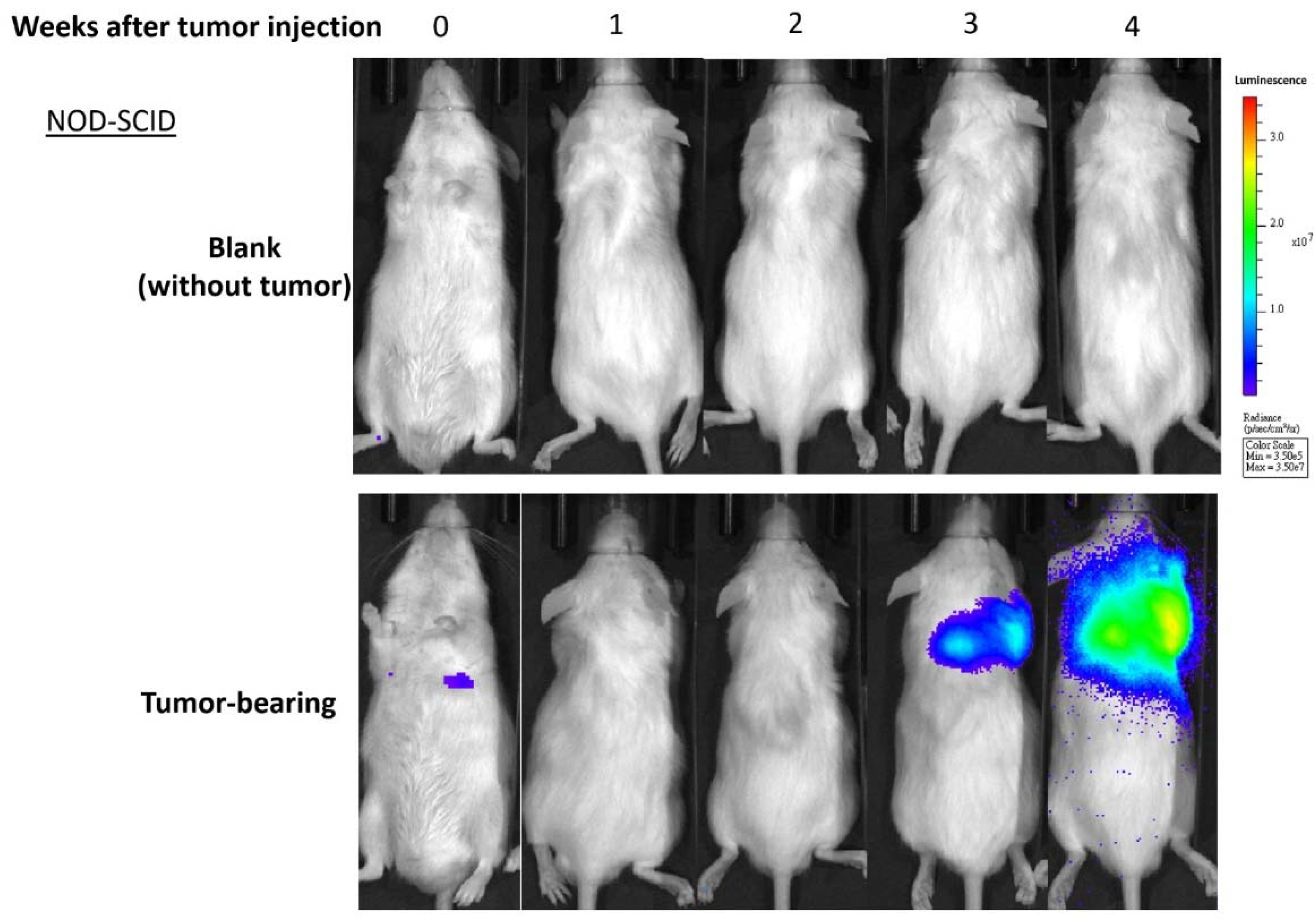
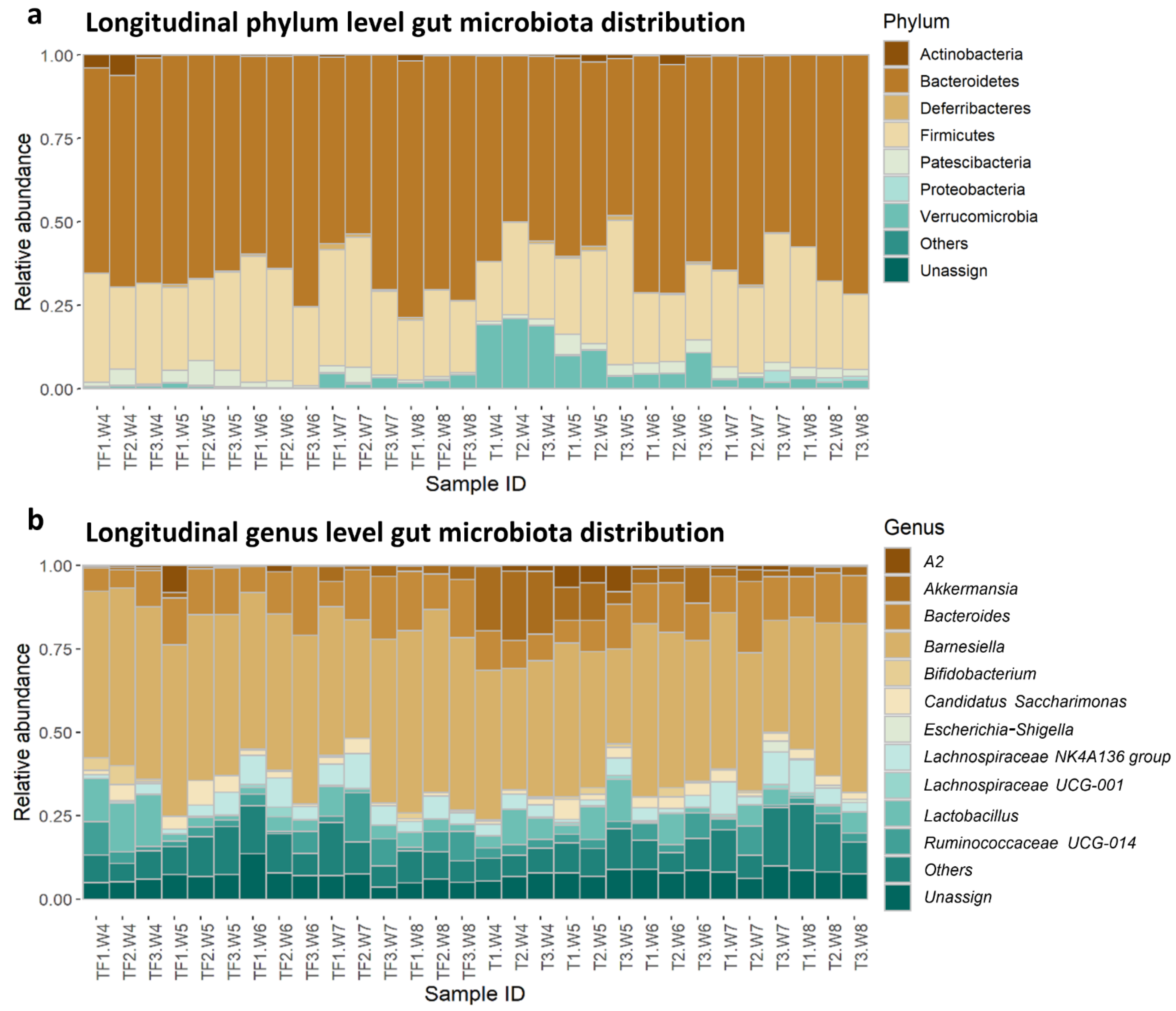
2.1. Longitudinal Microbiota Profiles in Cancer-Bearing (T) and Tumor-Free (TF) Mice
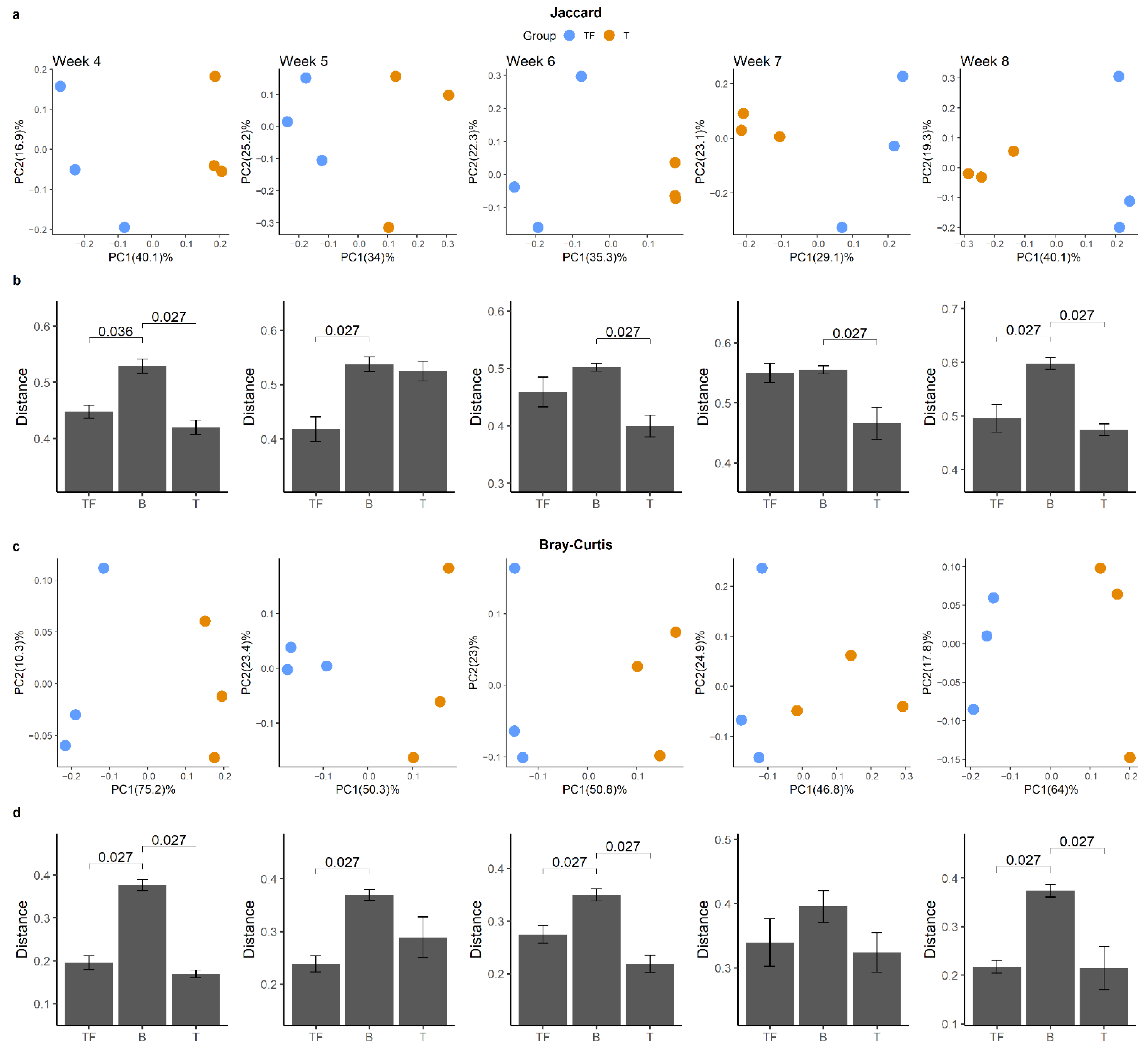
2.2. Distinct Microbiota Compositions in T and TF Groups
2.2.1. No Significant Difference in Alpha Diversity Indexes between T and TF Groups
2.2.2. Separation of T and TF Groups in Beta Diversity
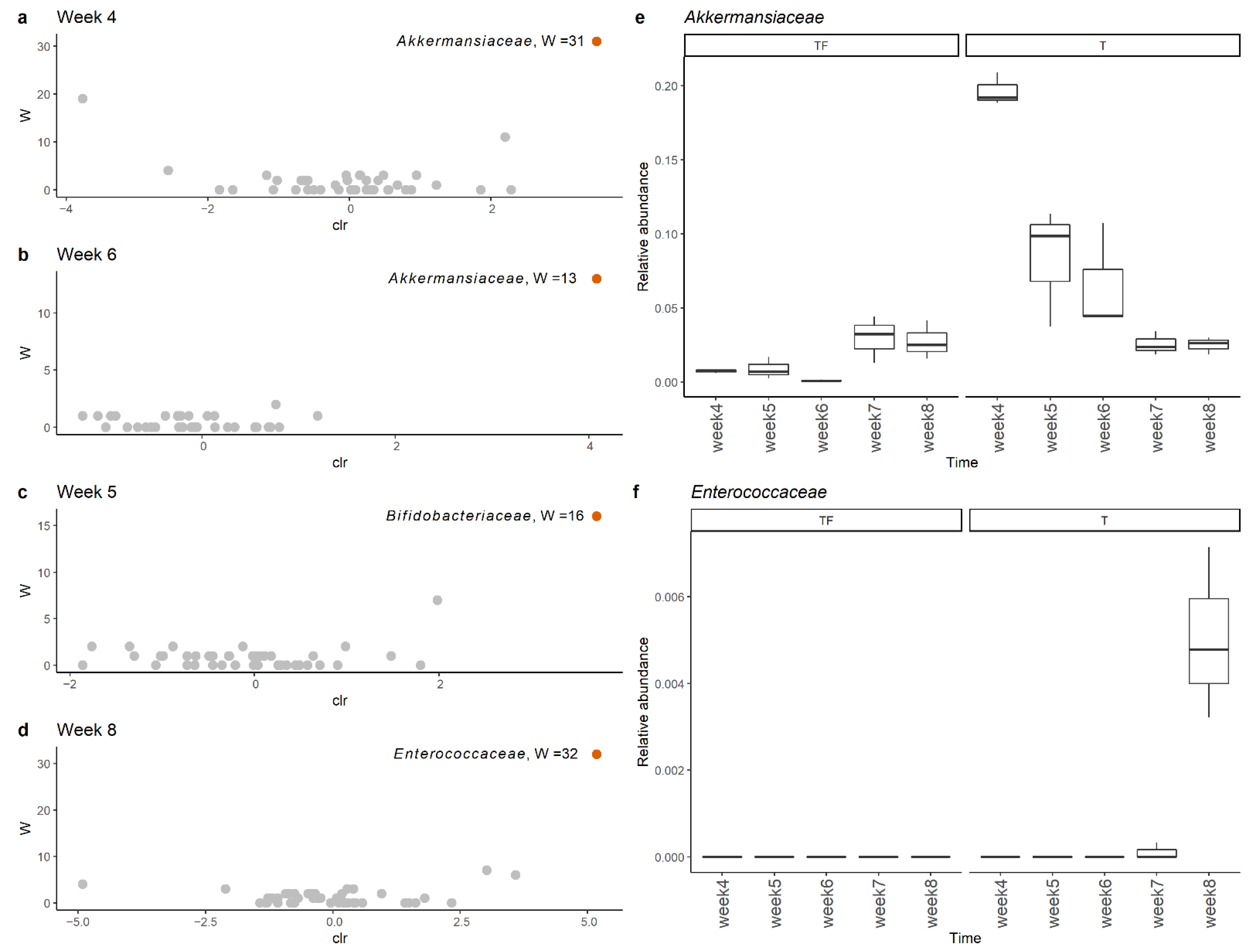
2.3. Detection of Different Abundant Bacteria Family between T and TF Groups
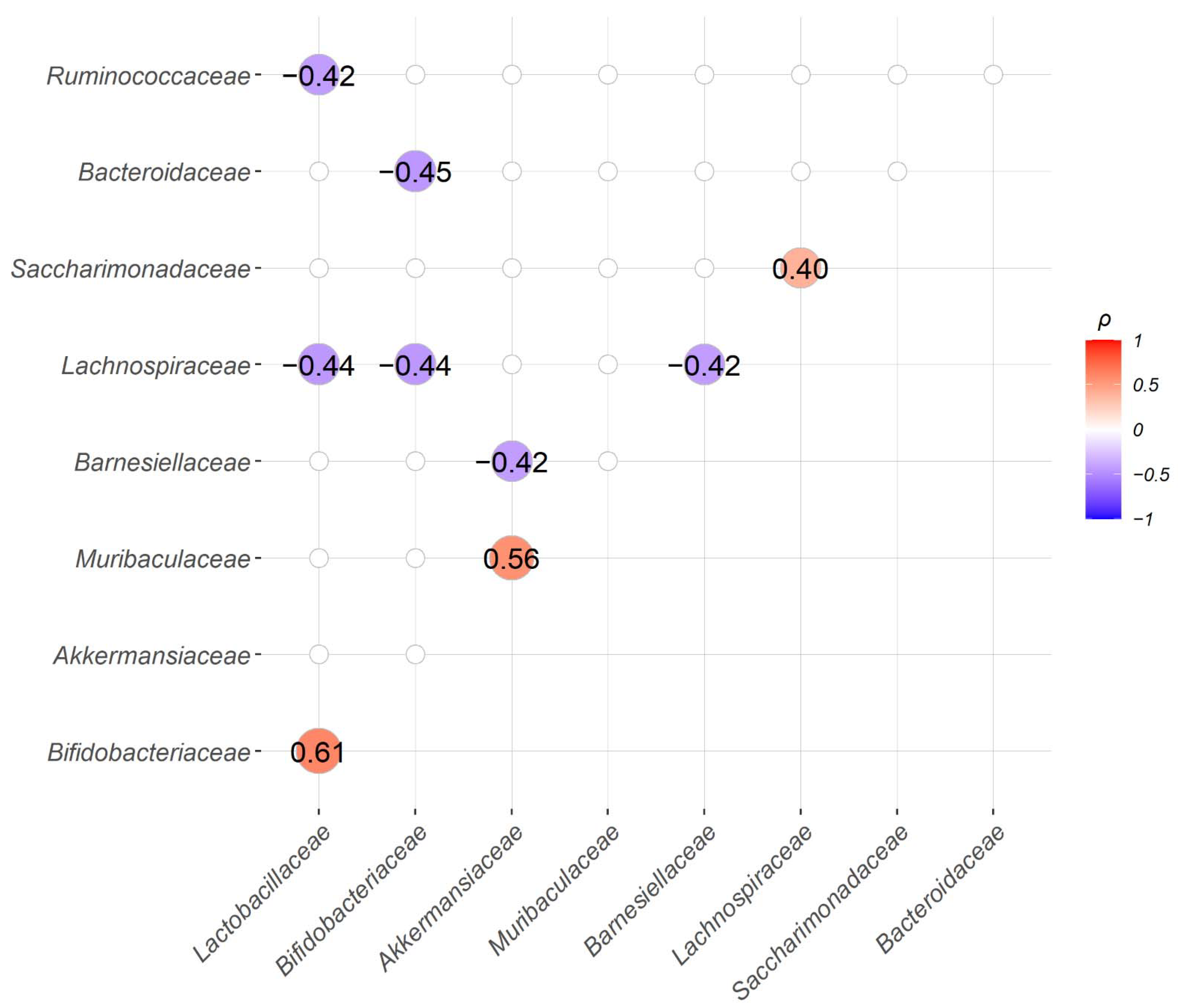
2.4. Correlation between the Abundance of Bacteria Families
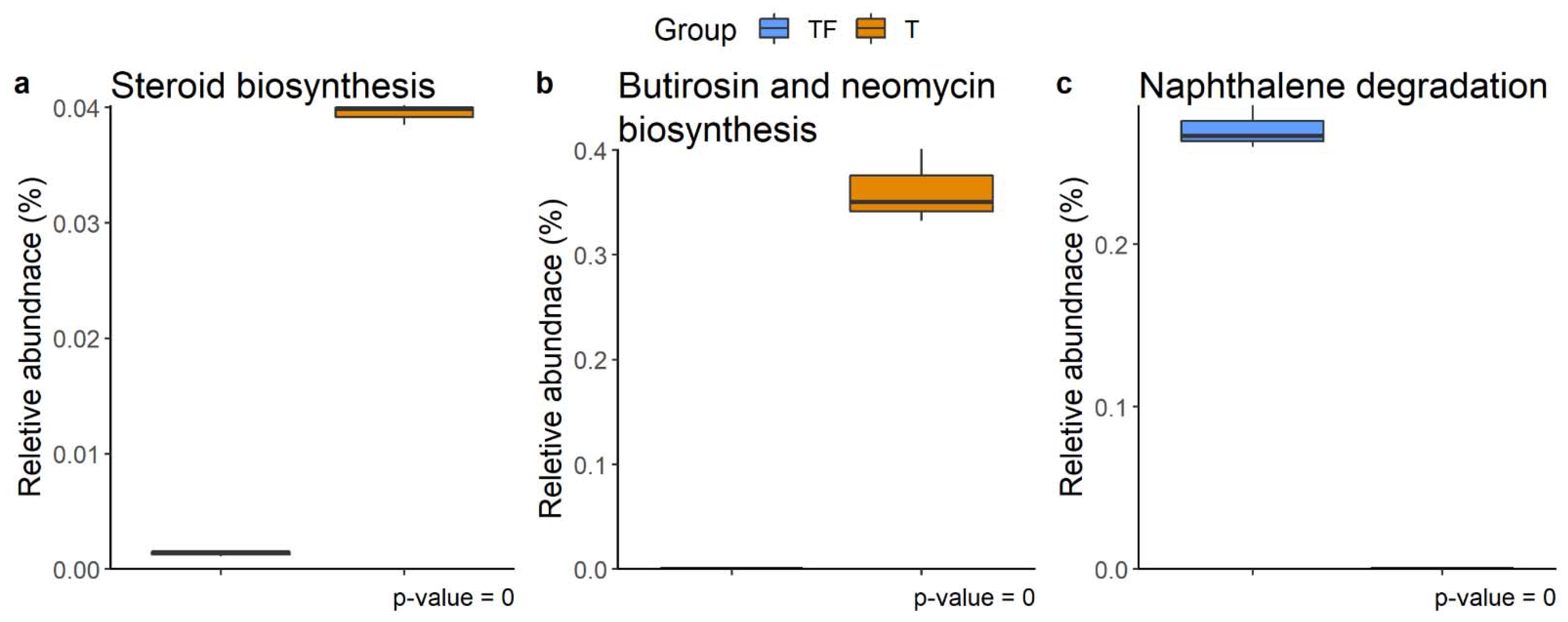
2.5. Function Prediction for Microbiota in T and TF Groups
3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.2. Sample Collection and DNA Extraction
4.3. 16S rRNA Gene Amplification and Sequencing
4.4. Sequence Preprocessing and Taxonomy Classification
4.5. Diversity and Correlation Analysis
4.6. Detection of Different Abundant Bacteria Families and Function Prediction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID * | Raw PE | Filtered Tags | Filtered Tags (%) | Merged Tags | Merged Tags (%) | Nonchimeric Tags | Effective Tags (%) |
|---|---|---|---|---|---|---|---|
| TF1.W4 | 123,903 | 79,394 | 64.08% | 72,872 | 58.81% | 64,776 | 52.28% |
| TF1.W5 | 86,941 | 55,172 | 63.46% | 50,744 | 58.37% | 45,633 | 52.49% |
| TF1.W6 | 101,821 | 65,428 | 64.26% | 58,992 | 57.94% | 54,234 | 53.26% |
| TF1.W7 | 138,003 | 88,307 | 63.99% | 80,465 | 58.31% | 73,880 | 53.54% |
| TF1.W8 | 98,923 | 61,695 | 62.37% | 55,449 | 56.05% | 50,946 | 51.50% |
| TF2.W4 | 117,882 | 75,321 | 63.90% | 69,764 | 59.18% | 62,166 | 52.74% |
| TF2.W5 | 112,773 | 66,447 | 58.92% | 61,495 | 54.53% | 55,681 | 49.37% |
| TF2.W6 | 105,421 | 65,833 | 62.45% | 59,271 | 56.22% | 54,147 | 51.36% |
| TF2.W7 | 118,204 | 76,548 | 64.76% | 71,142 | 60.19% | 66,869 | 56.57% |
| TF2.W8 | 88,600 | 55,839 | 63.02% | 50,175 | 56.63% | 45,956 | 51.87% |
| TF3.W4 | 112,476 | 72,692 | 64.63% | 66,330 | 58.97% | 58,931 | 52.39% |
| TF3.W5 | 95,308 | 60,193 | 63.16% | 55,567 | 58.30% | 50,921 | 53.43% |
| TF3.W6 | 102,185 | 64,955 | 63.57% | 58,532 | 57.28% | 52,719 | 51.59% |
| TF3.W7 | 92,117 | 56,548 | 61.39% | 51,256 | 55.64% | 47,089 | 51.12% |
| TF3.W8 | 89,963 | 58,077 | 64.56% | 52,419 | 58.27% | 48,176 | 53.55% |
| T1.W4 | 302,001 | 205,657 | 68.10% | 188,734 | 62.49% | 165,859 | 54.92% |
| T1.W5 | 112,046 | 68,051 | 60.73% | 62,878 | 56.12% | 57,506 | 51.32% |
| T1.W6 | 118,202 | 77,451 | 65.52% | 68,632 | 58.06% | 61,049 | 51.65% |
| T1.W7 | 94,575 | 53,614 | 56.69% | 48,405 | 51.18% | 46,262 | 48.92% |
| T1.W8 | 107,927 | 67,024 | 62.10% | 59,844 | 55.45% | 56,116 | 51.99% |
| T2.W4 | 132,767 | 83,571 | 62.95% | 76,393 | 57.54% | 69,621 | 52.44% |
| T2.W5 | 126,506 | 80,552 | 63.67% | 74,922 | 59.22% | 71,227 | 56.30% |
| T2.W6 | 124,477 | 79,001 | 63.47% | 70,664 | 56.77% | 64,270 | 51.63% |
| T2.W7 | 79,886 | 52,599 | 65.84% | 47,511 | 59.47% | 44,677 | 55.93% |
| T2.W8 | 105,825 | 69,527 | 65.70% | 62,486 | 59.05% | 57,150 | 54.00% |
| T3.W4 | 128,444 | 84,426 | 65.73% | 77,355 | 60.22% | 71,235 | 55.46% |
| T3.W5 | 108,635 | 68,023 | 62.62% | 62,412 | 57.45% | 59,490 | 54.76% |
| T3.W6 | 128,206 | 83,377 | 65.03% | 75,457 | 58.86% | 68,640 | 53.54% |
| T3.W7 | 76,829 | 46,952 | 61.11% | 42,923 | 55.87% | 41,842 | 54.46% |
| T3.W8 | 94,509 | 60,704 | 64.23% | 54,762 | 57.94% | 50,392 | 53.32% |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Fang, L.-Y.; Izumi, K.; Lai, K.-P.; Liang, L.; Li, L.; Miyamoto, H.; Lin, W.-J.; Chang, C. Infiltrating macrophages promote prostate tumorigenesis via modulating androgen receptor-mediated CCL4–STAT3 signaling. Cancer Res. 2013, 73, 5633–5646. [Google Scholar] [CrossRef] [Green Version]
- Sfanos, K.S.; Bruno, T.C.; Maris, C.H.; Xu, L.; Thoburn, C.J.; DeMarzo, A.M.; Meeker, A.K.; Isaacs, W.B.; Drake, C.G. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin. Cancer Res. 2008, 14, 3254–3261. [Google Scholar] [CrossRef] [Green Version]
- Vykhovanets, E.V.; MacLennan, G.T.; Vykhovanets, O.V.; Gupta, S. IL-17 Expression by macrophages is associated with proliferative inflammatory atrophy lesions in prostate cancer patients. Int. J. Clin. Exp. Pathol. 2011, 4, 552. [Google Scholar]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- De Marzo, A.M.; Platz, E.A.; Sutcliffe, S.; Xu, J.; Grönberg, H.; Drake, C.G.; Nakai, Y.; Isaacs, W.B.; Nelson, W.G. Inflammation in prostate carcinogenesis. Nat. Rev. Cancer 2007, 7, 256–269. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Trinchieri, G. Microbiota: A key orchestrator of cancer therapy. Nat. Rev. Cancer 2017, 17, 271–285. [Google Scholar] [CrossRef]
- Bashiardes, S.; Tuganbaev, T.; Federici, S.; Elinav, E. The Microbiome in Anti-Cancer Therapy. In Seminars in Immunology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 74–81. [Google Scholar]
- Lynch, S.V.; Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Jan, G.; Belzacq, A.; Haouzi, D.; Rouault, A.; Metivier, D.; Kroemer, G.; Brenner, C. Propionibacteria induce apoptosis of colorectal carcinoma cells via short-chain fatty acids acting on mitochondria. Cell Death Differ. 2002, 9, 179–188. [Google Scholar] [CrossRef]
- Paulos, C.M.; Wrzesinski, C.; Kaiser, A.; Hinrichs, C.S.; Chieppa, M.; Cassard, L.; Palmer, D.C.; Boni, A.; Muranski, P.; Yu, Z. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J. Clin. Investig. 2007, 117, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, M.; Del Carmen, S.; Cortes-Perez, N.G.; Lozano-Ojalvo, D.; Muñoz-Provencio, D.; Chain, F.; Langella, P.; de LeBlanc, A.d.M.; LeBlanc, J.G.; Bermúdez-Humarán, L.G. RETRACTED ARTICLE: Lactobacillus casei BL23 regulates T reg and Th17 T-cell populations and reduces DMH-associated colorectal cancer. J. Gastroenterol. 2016, 51, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Vivarelli, S.; Salemi, R.; Candido, S.; Falzone, L.; Santagati, M.; Stefani, S.; Torino, F.; Banna, G.L.; Tonini, G.; Libra, M. Gut microbiota and cancer: From pathogenesis to therapy. Cancers 2019, 11, 38. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, M. Structure and function of Helicobacter pylori CagA, the first-identified bacterial protein involved in human cancer. Proc. Jpn. Acad. Ser. B 2017, 93, 196–219. [Google Scholar] [CrossRef] [Green Version]
- Lara-Tejero, M.; Galán, J.E. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 2000, 290, 354–357. [Google Scholar] [CrossRef] [Green Version]
- Buti, L.; Spooner, E.; Van der Veen, A.G.; Rappuoli, R.; Covacci, A.; Ploegh, H.L. Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc. Natl. Acad. Sci. USA 2011, 108, 9238–9243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poutahidis, T.; Kleinewietfeld, M.; Erdman, S. Gut microbiota and the paradox of cancer immunotherapy. Front. Immunol. 2014, 5, 157. [Google Scholar] [CrossRef] [PubMed]
- Sfanos, K.S.; Yegnasubramanian, S.; Nelson, W.G.; De Marzo, A.M. The inflammatory microenvironment and microbiome in prostate cancer development. Nat. Rev. Urol. 2018, 15, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Sfanos, K.S.; Markowski, M.C.; Peiffer, L.B.; Ernst, S.E.; White, J.R.; Pienta, K.J.; Antonarakis, E.S.; Ross, A.E. Compositional differences in gastrointestinal microbiota in prostate cancer patients treated with androgen axis-targeted therapies. Prostate Cancer Prostatic Dis. 2018, 21, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Mollica, V.; Di Nunno, V.; Cimadamore, A.; Lopez-Beltran, A.; Cheng, L.; Santoni, M.; Scarpelli, M.; Montironi, R.; Massari, F. Molecular mechanisms related to hormone inhibition resistance in prostate cancer. Cells 2019, 8, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J.; Ganesh, K. Metastasis-Initiating Cells and Ecosystems. Cancer Discov. 2021, 11, 971–994. [Google Scholar] [CrossRef]
- Risson, E.; Nobre, A.R.; Maguer-Satta, V.; Aguirre-Ghiso, J.A. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat. Cancer 2020, 1, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ahn, E.H.; Kang, S.S.; Liu, X.; Alam, A.; Ye, K. Gut dysbiosis contributes to amyloid pathology, associated with C/EBPbeta/AEP signaling activation in Alzheimer’s disease mouse model. Sci. Adv. 2020, 6, eaba0466. [Google Scholar] [CrossRef]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbo, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, Y.; Wang, G.; Han, R.; Xie, X. Effects of chlorpyrifos on the gut microbiome and urine metabolome in mouse (Mus musculus). Chemosphere 2016, 153, 287–293. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Mayneris-Perxachs, J.; Arnoriaga-Rodriguez, M.; Luque-Cordoba, D.; Priego-Capote, F.; Perez-Brocal, V.; Moya, A.; Burokas, A.; Maldonado, R.; Fernandez-Real, J.M. Gut microbiota steroid sexual dimorphism and its impact on gonadal steroids: Influences of obesity and menopausal status. Microbiome 2020, 8, 136. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Ikegawa, S.; Alves, J.M.P.; Zhou, B.; Kobayashi, A.; Iida, T.; Mitamura, K.; Tanabe, G.; Serrano, M.; De Guzman, A.; et al. Clostridium scindens: A human gut microbe with a high potential to convert glucocorticoids into androgens. J. Lipid. Res. 2013, 54, 2437–2449. [Google Scholar] [CrossRef] [Green Version]
- Kwa, M.; Plottel, C.S.; Blaser, M.J.; Adams, S. The Intestinal Microbiome and Estrogen Receptor-Positive Female Breast Cancer. J. Natl. Cancer Inst. 2016, 108, djw029. [Google Scholar]
- Severi, G.; Morris, H.A.; MacInnis, R.J.; English, D.R.; Tilley, W.; Hopper, J.L.; Boyle, P.; Giles, G.G. Circulating steroid hormones and the risk of prostate cancer. Cancer Epidemiol. Biomark. Prev. 2006, 15, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Sharifi, N.; Auchus, R.J. Steroid biosynthesis and prostate cancer. Steroids 2012, 77, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.N.; Dong, Y.; Mu, Y.Z.; Yan, J.; Lu, M.; Zhu, Y.L.; Zhang, G.X. Identification of gastric microbiota biomarker for gastric cancer. Chin. Med. J. 2020, 133, 2765–2767. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liu, A.; Lu, X.; Zhang, Z.; Xue, Y.; Xu, J.; Zeng, S.; Xiong, Q.; Tan, H.; He, X.; et al. Dysbiosis signatures of the microbial profile in tissue from bladder cancer. Cancer Med. 2019, 8, 6904–6914. [Google Scholar] [CrossRef] [Green Version]
- Armour, C.R.; Nayfach, S.; Pollard, K.S.; Sharpton, T.J. A Metagenomic Meta-analysis Reveals Functional Signatures of Health and Disease in the Human Gut Microbiome. mSystems 2019, 4, e00332-18. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhao, L.; Han, L.; Fu, G.; Tuo, X.; Ma, S.; Li, Q.; Wang, Y.; Liang, D.; Tang, M.; et al. The differential distribution of bacteria between cancerous and noncancerous ovarian tissues in situ. J. Ovarian Res. 2020, 13, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, C.H.; Krych, L.; Buschard, K.; Metzdorff, S.B.; Nellemann, C.; Hansen, L.H.; Nielsen, D.S.; Frokiaer, H.; Skov, S.; Hansen, A.K. A maternal gluten-free diet reduces inflammation and diabetes incidence in the offspring of NOD mice. Diabetes 2014, 63, 2821–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, C.H.; Krych, L.; Nielsen, D.S.; Vogensen, F.K.; Hansen, L.H.; Sorensen, S.J.; Buschard, K.; Hansen, A.K. Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia 2012, 55, 2285–2294. [Google Scholar] [CrossRef] [Green Version]
- Roopchand, D.E.; Carmody, R.N.; Kuhn, P.; Moskal, K.; Rojas-Silva, P.; Turnbaugh, P.J.; Raskin, I. Dietary Polyphenols Promote Growth of the Gut Bacterium Akkermansia muciniphila and Attenuate High-Fat Diet-Induced Metabolic Syndrome. Diabetes 2015, 64, 2847–2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.R.; Han, K.; Han, Y.; Kang, N.; Shin, T.S.; Park, H.J.; Kim, H.; Kwon, W.; Lee, S.; Kim, Y.K.; et al. Microbiome Markers of Pancreatic Cancer Based on Bacteria-Derived Extracellular Vesicles Acquired from Blood Samples: A Retrospective Propensity Score Matching Analysis. Biology 2021, 10, 219. [Google Scholar] [CrossRef]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Ganesh, B.P.; Klopfleisch, R.; Loh, G.; Blaut, M. Commensal Akkermansia muciniphila exacerbates gut inflammation in Salmonella Typhimurium-infected gnotobiotic mice. PLoS ONE 2013, 8, e74963. [Google Scholar] [CrossRef]
- Dingemanse, C.; Belzer, C.; van Hijum, S.A.; Gunthel, M.; Salvatori, D.; den Dunnen, J.T.; Kuijper, E.J.; Devilee, P.; de Vos, W.M.; van Ommen, G.B.; et al. Akkermansia muciniphila and Helicobacter typhlonius modulate intestinal tumor development in mice. Carcinogenesis 2015, 36, 1388–1396. [Google Scholar] [CrossRef] [Green Version]
- Weir, T.L.; Manter, D.K.; Sheflin, A.M.; Barnett, B.A.; Heuberger, A.L.; Ryan, E.P. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS ONE 2013, 8, e70803. [Google Scholar] [CrossRef] [Green Version]
- Howe, C.; Kim, S.J.; Mitchell, J.; Im, E.; Kim, Y.S.; Kim, Y.S.; Rhee, S.H. Differential expression of tumor-associated genes and altered gut microbiome with decreased Akkermansia muciniphila confer a tumor-preventive microenvironment in intestinal epithelial Pten-deficient mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3746–3758. [Google Scholar] [CrossRef]
- Nagalingam, N.A.; Kao, J.Y.; Young, V.B. Microbial ecology of the murine gut associated with the development of dextran sodium sulfate-induced colitis. Inflamm. Bowel. Dis. 2011, 17, 917–926. [Google Scholar] [CrossRef]
- An, J.; Ha, E.M. Combination Therapy of Lactobacillus plantarum Supernatant and 5-Fluouracil Increases Chemosensitivity in Colorectal Cancer Cells. J. Microbiol. Biotechnol. 2016, 26, 1490–1503. [Google Scholar] [CrossRef]
- Luo, Z.W.; Xia, K.; Liu, Y.W.; Liu, J.H.; Rao, S.S.; Hu, X.K.; Chen, C.Y.; Xu, R.; Wang, Z.X.; Xie, H. Extracellular Vesicles from Akkermansia muciniphila Elicit Antitumor Immunity Against Prostate Cancer via Modulation of CD8(+) T Cells and Macrophages. Int. J. Nanomed. 2021, 16, 2949–2963. [Google Scholar] [CrossRef]
- Mira-Pascual, L.; Cabrera-Rubio, R.; Ocon, S.; Costales, P.; Parra, A.; Suarez, A.; Moris, F.; Rodrigo, L.; Mira, A.; Collado, M.C. Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. J. Gastroenterol. 2015, 50, 167–179. [Google Scholar] [CrossRef]
- Mima, K.; Nakagawa, S.; Sawayama, H.; Ishimoto, T.; Imai, K.; Iwatsuki, M.; Hashimoto, D.; Baba, Y.; Yamashita, Y.I.; Yoshida, N.; et al. The microbiome and hepatobiliary-pancreatic cancers. Cancer Lett. 2017, 402, 9–15. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Yang, L.; Peng, G.; Yang, K.; Mi, Y.; Hu, X.; Hao, X.; Jiao, Y.; Wang, X.; Wang, Y. The commensal consortium of the gut microbiome is associated with favorable responses to anti-programmed death protein 1 (PD-1) therapy in thoracic neoplasms. Cancer Biol. Med. 2021, 18, 1–13. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Geier, M.S.; Butler, R.N.; Howarth, G.S. Probiotics, prebiotics and synbiotics: A role in chemoprevention for colorectal cancer? Cancer Biol. Ther. 2006, 5, 1265–1269. [Google Scholar] [CrossRef] [Green Version]
- Konishi, H.; Fujiya, M.; Tanaka, H.; Ueno, N.; Moriichi, K.; Sasajima, J.; Ikuta, K.; Akutsu, H.; Tanabe, H.; Kohgo, Y. Probiotic-derived ferrichrome inhibits colon cancer progression via JNK-mediated apoptosis. Nat. Commun. 2016, 7, 12365. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianotti, L.; Morelli, L.; Galbiati, F.; Rocchetti, S.; Coppola, S.; Beneduce, A.; Gilardini, C.; Zonenschain, D.; Nespoli, A.; Braga, M. A randomized double-blind trial on perioperative administration of probiotics in colorectal cancer patients. World J. Gastroenterol. 2010, 16, 167–175. [Google Scholar] [CrossRef]
- Garber, K. First microbiome-based drug clears phase III, in clinical trial turnaround. Nat. Rev. Drug Discov. 2020, 19, 655–656. [Google Scholar] [CrossRef]
- Markle, J.G.; Frank, D.N.; Mortin-Toth, S.; Robertson, C.E.; Feazel, L.M.; Rolle-Kampczyk, U.; von Bergen, M.; McCoy, K.D.; Macpherson, A.J.; Danska, J.S. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013, 339, 1084–1088. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.M.; Sun, E.W.; Rogers, G.B.; Keating, D.J. The Influence of the Gut Microbiome on Host Metabolism Through the Regulation of Gut Hormone Release. Front. Physiol. 2019, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Sha, S.; Ni, L.; Stefil, M.; Dixon, M.; Mouraviev, V. The human gastrointestinal microbiota and prostate cancer development and treatment. Investig. Clin. Urol. 2020, 61 (Suppl. 1), S43–S50. [Google Scholar] [CrossRef]
- Tsai, C.H.; Tzeng, S.F.; Chao, T.K.; Tsai, C.Y.; Yang, Y.C.; Lee, M.T.; Hwang, J.J.; Chou, Y.C.; Tsai, M.H.; Cha, T.L.; et al. Metastatic Progression of Prostate Cancer Is Mediated by Autonomous Binding of Galectin-4-O-Glycan to Cancer Cells. Cancer Res. 2016, 76, 5756–5767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation Evaluation and Phylogenetic Diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Whittaker, R.H. Evolution and Measurement of Species Diversity. TAXON 1972, 21, 213–251. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, P.-Y.; Yang, Y.-C.; Wang, C.-I.; Hsiao, P.-W.; Chiang, H.-I.; Chen, T.-W. Increase in Akkermansiaceae in Gut Microbiota of Prostate Cancer-Bearing Mice. Int. J. Mol. Sci. 2021, 22, 9626. https://doi.org/10.3390/ijms22179626
Huang P-Y, Yang Y-C, Wang C-I, Hsiao P-W, Chiang H-I, Chen T-W. Increase in Akkermansiaceae in Gut Microbiota of Prostate Cancer-Bearing Mice. International Journal of Molecular Sciences. 2021; 22(17):9626. https://doi.org/10.3390/ijms22179626
Chicago/Turabian StyleHuang, Pin-Yu, Yu-Chih Yang, Chun-I Wang, Pei-Wen Hsiao, Hsin-I Chiang, and Ting-Wen Chen. 2021. "Increase in Akkermansiaceae in Gut Microbiota of Prostate Cancer-Bearing Mice" International Journal of Molecular Sciences 22, no. 17: 9626. https://doi.org/10.3390/ijms22179626
APA StyleHuang, P.-Y., Yang, Y.-C., Wang, C.-I., Hsiao, P.-W., Chiang, H.-I., & Chen, T.-W. (2021). Increase in Akkermansiaceae in Gut Microbiota of Prostate Cancer-Bearing Mice. International Journal of Molecular Sciences, 22(17), 9626. https://doi.org/10.3390/ijms22179626