
NK Cells Lose Their Cytotoxicity Function against Cancer Stem Cell-Rich Radiotherapy-Resistant Breast Cancer Cell Populations


Abstract
:1. Introduction
2. Results
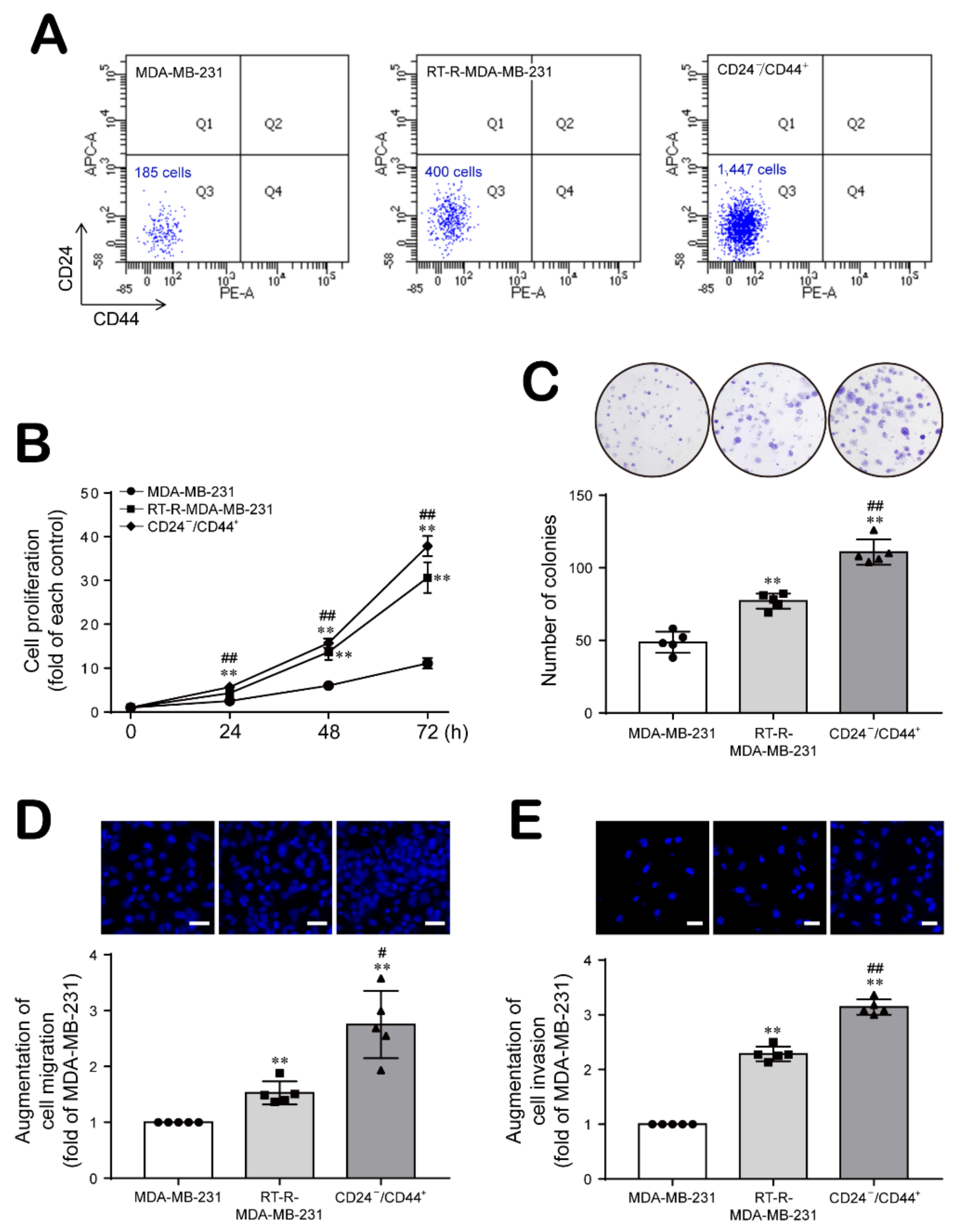
2.1. Breast Cancer Cells with Acquired Radioresistance Contain a Larger Population of Breast Cancer Stem Cells (BCSCs) and Increased Proliferation, Migration, and Invasion Abilities
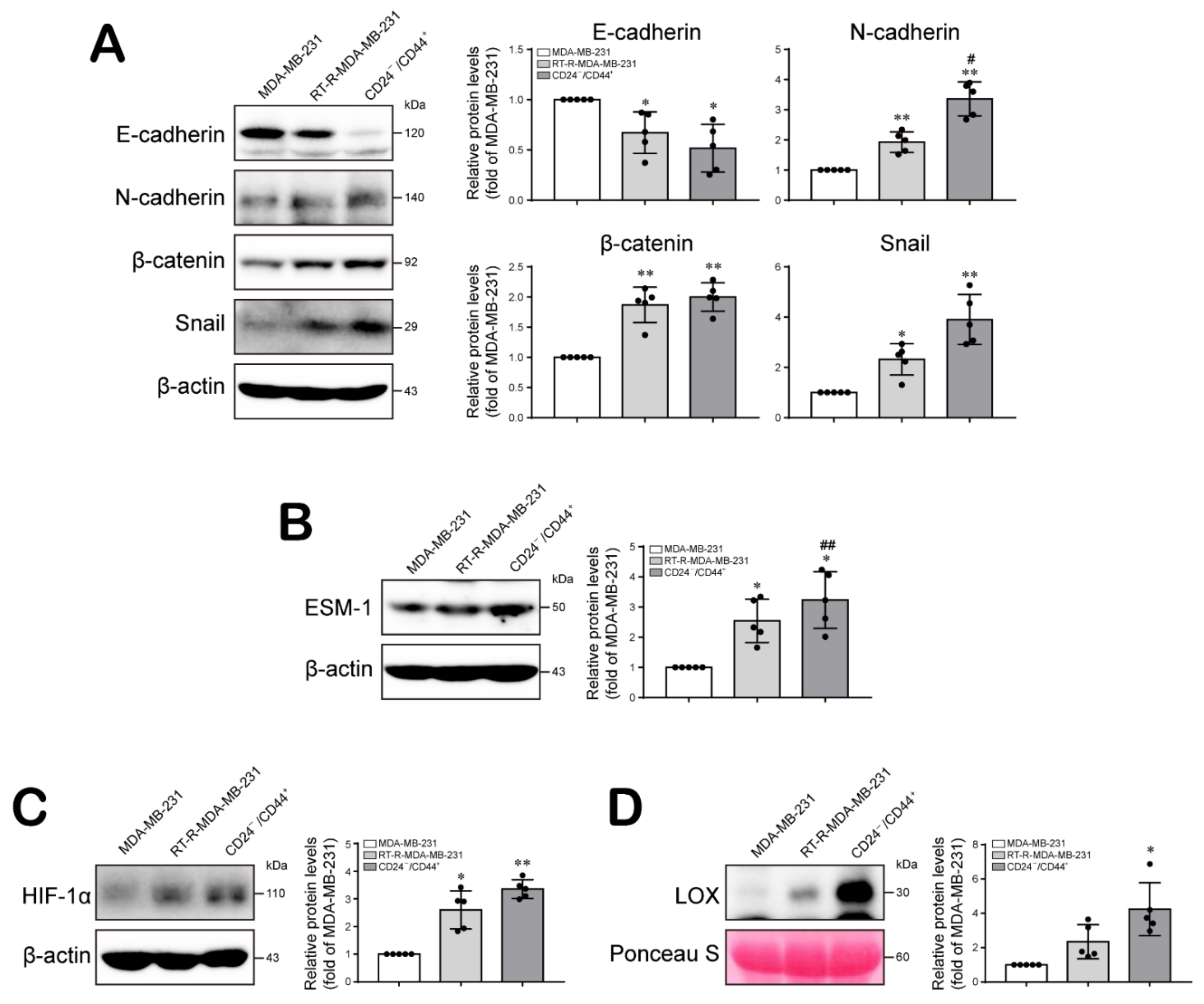
2.2. The Increased Population of CD24−/low/CD44+ Cells among RT-R-BCs Induces the Expression Levels of Tumor Progression-Related Proteins
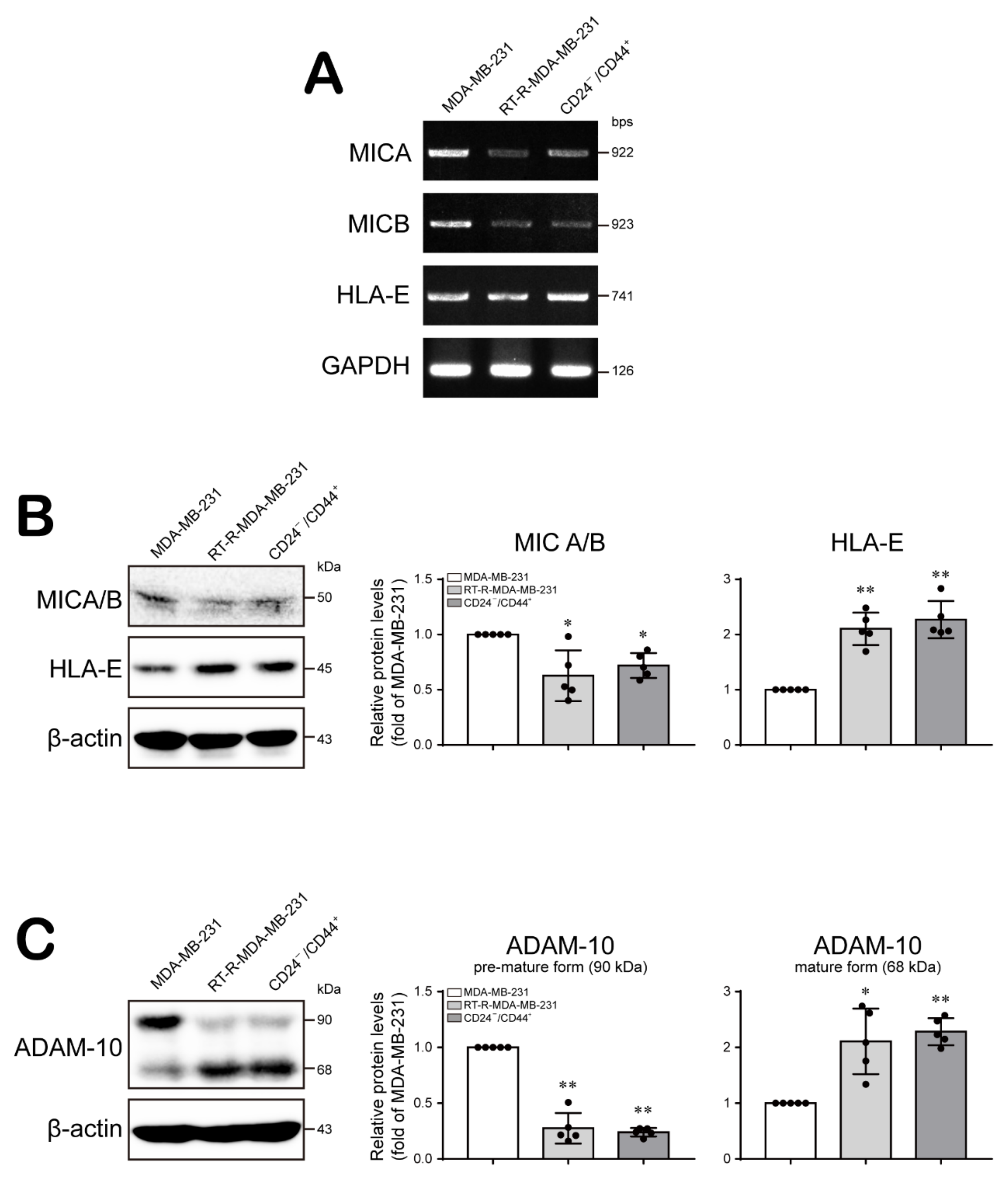
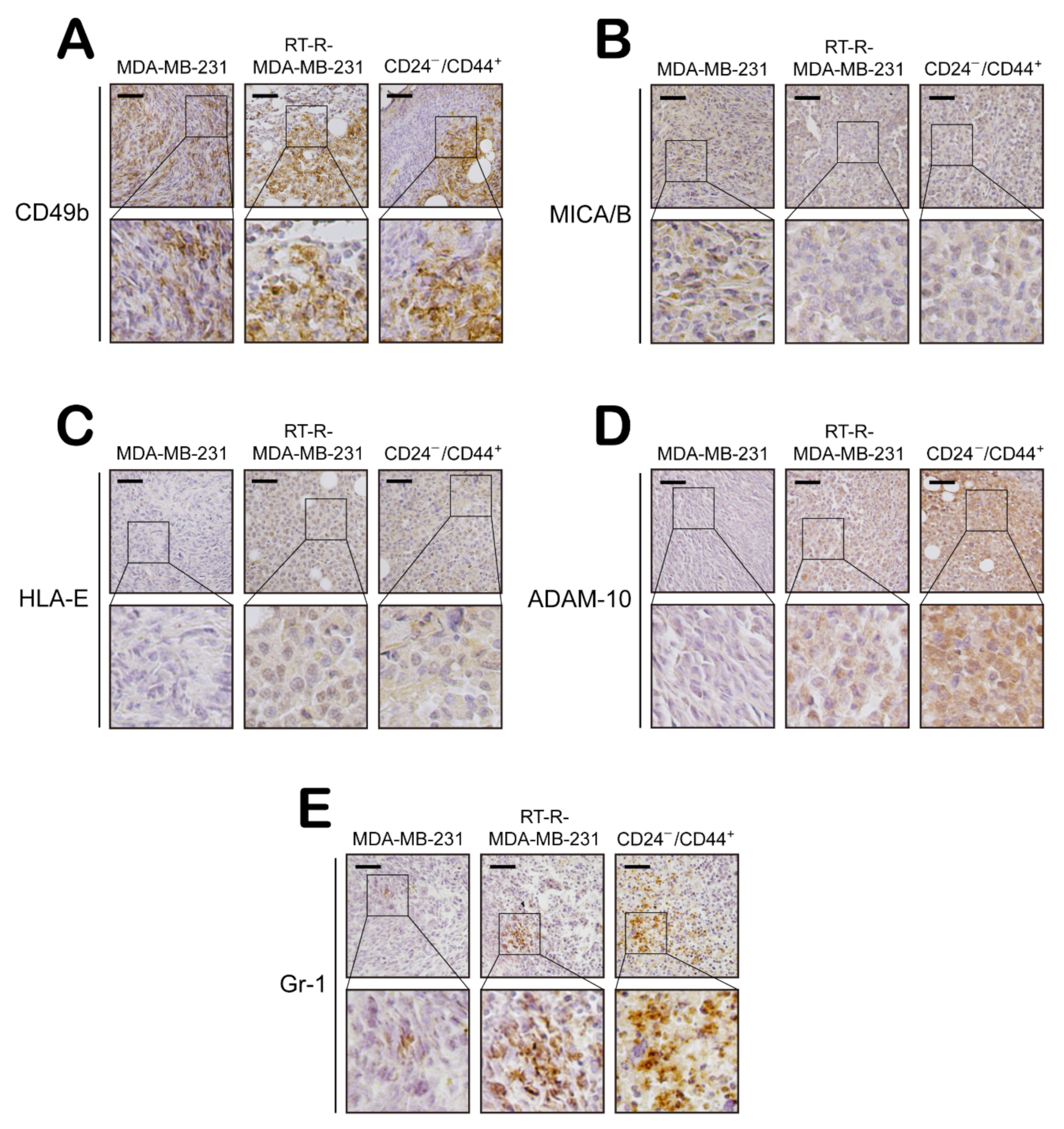
2.3. CD24−/low/CD44+ Cells Recruit NK Cells but Suppress Their Cytotoxicity through Modulation of the Levels of NK Cell Function-Related Ligands
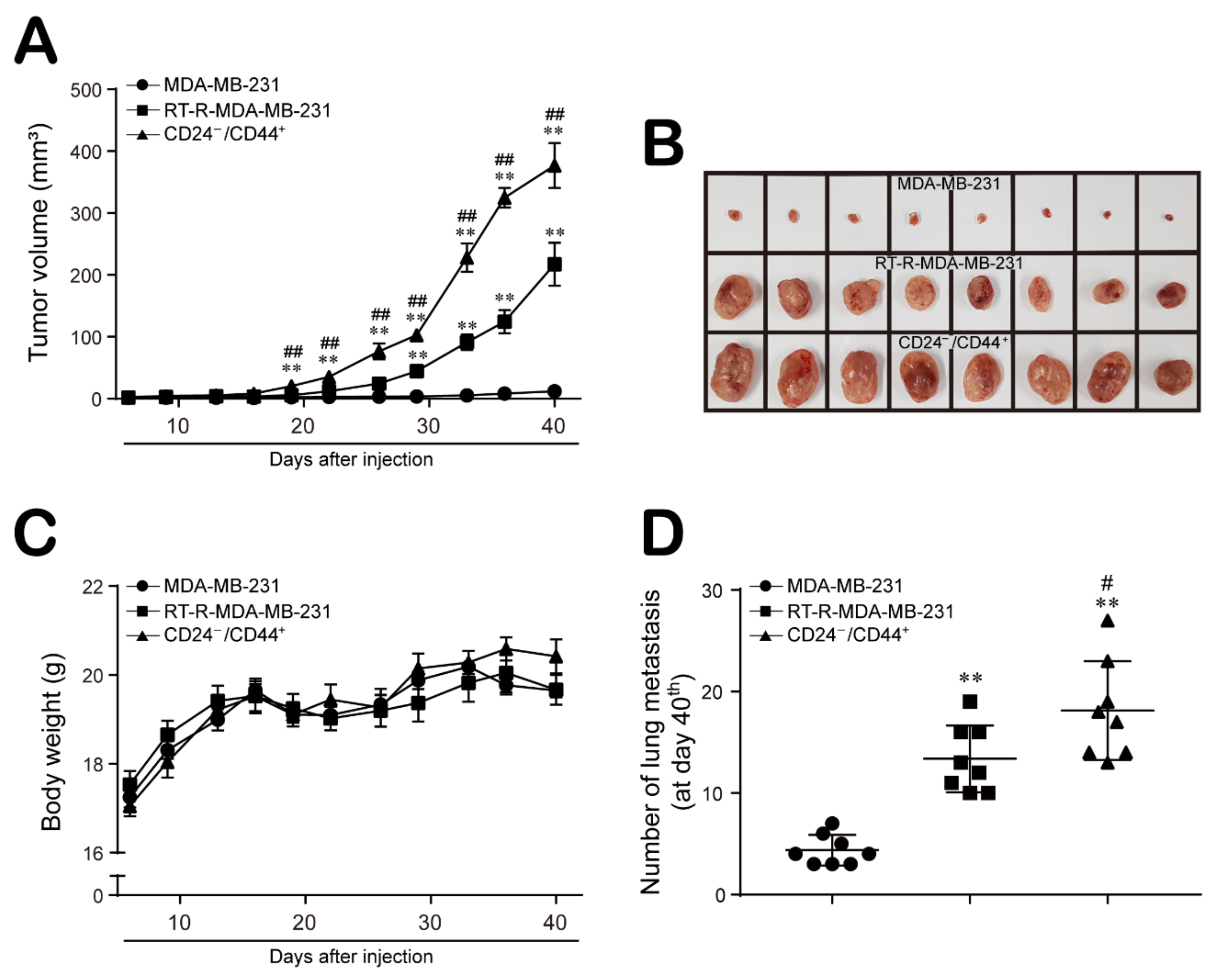
2.4. CD24−/low/CD44+ Cells Exhibit Enhanced Aggressive Features Supporting the Progression and Metastasis of Breast Cancer In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Establishment of RT-R-BC Cells
4.2. Isolation of CD24−/low CD44+ Cells from Breast Cancer Cells
4.3. Flow Cytometric Analysis to Compare the BCSC Population between MDA-MB-231 Cells and RT-R-MDA-MB-231 Cells
4.4. Cell Proliferation Assay
4.5. Colony Formation Assay
4.6. Cancer Cell Migration and Matrigel-Invasion Assays
4.7. Protein Extraction and Western Blot Analysis
4.8. Total RNA Extraction and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.9. NK Cell Migration Assay
4.10. Cytotoxicity Assay
4.11. Measurement of Extracellular Perforin and Granzyme B Levels
4.12. In Vivo Animal Study
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADAM-10 | A disintegrin and metalloproteinase domain containing protein-10 |
| ALDH-1 | Aldehyde dehydrogenase-1 |
| APCs | Antigen presenting cells |
| BC | Breast cancer |
| BCSCs | Breast cancer stem cells |
| CM | Conditioned medium |
| CSCs | Cancer stem cell-like cells |
| DAB | 3,3′-Diaminobenzidine |
| DAPI | 4′,6-Diamidine-2′-phenylindole dihydrochloride |
| ECL | Enhanced chemiluminescence |
| EMT | Epithelial–mesenchymal transition |
| ESM-1 | Endothelial cell specific molecule-1 |
| HER2 | Human epidermal growth factor receptor 2 |
| HIF-1α | Hypoxia-inducible factor-1α |
| HLA-E | HLA class I molecules-E |
| HRP | Horseradish peroxidase |
| IHC | Immunohistochemistry |
| LDH | Lactate dehydrogenase |
| LOX | Lysyl oxidase |
| MDSC | Myeloid-derived suppressor cells |
| MICA/B | MHC class I chain-related molecules A/B |
| NK cells | Natural killer cells |
| NLR | Neutrophil-to-lymphocyte ratio |
| NP-40 | Nonylphonoxypolyethoxypthanol-40 |
| ROS | Reactive oxygen species |
| RT | Radiotherapy |
| RT-R | Radiotherapy-resistant |
| RT-PCR | Reverse transcription-polymerase chain reaction |
| SDS-PAGE | Sodium dodecyl sulfate- polyacrylamide gel electrophoresis |
| TAMs | Tumor-associated macrophages |
| TME | Tumor microenvironment |
| TNBC | Triple-negative breast cancer |
| Tregs | Regulatory T cells |
References
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Boiko, A.D.; Razorenova, O.V.; van de Rijn, M.; Swetter, S.M.; Johnson, D.L.; Ly, D.P.; Butler, P.D.; Yang, G.P.; Joshua, B.; Kaplan, M.J.; et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 2010, 466, 133–137. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Dallas, N.A.; Xia, L.; Fan, F.; Gray, M.J.; Gaur, P.; van Buren, G., II; Samuel, S.; Kim, M.P.; Lim, S.J.; Ellis, L.M. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009, 69, 1951–1957. [Google Scholar] [CrossRef] [Green Version]
- Rosa, R.; D’Amato, V.; de Placido, S.; Bianco, R. Approaches for targeting cancer stem cells drug resistance. Expert Opin. Drug Discov. 2016, 11, 1201–1212. [Google Scholar] [CrossRef] [Green Version]
- Murayama, T.; Gotoh, N. Drug resistance mechanisms of cancer stem-like cells and their therapeutic potential as drug targets. Cancer Drug Resist. 2019, 2, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Berkey, F.J. Managing the adverse effects of radiation therapy. Am. Fam. Physician 2010, 82, 381–388. [Google Scholar]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front. Oncol. 2013, 3, 113. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Morgan, M.A.; Lawrence, T.S. Molecular pathways: Overcoming radiation resistance by targeting DNA damage response pathways. Clin. Cancer Res. 2015, 21, 2898–2904. [Google Scholar] [CrossRef] [Green Version]
- Philchenkov, A. Radiation-Induced cell death: Signaling and pharmacological modulation. Crit. Rev. Oncog. 2018, 23, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.S.; Jin, H.; Lee, J.S.; Park, S.W.; Chang, K.C.; Kang, K.M.; Jeong, B.K.; Kim, H.J. Radioresistant breast cancer cells exhibit increased resistance to chemotherapy and enhanced invasive properties due to cancer stem cells. Oncol. Rep. 2018, 40, 3752–3762. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.H.; Go, S.I.; Song, H.N.; Lee, A.; Kim, S.H.; Kang, J.H.; Jeong, B.K.; Kang, K.M.; Ling, H.; Lee, G.W. The prognostic impact of the neutrophil-to-lymphocyte ratio in patients with small-cell lung cancer. Br. J. Cancer 2014, 111, 452–460. [Google Scholar] [CrossRef]
- Joo, Y.N.; Jin, H.; Eun, S.Y.; Park, S.W.; Chang, K.C.; Kim, H.J. P2Y2R activation by nucleotides released from the highly metastatic breast cancer cell MDA-MB-231 contributes to pre-metastatic niche formation by mediating lysyl oxidase secretion, collagen crosslinking, and monocyte recruitment. Oncotarget 2014, 5, 9322–9334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Bassani, B.; Baci, D.; Gallazzi, M.; Poggi, A.; Bruno, A.; Mortara, L. Natural Killer Cells as Key Players of Tumor Progression and Angiogenesis: Old and Novel Tools to Divert Their Pro-Tumor Activities into Potent Anti-Tumor Effects. Cancers 2019, 11, 461. [Google Scholar] [CrossRef] [Green Version]
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front. Immunol. 2020, 10, 3038. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Kunimasa, K.; Goto, T. Immunosurveillance and Immunoediting of Lung Cancer: Current Perspectives and Challenges. Int. J. Mol. Sci. 2020, 21, 597. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Jin, H.; Rugira, T.; Ko, Y.S.; Park, S.W.; Yun, S.P.; Kim, H.J. ESM-1 Overexpression is Involved in Increased Tumorigenesis of Radiotherapy-Resistant Breast Cancer Cells. Cancers 2020, 12, 1363. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.Z.; Terunuma, H.; Takada, M.; Tanaka, Y.; Abe, H.; Sata, T.; Toi, M.; Yamamoto, N. Role of natural killer cells in hormone-independent rapid tumor formation and spontaneous metastasis of breast cancer cells in vivo. Breast Cancer Res. Treat. 2007, 104, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Mamessier, E.; Sylvain, A.; Bertucci, F.; Castellano, R.; Finetti, P.; Houvenaeghel, G.; Charaffe-Jaufret, E.; Birnbaum, D.; Moretta, A.; Olive, D. Human breast tumor cells induce self-tolerance mechanisms to avoid NKG2D-mediated and DNAM-mediated NK cell recognition. Cancer Res. 2011, 71, 6621–6632. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wang, Q.; Wang, Z.; Jiang, J.; Yu, S.C.; Ping, Y.F.; Yang, J.; Xu, S.L.; Ye, X.Z.; Xu, C.; et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res. 2014, 74, 5746–5757. [Google Scholar] [CrossRef] [Green Version]
- Krzewski, K.; Coligan, J.E. Human NK cell lytic granules and regulation of their exocytosis. Front. Immunol. 2012, 3, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay-McLean, A.; Coenraads, S.; Kiani, Z.; Dupuy, F.P.; Bernard, N.F. Expression of ligands for activating natural killer cell receptors on cell lines commonly used to assess natural killer cell function. BMC Immunol. 2019, 20, 8. [Google Scholar] [CrossRef] [Green Version]
- Zingoni, A.; Vulpis, E.; Loconte, L.; Santoni, A. NKG2D Ligand Shedding in Response to Stress: Role of ADAM10. Front. Immunol. 2020, 11, 447. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Eun, S.Y.; Lee, J.S.; Park, S.W.; Lee, J.H.; Chang, K.C.; Kim, H.J. P2Y2 receptor activation by nucleotides released from highly metastatic breast cancer cells increases tumor growth and invasion via crosstalk with endothelial cells. Breast Cancer Res. 2014, 16, R77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Tudoran, O.M.; Balacescu, O.; Berindan-Neagoe, I. Breast cancer stem-like cells: Clinical implications and therapeutic strategies. Clujul Med. 2016, 89, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Koch, U.; Krause, M.; Baumann, M. Cancer stem cells at the crossroads of current cancer therapy failures—Radiation oncology perspective. Semin. Cancer Biol. 2010, 20, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Woodward, W.A.; Chen, M.S.; Behbod, F.; Alfaro, M.P.; Buchholz, T.A.; Rosen, J.M. Wnt/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc. Natl. Acad. Sci. USA 2007, 104, 618–623. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Diophode, R.Y.; Bapat, S.A. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, C.M.; Kuperwasser, C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.Y.; Lee, H.E.; Li, H.; Shipitsin, M.; Gelman, R.; Polyak, K. Heterogeneity for stem cell-related markers according to tumor subtype and histologic stage in breast cancer. Clin. Cancer Res. 2010, 16, 876–887. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Vacca, P.; del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef]
- Kaiser, B.K.; Barahmand-Pour, F.; Paulsene, W.; Medley, S.; Geraghty, D.E.; Strong, R.K. Interactions between NKG2x immunoreceptors and HLA-E ligands display overlapping affinities and thermodynamics. J. Immunol. 2005, 174, 2878–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brostjan, C.; Bellón, T.; Sobanov, Y.; López-Botet, M.; Hofer, E. Differential expression of inhibitory and activating CD94/NKG2 receptors on NK cell clones. J. Immunol. Methods 2002, 264, 109–119. [Google Scholar] [CrossRef]
- Liu, P.C.; Liu, X.; Li, Y.; Covington, M.; Wynn, R.; Huber, R.; Hillman, M.; Yang, G.; Ellis, D.; Marando, C.; et al. Identification of ADAM10 as a major source of HER2 ectodomain sheddase activity in HER2 overexpressing breast cancer cells. Cancer Biol. Ther. 2006, 5, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Feldinger, K.; Generali, D.; Kramer-Marek, G.; Gijsen, M.; Ng, T.B.; Wong, J.H.; Strina, C.; Cappelletti, M.; Andreis, D.; Li, J.L.; et al. ADAM10 mediates trastuzumab resistance and is correlated with survival in HER2 positive breast cancer. Oncotarget 2014, 5, 6633–6646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Lin, L.; Li, X.; Lu, A.; Hou, C.; Wu, Q.; Hu, X.; Zhou, Z.; Chen, Z.; Tang, F. ADAM10 is involved in the oncogenic process and chemo-resistance of triple-negative breast cancer via regulating Notch1 signaling pathway, CD44 and PrPc. Cancer Cell Int. 2021, 21, 32. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid Derived Suppressor Cells Interactions with Natural Killer Cells and Pro-angiogenic Activities: Roles in Tumor Progression. Front. Immunol. 2019, 10, 771. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortin, C.; Huang, X.; Yang, Y. NK cell response to vaccinia virus is regulated by myeloid-derived suppressor cells. J. Immunol. 2012, 189, 1843–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence |
|---|---|
| hMICA | Forward: 5′-TCCTGCTTCTGGCTGGCATC-3′ Reverse: 5′-GCAGAAACATGGAATGTCTG-3′ |
| hMICB | Forward: 5′-CTGCTGTTTCTGGCCGTCGC-3′ Reverse: 5′-GAAACATATGGAAAGTCTGTC-3′ |
| hHLA-E | Forward: 5′-GTGAATCTGCGGACGCTGCG-3′ Reverse: 5′-CTTAGAGTAGCTCCCTCCTT-3′ |
| hGAPDH | Forward: 5′-TCAACAGCGACACCCACTCC-3′ Reverse: 5′-TGAGGTCCACCACCCTGTTG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, H.; Kim, H.J. NK Cells Lose Their Cytotoxicity Function against Cancer Stem Cell-Rich Radiotherapy-Resistant Breast Cancer Cell Populations. Int. J. Mol. Sci. 2021, 22, 9639. https://doi.org/10.3390/ijms22179639
Jin H, Kim HJ. NK Cells Lose Their Cytotoxicity Function against Cancer Stem Cell-Rich Radiotherapy-Resistant Breast Cancer Cell Populations. International Journal of Molecular Sciences. 2021; 22(17):9639. https://doi.org/10.3390/ijms22179639
Chicago/Turabian StyleJin, Hana, and Hye Jung Kim. 2021. "NK Cells Lose Their Cytotoxicity Function against Cancer Stem Cell-Rich Radiotherapy-Resistant Breast Cancer Cell Populations" International Journal of Molecular Sciences 22, no. 17: 9639. https://doi.org/10.3390/ijms22179639
APA StyleJin, H., & Kim, H. J. (2021). NK Cells Lose Their Cytotoxicity Function against Cancer Stem Cell-Rich Radiotherapy-Resistant Breast Cancer Cell Populations. International Journal of Molecular Sciences, 22(17), 9639. https://doi.org/10.3390/ijms22179639