
The Ubiquitin System: An Emerging Therapeutic Target for Lung Cancer

 ,
,  ,
,  and
and
Abstract
:1. Introduction
1.1. Lung Cancer
1.2. Small Cell Lung Cancer (SCLC)
1.3. Nonsmall Cell Lung Cancer (NSCLC)
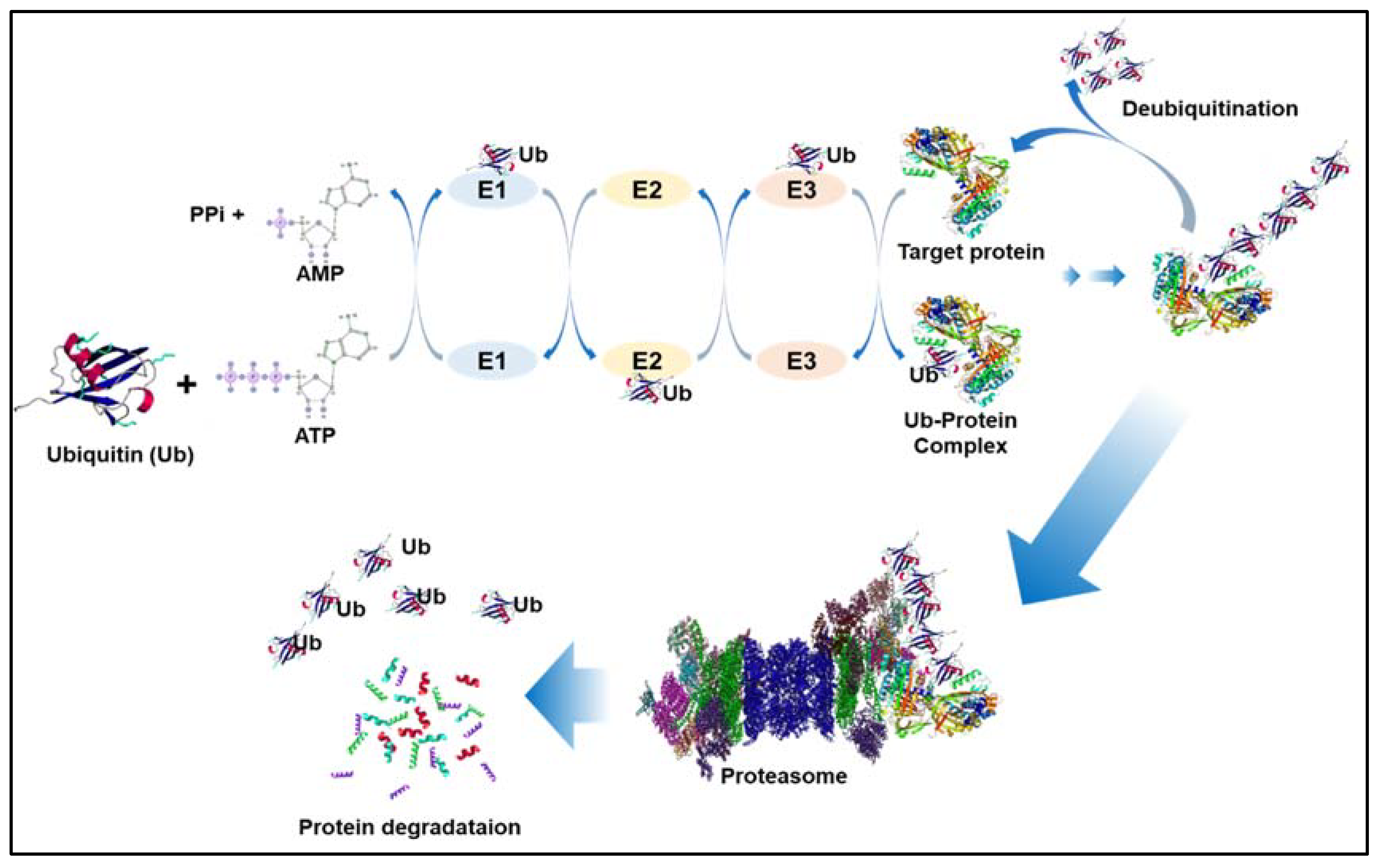
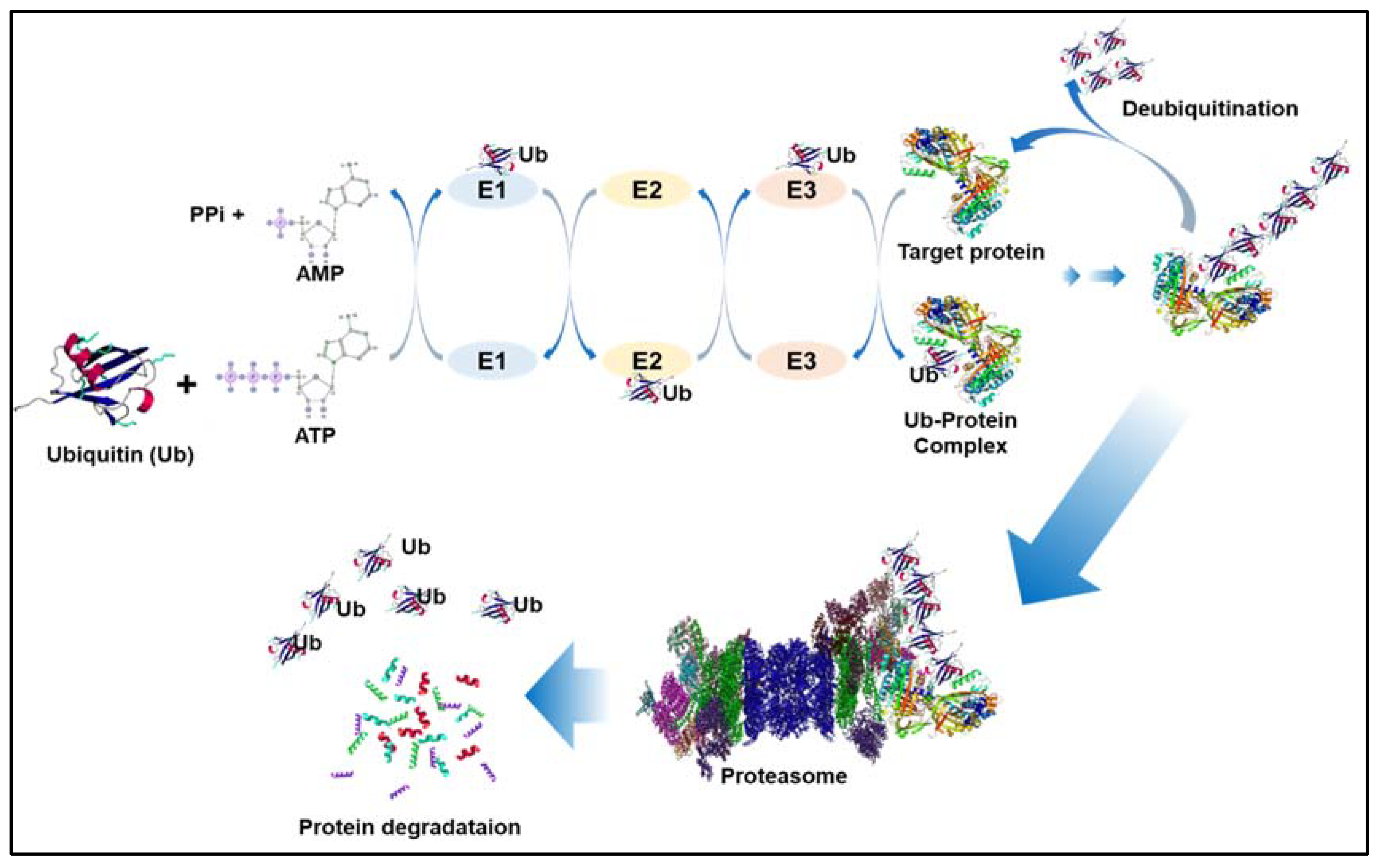
2. Ubiquitination and Deubiquitination
3. Ubiquitination, Deubiquitination and Lung Cancer
4. Ubiquitination and Deubiquitination-Mediated Therapy for Lung Cancer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cao, B.; Bray, F.; Ilbawi, A.; Soerjomataram, I. Effect on longevity of one-third reduction in premature mortality from non-communicable diseases by 2030: A global analysis of the Sustainable Development Goal health target. Lancet Glob. Health 2018, 6, e1288–e1296. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- WHO. WHO Report on Cancer: Setting Priorities, Investing Wisely and Providing Care for All; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Collins, L.G.; Haines, C.; Perkel, R.; Enck, R.E. Lung cancer: Diagnosis and management. Am. Fam. Physician 2007, 75, 56–63. [Google Scholar] [PubMed]
- Nasim, F.; Sabath, B.F.; Eapen, G.A. Lung Cancer. Med. Clin. N. Am. 2019, 103, 463–473. [Google Scholar] [CrossRef]
- Dai, J.; Yang, P.; Cox, A.; Jiang, G. Lung cancer and chronic obstructive pulmonary disease: From a clinical perspective. Oncotarget 2017, 8, 18513–18524. [Google Scholar] [CrossRef] [Green Version]
- Travis, W.D. Pathology of lung cancer. Clin. Chest Med. 2011, 32, 669–692. [Google Scholar] [CrossRef]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non–Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef] [PubMed]
- Raso, M.; Bota-Rabassedas, N.; Wistuba, I. Pathology and Classification of SCLC. Cancers 2021, 13, 820. [Google Scholar] [CrossRef] [PubMed]
- Zakowski, M.F. Pathology of small cell carcinoma of the lung. Semin. Oncol. 2003, 30, 3–8. [Google Scholar] [CrossRef]
- Ruiz-Cordero, R.; Devine, W.P. Targeted Therapy and Checkpoint Immunotherapy in Lung Cancer. Surg. Pathol. Clin. 2020, 13, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Chanvorachote, P.; Sriratanasak, N.; Nonpanya, N. C-myc Contributes to Malignancy of Lung Cancer: A Potential Anticancer Drug Target. Anticancer. Res. 2020, 40, 609–618. [Google Scholar] [CrossRef]
- Ray, M.R.; Jablons, D.; He, B. Lung cancer therapeutics that target signaling pathways: An update. Expert Rev. Respir. Med. 2010, 4, 631–645. [Google Scholar] [CrossRef] [Green Version]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B. The 2015 World Health Organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, X.; Lu, M. Abnormal Ubiquitination of Ubiquitin-Proteasome System in Lung Squamous Cell Carcinomas. In Ubiquitin-Proteasome Pathway; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Rodriguez-Canales, J.; Parra-Cuentas, E.; Wistuba, I.I. Diagnosis and Molecular Classification of Lung Cancer. Lung Cancer 2016, 170, 25–46. [Google Scholar] [CrossRef]
- Brambilla, E.; Gazdar, A. Pathogenesis of lung cancer signalling pathways: Roadmap for therapies. Eur. Respir. J. 2009, 33, 1485–1497. [Google Scholar] [CrossRef]
- Zhang, X.; Linder, S.; Bazzaro, M. Drug Development Targeting the Ubiquitin–Proteasome System (UPS) for the Treatment of Human Cancers. Cancers 2020, 12, 902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyer, M.L.; Milhollen, M.A.; Ciavarri, J.; Fleming, P.; Traore, T.; Sappal, D.; Huck, J.; Shi, J.; Gavin, J.; Brownell, J.; et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat. Med. 2018, 24, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Meng, T.; Cui, S.; Feng, L.; Liu, D.; Pang, Q.; Wang, P. Ubiquitination of Nonhistone Proteins in Cancer Development and Treatment. Front. Oncol. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, M.; Nicholson, B.; Kessler, B. Pharmacological targets in the ubiquitin system offer new ways of treating cancer, neurodegenerative disorders and infectious diseases. Expert Rev. Mol. Med. 2011, 13, e35. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Cho, J.; Song, E.J. Ubiquitin–proteasome system (UPS) as a target for anticancer treatment. Arch. Pharmacal Res. 2020, 43, 1144–1161. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herhaus, L.; Dikic, I. Expanding the ubiquitin code through post-translational modification. EMBO Rep. 2015, 16, 1071–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Ye, Y. Polyubiquitin chains: Functions, structures, and mechanisms. Experientia 2008, 65, 2397–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular functions and molecular mechanisms of non-lysine ubiquitination. Open Biol. 2019, 9, 190147. [Google Scholar] [CrossRef] [Green Version]
- Buetow, L.; Huang, D.T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016, 17, 626. [Google Scholar] [CrossRef] [Green Version]
- Berndsen, C.; Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.-H.; Chen, Y.-H.; Huang, T.-Y. Ubiquitin-mediated regulation of autophagy. J. Biomed. Sci. 2019, 26, 1–12. [Google Scholar] [CrossRef]
- Brinkmann, K.; Schell, M.; Hoppe, T.; Kashkar, H. Regulation of the DNA damage response by ubiquitin conjugation. Front. Genet. 2015, 6, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Ronai, Z.A. Dysregulation of ubiquitin ligases in cancer. Drug Resist. Updat. 2015, 23, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, A.J.; Hoffiz, Y.C.; Charles, A.J.; Zhu, Y.; Mabb, A.M. A Comprehensive Atlas of E3 Ubiquitin Ligase Mutations in Neurological Disorders. Front. Genet. 2018, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, N.; Lagopati, N.; Balourdas, D.-I.; Nikolaou, M.; Papalampros, A.; Vasileiou, P.V.S.; Myrianthopoulos, V.; Kotsinas, A.; Shiloh, Y.; Liontos, M.; et al. The Role of E3, E4 Ubiquitin Ligase (UBE4B) in Human Pathologies. Cancers 2019, 12, 62. [Google Scholar] [CrossRef] [Green Version]
- Bui, Q.; Hong, J.; Kwak, M.; Lee, J.; Lee, P. Ubiquitin-Conjugating Enzymes in Cancer. Cells 2021, 10, 1383. [Google Scholar] [CrossRef]
- Rodríguez-Alonso, A.; Casas-Pais, A.; Roca-Lema, D.; Graña, B.; Romay, G.; Figueroa, A. Regulation of Epithelial–Mesenchymal Plasticity by the E3 Ubiquitin-Ligases in Cancer. Cancers 2020, 12, 3093. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. The Ubiquitin System, Autophagy, and Regulated Protein Degradation. Annu. Rev. Biochem. 2017, 86, 123–128. [Google Scholar] [CrossRef]
- Khalil, R. Ubiquitin-Proteasome Pathway and Muscle Atrophy. Adv. Exp. Med. Biol. 2018, 1088, 235–248. [Google Scholar] [CrossRef]
- Sun, Y. E3 Ubiquitin Ligases as Cancer Targets and Biomarkers. Neoplasia 2006, 8, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.; Wang, Q.; Cai, R.; Yuan, H.; Xu, M. The ubiquitin system: Orchestrating cellular signals in non-small-cell lung cancer. Cell. Mol. Biol. Lett. 2020, 25, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.; Zhou, Z.; Shah, A.A.; Zou, H.; Tao, J.; Chen, Q.; Wan, Y. The emerging role of deubiquitinating enzymes in genomic integrity, diseases, and therapeutics. Cell Biosci. 2016, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Confalonieri, S.; Quarto, M.; Goisis, G.; Nuciforo, P.; Donzelli, M.; Jodice, G.; Pelosi, G.; Viale, G.; Pece, S.; Di Fiore, P.P. Alterations of ubiquitin ligases in human cancer and their association with the natural history of the tumor. Oncogene 2009, 28, 2959–2968. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Liu, Z.; Yang, Q. The role of ubiquitination and deubiquitination in cancer metabolism. Mol. Cancer 2020, 19, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Satija, Y.K.; Bhardwaj, A.; Das, S. A portrayal of E3 ubiquitin ligases and deubiquitylases in cancer. Int. J. Cancer 2013, 133, 1–10. [Google Scholar] [CrossRef]
- Van Tol, B.D.; Geurink, P.P.; Ovaa, H. A MALDI-TOF Approach to Ubiquitin Ligase Activity. Cell Chem. Biol. 2018, 25, 1053–1055. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Dexheimer, T.S.; Ai, Y.; Liang, Q.; Villamil, M.A.; Inglese, J.; Maloney, D.J.; Jadhav, A.; Simeonov, A.; Zhuang, Z. Selective and Cell-Active Inhibitors of the USP1/ UAF1 Deubiquitinase Complex Reverse Cisplatin Resistance in Non-small Cell Lung Cancer Cells. Chem. Biol. 2011, 18, 1390–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Lu, J.; Zhang, Q.-W.; Zhao, W.; Guo, J.-H.; Liu, S.-L.; Wu, Y.-L.; Jiang, B.; Gao, F.-H. USP7 promotes cell proliferation through the stabilization of Ki-67 protein in non-small cell lung cancer cells. Int. J. Biochem. Cell Biol. 2016, 79, 209–221. [Google Scholar] [CrossRef]
- Byun, S.; Lee, S.-Y.; Lee, J.; Jeong, C.-H.; Farrand, L.; Lim, S.; Reddy, K.; Kim, J.Y.; Lee, M.-H.; Lee, H.J.; et al. USP8 Is a Novel Target for Overcoming Gefitinib Resistance in Lung Cancer. Clin. Cancer Res. 2013, 19, 3894–3904. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Liu, C.; Bai, C.; Han, Y.-P.; Cho, W.C.S.; Li, Q. Over-Expression of Deubiquitinating Enzyme USP14 in Lung Adenocarcinoma Promotes Proliferation through the Accumulation of β-Catenin. Int. J. Mol. Sci. 2013, 14, 10749–10760. [Google Scholar] [CrossRef]
- McFarlane, C.; McFarlane, S.; Paul, I.; Arthur, K.; Scheaff, M.; Kerr, K.; Stevenson, M.; Fennell, D.A.; Johnston, J.A. The deubiquitinating enzyme USP17 is associated with nonsmall cell lung cancer (NSCLC) recurrence and metastasis. Oncotarget 2013, 4, 1836–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Liu, N.; Zhao, Y.; Zhu, X.; Wang, C.; Liu, Q.; Gao, C.; Zhao, X.; Li, J. Oncogenic USP22 supports gastric cancer growth and metastasis by activating c-Myc/NAMPT/SIRT1-dependent FOXO1 and YAP signaling. Aging 2019, 11, 9643–9660. [Google Scholar] [CrossRef]
- Hu, J.; Yang, D.; Zhang, H.; Liu, W.; Zhao, Y.; Lu, H.; Meng, Q.; Pang, H.; Chen, X.; Liu, Y.; et al. USP22 promotes tumor progression and induces epithelial–mesenchymal transition in lung adenocarcinoma. Lung Cancer 2015, 88, 239–245. [Google Scholar] [CrossRef]
- Sun, X.-X.; He, X.; Yin, L.; Komada, M.; Sears, R.C.; Dai, M.-S. The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc. Natl. Acad. Sci. USA 2015, 112, 3734–3739. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Deng, Q.; Jiang, C.; Wang, X.; Niu, T.; Li, H.; Chen, T.; Jin, J.; Pan, W.; Cai, X.; et al. USP37 directly deubiquitinates and stabilizes c-Myc in lung cancer. Oncogene 2014, 34, 3957–3967. [Google Scholar] [CrossRef]
- Lin, Z.; Xiong, L.; Lin, Q. Ubiquitin-specific protease 39 is overexpressed in human lung cancer and promotes tumor cell proliferation in vitro. Mol. Cell. Biochem. 2016, 422, 97–107. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, W.; Zhang, R.; Zhang, Y.; Ma, H. Ubiquitin-specific protease 44 inhibits cell growth by suppressing AKT signaling in non-small cell lung cancer. Kaohsiung J. Med. Sci. 2019, 35, 535–541. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Han, B.; Lu, H.; Zhao, Y.; Chen, X.; Meng, Q.; Cao, M.; Cai, L.; Hu, J. USP22 promotes resistance to EGFR-TKIs by preventing ubiquitination-mediated EGFR degradation in EGFR-mutant lung adenocarcinoma. Cancer Lett. 2018, 433, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhang, G.; Li, X.; Ma, Q.; Cheng, W.; Wang, W.; Zhang, B.; Hu, T.; Song, G. Knocking down USP39 Inhibits the Growth and Metastasis of Non-Small-Cell Lung Cancer Cells through Activating the p53 Pathway. Int. J. Mol. Sci. 2020, 21, 8949. [Google Scholar] [CrossRef]
- Sriratanasak, N.; Petsri, K.; Laobuthee, A.; Wattanathana, W.; Vinayanuwattikun, C.; Luanpitpong, S.; Chanvorachote, P. Novel c-Myc–Targeting Compound N, N-Bis (5-Ethyl-2-Hydroxybenzyl) Methylamine for Mediated c-Myc Ubiquitin-Proteasomal Degradation in Lung Cancer Cells. Mol. Pharmacol. 2020, 98, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Z.; Zhang, L.; Yang, Z.; Chen, X.; Luo, J.; Zhou, Z.; Mei, X.; Yu, X.; Shao, Z.; et al. Targeting deubiquitinase USP28 for cancer therapy. Cell Death Dis. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Zaugg, K.; Mak, T.W.; Elledge, S.J. A Role for the Deubiquitinating Enzyme USP28 in Control of the DNA-Damage Response. Cell 2006, 126, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Xu, B.; Qiang, Y.; Huang, H.; Wang, C.; Li, D.; Qian, J. Overexpression of deubiquitinating enzyme USP 28 promoted non-small cell lung cancer growth. J. Cell. Mol. Med. 2015, 19, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, E.J.; Pinto-Fernandez, A.; Turnbull, A.P.; Lan, L.; Charlton, T.M.; Scott, H.C.; Damianou, A.; Vere, G.; Riising, E.M.; Costa, C.D.; et al. USP28 deletion and small molecule inhibition destabilises c-Myc and elicits regression of squamous cell lung carcinoma. bioRxiv 2021. [Google Scholar] [CrossRef]
- Li, P.; Huang, Z.; Wang, J.; Chen, W.; Huang, J. Ubiquitin-specific peptidase 28 enhances STAT3 signaling and promotes cell growth in non-small-cell lung cancer. Onco Targets Ther. 2019, 12, 1603–1611. [Google Scholar] [CrossRef] [Green Version]
- Ong, J.Y.; Torres, J.Z. E3 ubiquitin ligases in cancer and their pharmacological targeting. In Ubiquitin Proteasome System-Current Insights into Mechanism Cellular Regulation and Disease; IntechOpen: London, UK, 2019. [Google Scholar]
- Gu, J.; Mao, W.; Ren, W.; Xu, F.; Zhu, Q.; Lu, C.; Lin, Z.; Zhang, Z.; Chu, Y.; Liu, R.; et al. Ubiquitin-protein ligase E3C maintains non-small-cell lung cancer stemness by targeting AHNAK-p53 complex. Cancer Lett. 2018, 443, 125–134. [Google Scholar] [CrossRef]
- Hong, S.-Y.; Kao, Y.-R.; Lee, T.-C.; Wu, C.-W. Upregulation of E3 Ubiquitin Ligase CBLC Enhances EGFR Dysregulation and Signaling in Lung Adenocarcinoma. Cancer Res. 2018, 78, 4984–4996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Benchimol, S. Pirh2, a p53-Induced Ubiquitin-Protein Ligase, Promotes p53 Degradation. Cell 2003, 112, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Snoek, B.C.; De Wilt, L.H.; Jansen, G.; Peters, G.J. Role of E3 ubiquitin ligases in lung cancer. World J. Clin. Oncol. 2013, 4, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Gao, L.; Druhan, L.J.; Zhu, W.-G.; Morrison, C.; Otterson, G.A.; Villalona-Calero, M.A. Expression of Pirh2, a Newly Identified Ubiquitin Protein Ligase, in Lung Cancer. J. Natl. Cancer Inst. 2004, 96, 1718–1721. [Google Scholar] [CrossRef] [Green Version]
- Daks, A.; Petukhov, A.; Fedorova, O.; Shuvalov, O.; Merkulov, V.; Vasileva, E.; Antonov, A.; Barlev, N.A. E3 ubiquitin ligase Pirh2 enhances tumorigenic properties of human non-small cell lung carcinoma cells. Genes Cancer 2016, 7, 383–393. [Google Scholar] [CrossRef]
- Bernassola, F.; Chillemi, G.; Melino, G. HECT-Type E3 Ubiquitin Ligases in Cancer. Trends Biochem. Sci. 2019, 44, 1057–1075. [Google Scholar] [CrossRef]
- Qiao, X.; Liu, Y.; Prada, M.L.; Mohan, A.K.; Gupta, A.; Jaiswal, A.; Sharma, M.; Merisaari, J.; Haikala, H.M.; Talvinen, K.; et al. UBR5 Is Coamplified with MYC in Breast Tumors and Encodes an Ubiquitin Ligase That Limits MYC-Dependent Apoptosis. Cancer Res. 2020, 80, 1414–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammoudi, N.; Ahmed, K.B.R.; Garcia-Prieto, C.; Huang, P. Metabolic alterations in cancer cells and therapeutic implications. Chin. J. Cancer 2011, 30, 508–525. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Grossman, S.R. Ubiquitin becomes ubiquitous in cancer. Cancer Biol. Ther. 2010, 10, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bednash, J.S.; Mallampalli, R.K. Targeting Deubiquitinases in Cancer. Methods Mol. Biol. 2018, 1731, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Obrist, F.; Manic, G.; Kroemer, G.; Vitale, I.; Galluzzi, L. Trial Watch: Proteasomal inhibitors for anticancer therapy. Mol. Cell. Oncol. 2014, 2, e974463. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Sidhu, S.S. Development of inhibitors in the ubiquitination cascade. FEBS Lett. 2013, 588, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Bielskienė, K.; Bagdonienė, L.; Mozūraitienė, J.; Kazbarienė, B.; Janulionis, E. E3 ubiquitin ligases as drug targets and prognostic biomarkers in melanoma. Medicina 2015, 51, 1–9. [Google Scholar] [CrossRef]
- Tan, Y.-H.C.; Krishnaswamy, S.; Nandi, S.; Kanteti, R.; Vora, S.; Onel, K.; Hasina, R.; Lo, F.-Y.; El-Hashani, E.; Cervantes, G.; et al. CBL Is Frequently Altered in Lung Cancers: Its Relationship to Mutations in MET and EGFR Tyrosine Kinases. PLoS ONE 2010, 5, e8972. [Google Scholar] [CrossRef]
- Drakos, E.; Singh, R.R.; Rassidakis, G.Z.; Schlette, E.; Li, J.; Claret, F.X.; Ford, R.J.; Vega, F.; Medeiros, L.J. Activation of the p53 pathway by the MDM2 inhibitor nutlin-3a overcomes BCL2 overexpression in a preclinical model of diffuse large B-cell lymphoma associated with t(14;18)(q32;q21). Leukemia 2011, 25, 856–867. [Google Scholar] [CrossRef] [Green Version]
- Shangary, S.; Wang, S. Small-Molecule Inhibitors of the MDM2-p53 Protein-Protein Interaction to Reactivate p53 Function: A Novel Approach for Cancer Therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Hai, J.; Sakashita, S.; Allo, G.; Ludkovski, O.; Ng, C.; Shepherd, F.A.; Tsao, M. Inhibiting MDM2-p53 Interaction Suppresses Tumor Growth in Patient-Derived Non–Small Cell Lung Cancer Xenograft Models. J. Thorac. Oncol. 2015, 10, 1172–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.-J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.-H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled α−helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3445–E3454. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Tao, Z.; Le Blanc, J.M.; Zaorsky, N.G.; Sun, Y.; Vuagniaux, G.; Dicker, A.P.; Lu, B. Debio 1143, an antagonist of multiple inhibitor-of-apoptosis proteins, activates apoptosis and enhances radiosensitization of non-small cell lung cancer cells in vitro. Am. J. Cancer Res. 2014, 4, 943–951. [Google Scholar]
- Lee, S.-H.; Lee, J.-Y.; Jung, C.L.; Bae, I.H.; Suh, K.H.; Gil Ahn, Y.; Jin, D.-H.; Kim, T.W.; Suh, Y.-A.; Jang, S.J. A novel antagonist to the inhibitors of apoptosis (IAPs) potentiates cell death in EGFR-overexpressing non-small-cell lung cancer cells. Cell Death Dis. 2014, 5, e1477. [Google Scholar] [CrossRef] [PubMed]
- Sikic, B.I.; Eckhardt, S.G.; Gallant, G.; Burris, H.A.; Camidge, D.R.; Colevas, A.D.; Jones, S.F.; Messersmith, W.A.; Wakelee, H.A.; Li, H.; et al. Safety, pharmacokinetics (PK), and pharmacodynamics (PD) of HGS1029, an inhibitor of apoptosis protein (IAP) inhibitor, in patients (Pts) with advanced solid tumors: Results of a phase I study. J. Clin. Oncol. 2011, 29, 3008. [Google Scholar] [CrossRef]
- Mata-Cantero, L.; Lobato-Gil, S.; Aillet, F.; Lang, V.; Rodriguez, M.S. The Ubiquitin-Proteasome System (UPS) as a Cancer Drug Target: Emerging Mechanisms and Therapeutics. Stress Response Pathw. Cancer 2014, 225–264. [Google Scholar] [CrossRef]
- Rape, M. Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell Biol. 2017, 19, 59–70. [Google Scholar] [CrossRef]
- Jiang, J.; Thyagarajan-Sahu, A.; Krchnak, V.; Jedinak, A.; Sandusky, G.E.; Sliva, D. NAHA, a Novel Hydroxamic Acid-Derivative, Inhibits Growth and Angiogenesis of Breast Cancer In Vitro and In Vivo. PLoS ONE 2012, 7, e34283. [Google Scholar] [CrossRef] [Green Version]
- Bjorklund, C.C.; Lu, L.; Kang, J.; Hagner, P.; Havens, C.G.; Amatangelo, M.; Wang, M.; Ren, Y.; Couto, S.S.; Breider, M.; et al. Rate of CRL4CRBN substrate Ikaros and Aiolos degradation underlies differential activity of lenalidomide and pomalidomide in multiple myeloma cells by regulation of c-Myc and IRF4. Blood Cancer J. 2015, 5, e354. [Google Scholar] [CrossRef] [PubMed]
- Weathington, N.M.; Mallampalli, R.K. Emerging therapies targeting the ubiquitin proteasome system in cancer. J. Clin. Investig. 2014, 124, 6–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- DeCicco, K.L.; Tanaka, T.; Andreola, F.; De Luca, L.M. The effect of thalidomide on non-small cell lung cancer (NSCLC) cell lines: Possible involvement in the PPAR pathway. Carcinogenesis 2004, 25, 1805–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paludo, J.; Mikhael, J.R.; LaPlant, B.R.; Halvorson, A.E.; Kumar, S.; Gertz, M.A.; Hayman, S.R.; Buadi, F.K.; Dispenzieri, A.; Lust, J.A.; et al. Pomalidomide, bortezomib, and dexamethasone for patients with relapsed lenalidomide-refractory multiple myeloma. Blood 2017, 130, 1198–1204. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Bruins, L.A.; Schmidt, J.E.; Van Wier, S.; Chang, X.-B.; Bjorklund, C.C.; Fonseca, R.; Bergsagel, P.L.; et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011, 118, 4771–4779. [Google Scholar] [CrossRef] [PubMed]
- Lopezgirona, A.; Mendy, D.; Ito, T.; Miller, K.H.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Schmidt-Wolf, I.G. Lenalidomide in multiple myeloma. Expert Rev. Anticancer Ther. 2015, 15, 491–497. [Google Scholar] [CrossRef]
- Kim, K.; An, S.; Cha, H.J.; Choi, Y.M.; Choi, S.J.; An, I.-S.; Lee, H.G.; Min, Y.H.; Lee, S.-J.; Bae, S. Lenalidomide induces apoptosis and alters gene expression in non-small cell lung cancer cells. Oncol. Lett. 2012, 5, 588–592. [Google Scholar] [CrossRef]
- Liang, Q.; Dexheimer, T.S.; Zhang, P.; Rosenthal, A.S.; Villamil, M.A.; You, C.; Zhang, Q.; Chen, J.; Ott, C.A.; Sun, H.; et al. A selective USP1–UAF1 inhibitor links deubiquitination to DNA damage responses. Nat. Chem. Biol. 2014, 10, 298–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitta, K.; Paulus, A.; Akhtar, S.; Blake, M.K.K.; Caulfield, T.R.; Novak, A.J.; Ansell, S.M.; Advani, P.; Ailawadhi, S.; Asher, C.-K.; et al. Targeted inhibition of the deubiquitinating enzymes, USP14 and UCHL5, induces proteotoxic stress and apoptosis in Waldenström macroglobulinaemia tumour cells. Br. J. Haematol. 2015, 169, 377–390. [Google Scholar] [CrossRef]
- D’Arcy, P.; Brnjic, S.; Olofsson, M.H.; Fryknäs, M.; Lindsten, K.; De Cesare, M.; Perego, P.; Sadeghi, B.; Hassan, M.; Larsson, R.; et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 2011, 17, 1636–1640. [Google Scholar] [CrossRef]
- Jin, J.-O.; Lee, G.D.; Nam, S.H.; Lee, T.H.; Kang, D.H.; Yun, J.K.; Lee, P.C.-W. Sequential ubiquitination of p53 by TRIM28, RLIM, and MDM2 in lung tumorigenesis. Cell Death Differ. 2020, 28, 1790–1803. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fang, S.; Jensen, J.P.; Weissman, A.M.; Ashwell, J.D. Ubiquitin Protein Ligase Activity of IAPs and Their Degradation in Proteasomes in Response to Apoptotic Stimuli. Science 2000, 288, 874–877. [Google Scholar] [CrossRef] [PubMed]
- Micel, L.N.; Tentler, J.J.; Smith, P.G.; Eckhardt, G.S. Role of Ubiquitin Ligases and the Proteasome in Oncogenesis: Novel Targets for Anticancer Therapies. J. Clin. Oncol. 2013, 31, 1231–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streich, F.C.; Lima, C.D. Structural and Functional Insights to Ubiquitin-Like Protein Conjugation. Annu. Rev. Biophys. 2014, 43, 357–379. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccarelli, D.; Tang, X.; Pelletier, B.; Orlicky, S.; Xie, W.; Plantevin, V.; Neculai, D.; Chou, Y.-C.; Ogunjimi, A.; Al-Hakim, A.; et al. An Allosteric Inhibitor of the Human Cdc34 Ubiquitin-Conjugating Enzyme. Cell 2011, 145, 1075–1087. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Sun, Y. Targeting CDC34 E2 ubiquitin conjugating enzyme for lung cancer therapy. EBioMedicine 2020, 54, 102718. [Google Scholar] [CrossRef]
- Majeed, S.; Aparnathi, M.K.; Song, L.; Schimmer, A.D.; Tsao, M.S.; Liu, G.; Lok, B.H. Abstract 2699: Targeting an ubiquitin-activating enzyme in small-cell lung cancer (SCLC). In Proceedings of the AACR Annual Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar] [CrossRef]
- Morrow, J.; Lin, H.-K.; Sun, S.-C.; Zhang, S. Targeting ubiquitination for cancer therapies. Futur. Med. Chem. 2015, 7, 2333–2350. [Google Scholar] [CrossRef] [Green Version]
- Dale, B.; Cheng, M.; Park, K.-S.; Kaniskan, H.; Xiong, Y.; Jin, J. Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 1–17. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Characteristics | Small Cell Carcinoma | Non-Small Cell Carcinoma |
|---|---|---|
| Cell size | Small cell | Larger cell |
| Location | Peribronchial location | Any part of the lung |
| Metastasizes/Spread | Fast spread | Slow spread |
| Fatality | More, if untreated | Less |
| Type | Two types: small cell carcinoma and combined small cell carcinoma | Adenocarcinoma and squamous cell carcinoma |
| Diagnosis | In early stage of carcinoma | In late stage of carcinoma |
| Treatment | Fast | Slow, due to late examination |
| Incidence of total lung cancer | 15% | 85% |
| UPS | Target/Function | References |
|---|---|---|
| USP1 | Involvement in translesion synthesis and DNA damage | [49] |
| USP7 | Ki-67 antigen (Ki-67) | [50] |
| USP8 | Regulate RTKs including EGFR and ERBB2 | [51] |
| USP14 | Promotes NSCLC cell proliferation by β-catenin accumulation | [52] |
| USP17 | Deubiquitinates p53 and Mdm2 to alter the stability and activity of p53 | [53] |
| USP22 | Promotes tumor progression and induces epithelial-mesenchymal transition (EMT) | [54,55] |
| USP28 | Stabilization of the oncoprotein c-Myc | [11] |
| USP36 | Regulate and stabilize c-Myc | [56] |
| USP37 | Directly stabilizes c-Myc | [57] |
| USP39 | Akt, mTOR, p53, and PARP signaling pathways | [58] |
| USP44 | Akt signalling | [59] |
| Compounds | Target | Modes of Action | References |
|---|---|---|---|
| Nutlin-3a | MDM2 | Competitively binds to the Mdm2-P53 interaction region, activating the P53 pathway and causing cell cycle arrest, cell death, and growth inhibition. | [19,84,85] |
| RG7388 | MDM2 | The derivatives of nutlin-3a inhibit the Mdm2-P53 binding site | [86] |
| RG7112 | MDM2/MDMX | Restoration of p53 activity by inhibiting the p53-MDM2 interaction | [86,87] |
| ATSP-7041 | Dual inhibition of MDM2 and MDMX | Inhibitor of MDM2 and MDMX for p53-dependent processes | [88] |
| AT-406 (also known as Debio 1143 or SM-406) | XIAP, IAP1 and IAP2 | Suppresses the inhibitor of apoptosis protein | [89] |
| TL-32711 | IAP | Suppresses the inhibitor of apoptosis protein | [90] |
| HGS-1029 | IAP2 | Suppresses the inhibitor of apoptosis protein | [91,92] |
| TAK-243 (MLN7243) | UBA1 | Causes depletion of cellular ubiquitin conjugates resulting in disruption of signaling events | [93] |
| NAHA (Novel Hydroxamic Acid-Derivative) | Cdc20 | Decreases the expression of Cdc20. Inhibits tumor proliferation in vitro and in vivo associated with the initiation of apoptosis | [94] |
| Thalidomide | Multiple target Cereblon (CRBN) | Significantly increases PPAR (Peroxisome proliferator-activated receptor) gamma protein expression, significantly increased PPRE (PPAR response element) reporter activity and decreases NFkB reporter activity in LCC (large cell carcinoma) cells | [95,96,97,98] |
| Pomalidomide | CRBN | Suppresses CRBN E3 activity, reducing c-Myc and IRF4, suppressing MM cell transcriptional activity | [96,99,100] |
| Lenalidomide | CRBN | Induces apoptosis and modifies gene expression in NSCLC cells | [95,100,101,102,103] |
| Pimozide and GW7647 | USP1/UAF1 complex | Involved in translation synthesis and DNA damage response in NSCLC | [49,104] |
| b-AP15 (known as VLX1500) | UCHL5 (ubiquitin C-terminal hydrolase 5) and USP14 | Induces tumor cell apoptosis and inhibits tumor progression | [43,105,106] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, J.-O.; Puranik, N.; Bui, Q.T.; Yadav, D.; Lee, P.C.-W. The Ubiquitin System: An Emerging Therapeutic Target for Lung Cancer. Int. J. Mol. Sci. 2021, 22, 9629. https://doi.org/10.3390/ijms22179629
Jin J-O, Puranik N, Bui QT, Yadav D, Lee PC-W. The Ubiquitin System: An Emerging Therapeutic Target for Lung Cancer. International Journal of Molecular Sciences. 2021; 22(17):9629. https://doi.org/10.3390/ijms22179629
Chicago/Turabian StyleJin, Jun-O, Nidhi Puranik, Quyen Thu Bui, Dhananjay Yadav, and Peter Chang-Whan Lee. 2021. "The Ubiquitin System: An Emerging Therapeutic Target for Lung Cancer" International Journal of Molecular Sciences 22, no. 17: 9629. https://doi.org/10.3390/ijms22179629
APA StyleJin, J.-O., Puranik, N., Bui, Q. T., Yadav, D., & Lee, P. C.-W. (2021). The Ubiquitin System: An Emerging Therapeutic Target for Lung Cancer. International Journal of Molecular Sciences, 22(17), 9629. https://doi.org/10.3390/ijms22179629