
The Roles of c-Jun N-Terminal Kinase (JNK) in Infectious Diseases


Abstract
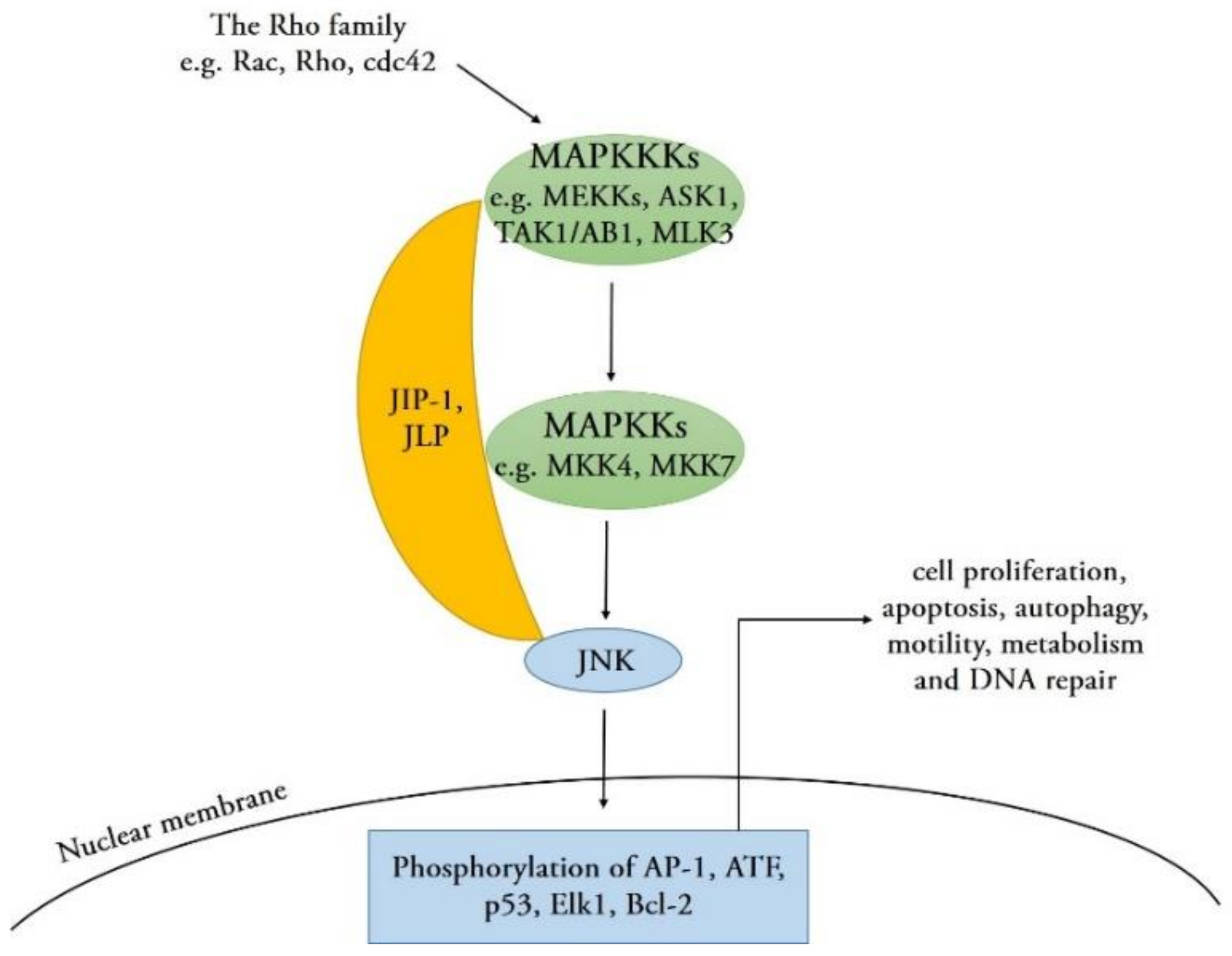
:1. Introduction
2. JNK Signaling in Apoptosis and Autophagy
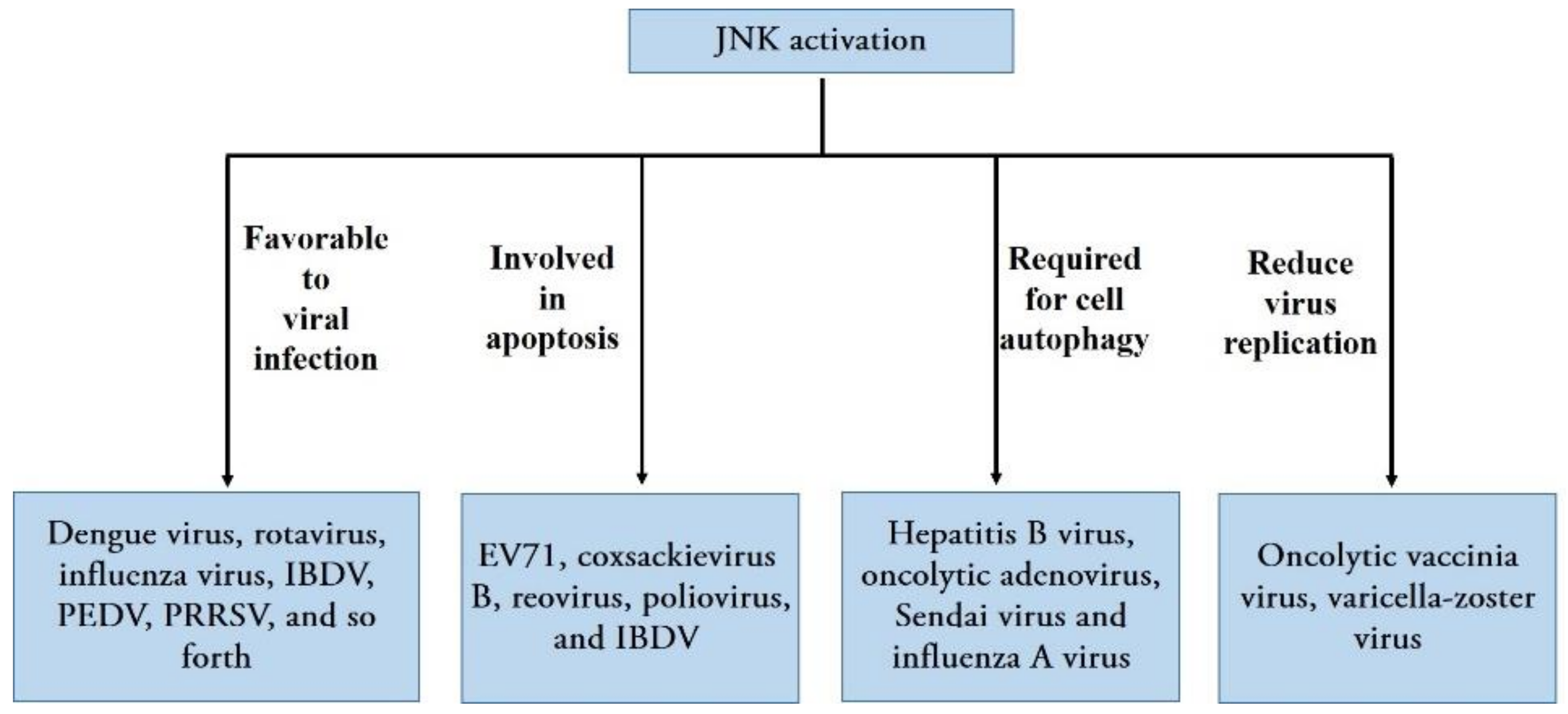
3. Role of JNK Signaling Pathway in Viral Diseases
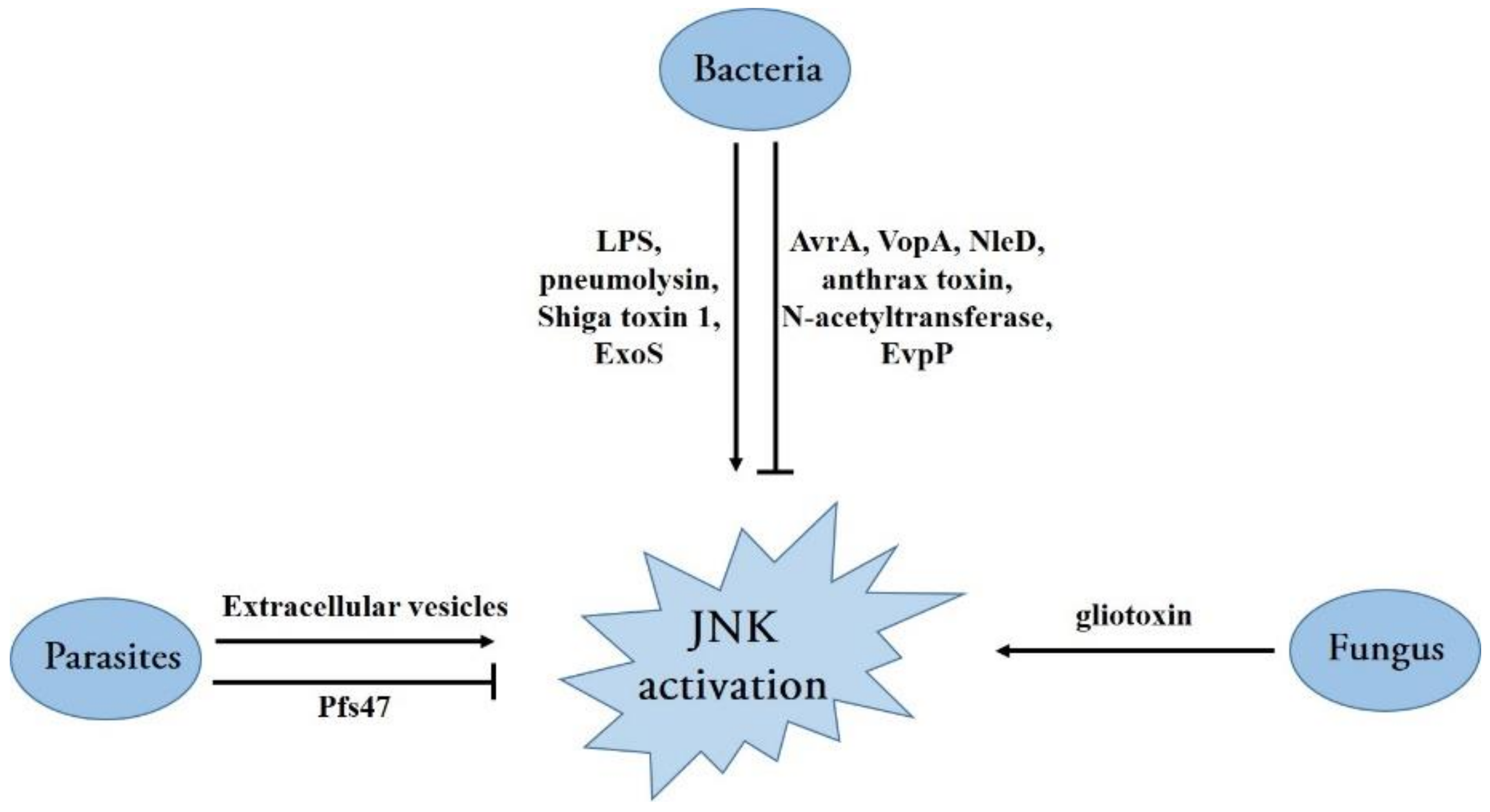
4. The Role of JNK Signaling in Bacterial, Fungal and Parasitic Infections
5. JNK Signaling Inhibitors as Therapeutic Agents against Infectious Diseases
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-activated protein kinase: Conservation of a three-kinase module from yeast to human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef]
- Fanger, G.R.; Gerwins, P.; Widmann, C.; Jarpe, M.B.; Johnson, G.L. MEKKs, GCKs, MLKs, PAKs, TAKs, and Tpls: Up-stream regulators of the c-Jun amino-terminal kinases? Curr. Opin. Genet. Dev. 1997, 7, 67–74. [Google Scholar] [CrossRef]
- Siow, Y.L.; Kalmar, G.B.; Sanghera, J.S.; Tai, G.; Oh, S.S.; Pelech, S.L. Identification of two essential phosphorylated threonine residues in the catalytic domain of Mekk1. Indirect activation by Pak3 and protein kinase C. J. Biol. Chem. 1997, 272, 7586–7594. [Google Scholar] [CrossRef] [Green Version]
- Gartner, A.; Nasmyth, K.; Ammerer, G. Signal transduction in Saccharomyces cerevisiae requires tyrosine and threonine phosphorylation of FUS3 and KSS1. Genes Dev. 1992, 6, 1280–1292. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Lee, C.; Kim, Y.; Jeon, J.H. JNK and p38 mitogen-activated protein kinase pathways contribute to porcine epidemic diarrhea virus infection. Virus Res. 2016, 222, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, U.K.; Kini, S.G.; Garg, V.; Agrawal, S.; Tomar, P.K.; Pathak, P.; Chaudhary, A.; Gupta, P.; Malik, A.; et al. JNK pathway signaling: A novel and smarter therapeutic targets for various biological diseases. Future Med. Chem. 2015, 7, 2065–2086. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. The functional contrariety of JNK. Mol. Carcinog. 2007, 46, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Dérijard, B.; Davis, R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.; Hu, H.; Wang, R.; Zhou, Y.; Zhang, L.; Wang, C.; Wang, Q. JNK1 induces Notch1 expression to regulate genes governing photoreceptor production. Cells 2019, 8, 970. [Google Scholar] [CrossRef] [Green Version]
- Kallunki, T.; Su, B.; Tsigelny, I.; Sluss, H.K.; Dérijard, B.; Moore, G.; Davis, R.; Karin, M. JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 1994, 8, 2996–3007. [Google Scholar] [CrossRef] [Green Version]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal kinase (JNK) in obesity and type 2 diabetes. Cells 2020, 9, 706. [Google Scholar]
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK signaling pathway in inflammatory skin disorders and cancer. Cells 2020, 9, 857. [Google Scholar] [CrossRef] [Green Version]
- Bennett, B.L. c-Jun N-terminal kinase-dependent mechanisms in respiratory disease. Eur. Respir. J. 2006, 28, 651–661. [Google Scholar] [CrossRef]
- Antoniou, X.; Falconi, M.; Di-Marino, D.; Borsello, T. JNK3 as a therapeutic target for neurodegenerative diseases. J. Alzheimers Dis. 2011, 24, 633–642. [Google Scholar] [CrossRef]
- Kumar, R.; Khandelwal, N.; Thachamvally, R.; Tripathi, B.N.; Barua, S.; Kashyap, S.K.; Maherchandani, S.; Kumar, N. Role of MAPK/MNK1 signaling in virus replication. Virus Res. 2018, 253, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Farahvash, A.; Douda, D.N.; Licht, J.C.; Grasemann, H.; Sweezey, N.; Palaniyar, N. JNK activation turns on LPS- and Gram-negative bacteria-induced NADPH oxidase-dependent suicidal NETosis. Sci. Rep. 2017, 7, 3409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Guo, Y.; Jiang, C.; Chang, Q.; Zhang, S.; Luo, T.; Zhang, B.; Jia, X.; Huang, M.; Dong, C.; et al. JNK1 negatively controls antifungal innate immunity by suppressing CD23 expression. Nat. Med. 2017, 23, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Lizundia, R.; Chaussepied, M.; Huerre, M.; Werling, D.; Di-Santo, J.P.; Langsley, G. c-Jun NH2-terminal kinase/c-Jun signaling promotes survival and metastasis of B lymphocytes transformed by Theileria. Cancer Res. 2006, 66, 6105–6110. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.M.; Onésime, D.; Reddy, C.D.; Dhanasekaran, N.; Reddy, E.P. JLP: A scaffolding protein that tethers JNK/p38MAPK signaling modules and transcription factors. Proc. Natl. Acad. Sci. USA 2002, 99, 14189–14194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadee, D.N.; Kyriakis, J.M. Activation of SAPK/JNKs in vitro. Methods Mol. Biol. 2010, 661, 59–73. [Google Scholar] [PubMed]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 344, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Meyer, C.F.; Tan, T.H. Persistent activation of c-Jun N-terminal kinase 1 (JNK1) in gamma radiation-induced apoptosis. J. Biol. Chem. 1996, 271, 631–634. [Google Scholar] [CrossRef] [Green Version]
- Tournier, C.; Hess, P.; Yang, D.D.; Xu, J.; Turner, T.K.; Nimnual, A.; Bar-Sagi, D.; Jones, S.N.; Flavell, R.A.; Davis, R.J. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000, 288, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.D.; Kuan, C.Y.; Whitmarsh, A.J.; Rincón, M.; Zheng, T.S.; Davis, R.J.; Rakic, P.; Flavell, R.A. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997, 389, 865–870. [Google Scholar] [CrossRef]
- Tang, R.X.; Kong, F.Y.; Fan, B.F.; Liu, X.M.; You, H.J.; Zhang, P.; Zheng, K.Y. HBx activates FasL and mediates HepG2 cell apoptosis through MLK3-MKK7-JNK signal module. World J. Gastroenterol. 2012, 18, 1485–1495. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Tu, Q.Q.; Zheng, R.Y.; Li, J.; Hu, L.; Chang, Y.X.; Li, L.; Li, M.H.; Wang, R.Y.; Huang, D.D.; Wu, M.C.; et al. Palmitic acid induces autophagy in hepatocytes via JNK2 activation. Acta Pharmacol. Sin. 2014, 35, 504–512. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.R.; Jiang, H.; Piya, S.; Lu, Z.; Gomez-Manzano, C.; Fueyo, J. The role of JNK isoforms in adenovirus-induced autophagy: Implications for cancer immunotherapy using oncolytic adenoviruses. Cancer Res. 2013, 73. [Google Scholar]
- Zhou, F.; Yang, Y.; Xing, D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011, 278, 403–413. [Google Scholar] [CrossRef]
- Lorin, S.; Pierron, G.; Ryan, K.M.; Codogno, P.; Djavaheri-Mergny, M. Evidence for the interplay between JNK and p53-DRAM signaling pathways in the regulation of autophagy. Autophagy 2010, 6, 153–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasickova, P.; Dvorska, L.; Lorencova, A.; Pavlik, I. Viruses as a cause of foodborne diseases: A review of the literature. Vet. Med. 2005, 50, 89–104. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Zhu, S.; Ruan, G.; Hou, L.; Wang, J.; Wang, B.; Liu, J. Infectious bursal disease virus-induced activation of JNK signaling pathway is required for virus replication and correlates with virus-induced apoptosis. Virology 2011, 420, 156–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Manna, S.K.; Dhawan, S.; Aggarwal, B.B. HIV-Tat protein activates cJun N-terminal kinase and activator protein-1. J. Immunol. 1998, 161, 776–781. [Google Scholar]
- Huttunen, P.; Hyypia, T.; Vihinen, P.; Nissinen, L.; Heino, J. Echovirus 1 infection induces both stress- and growth-activated mitogen-activated protein kinase pathways and regulates the transcription of cellular immediate-early genes. Virology 1998, 250, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachos, G.; Clements, B.; Conner, J. Herpes simplex virus type 1 infection stimulates p38/c-Jun N-terminal mitogen-activated protein kina se pathways and activates transcription factor AP-1. J. Biol. Chem. 1999, 274, 5097–5103. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Xie, J.; Ye, F.; Gao, S.J. Modulation of Kaposi’s sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J. Virol. 2006, 80, 5371–5382. [Google Scholar] [CrossRef] [Green Version]
- Zapata, H.J.; Nakatsugawa, M.; Moffat, J.F. Varicella-zoster virus infection of human fibroblast cells activates the c-Jun N-terminal kinase pathway. J. Virol. 2007, 81, 977–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceballos-Olvera, I.; Chávez-Salinas, S.; Medina, F.; Ludert, J.E.; del-Angel, R.M. JNK phosphorylation, induced during dengue virus infection, is important for viral infection and requires the presence of cholesterol. Virology 2010, 396, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Holloway, G.; Coulson, B.S. Rotavirus activates JNK and p38 signaling pathways in intestinal cells, leading to AP-1-driven transcriptional responses and enhanced virus replication. J. Virol. 2006, 80, 10624–10633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ruan, T.; Sheng, T.; Wang, J.; Sun, J.; Wang, J.; Prinz, R.A.; Peng, D.; Liu, X.; Xu, X. Role of c-Jun terminal kinase (JNK) activation in influenza A virus-induced autophagy and replication. Virology 2019, 526, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, C. Stress-activated protein kinases are involved in porcine reproductive and respiratory syndrome virus infection and modulate virus-induced cytokine production. Virology 2012, 427, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, H.; Weng, S.; Li, C.; He, J. White spot syndrome virus establishes a novel IE1/JNK/c-Jun positive feedback loop to drive replication. iScience 2020, 23, 100752. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.Y.; Huang, W.R.; Chi, P.I.; Chiu, H.C.; Liu, H.J. Cell entry of bovine ephemeral fevvier rus requires activation of Src-JNK-AP1 and PI3K-Akt-NF-κB pathways as well as Cox-2-mediated PGE2/EP receptor signaling to enhance clathrin-mediated virus endocytosis. Cell. Microbiol. 2015, 17, 967–987. [Google Scholar] [CrossRef] [PubMed]
- Autret, A.; Martin-Latil, S.; Mousson, L.; Wirotius, A.; Petit, F.; Arnoult, D.; Colbère-Garapin, F.; Estaquier, J.; Blondel, B. Poliovirus induces Bax-dependent cell death mediated by c-Jun NH2-terminal kinase. J. Virol. 2007, 81, 7504–7516. [Google Scholar] [CrossRef] [Green Version]
- Nacken, W.; Wixler, V.; Ehrhardt, C.; Ludwig, S. Influenza A virus NS1 protein-induced JNK activation and apoptosis are not functionally linked. Cell. Microbiol. 2017, 19, e12721. [Google Scholar] [CrossRef] [Green Version]
- Eliopoulos, A.G.; Young, L.S. Activation of the cJun N-terminal kinase (JNK) pathway by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1). Oncogene 1998, 16, 1731–1742. [Google Scholar] [CrossRef] [Green Version]
- Hargett, D.; Mclean, T.; Bachenheimer, S.L. Herpes simplex virus ICP27 activation of stress kinases JNK and p38. J. Virol. 2005, 79, 8348–8360. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Wu, R.F.; Xu, Y.C.; Flores, S.C.; Terada, L.S. HIV Tat activates c-Jun amino-terminal kinase through an oxidant-dependent mechanism. Virology 2001, 286, 62–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.F.; Ma, Z.; Myers, D.P.; Terada, L.S. HIV-1 Tat activates dual Nox pathways leading to independent activation of ERK and JNK MAP kinases. J. Biol. Chem. 2007, 282, 37412–37419. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Zhang, H.; Li, F.; Pan, Z.; Wu, Z.; Wang, Y.; Cui, Y. Activation of PI3K/Akt pathway limits JNK-mediated apoptosis during EV71 infection. Virus Res. 2014, 192, 74–84. [Google Scholar] [CrossRef]
- Kim, S.M.; Park, J.H.; Chung, S.K.; Kim, J.Y.; Hwang, H.Y.; Chung, K.C.; Jo, I.; Park, S.I.; Nam, J.H. Coxsackievirus B3 infection induces cyr61 activation via JNK to mediate cell death. J. Virol. 2004, 78, 13479–13488. [Google Scholar] [CrossRef] [Green Version]
- Clarke, P.; Meintzer, S.M.; Wang, Y.; Moffitt, L.A.; Richardson-Burns, S.M.; Johnson, G.L.; Tyler, K.L. JNK regulates the release of proapoptotic mitochondrial factors in reovirus-infected cells. J. Virol. 2004, 78, 13132–13138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Shi, M.; Zhang, L.; Li, Y.; Sun, J.; Zhang, L.; Wang, X.; Xu, X.; Zhang, X.; Mao, Y.; et al. Activation of JNK1/2 and p38 MAPK signaling pathways promotes enterovirus 71 infection in immature dendritic cells. BMC Microbiol. 2014, 14, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitomo, S.; Omatsu, T.; Tsuchiaka, S.; Nagai, M.; Furuya, T.; Mizutani, T. Activation of c-Jun N-terminal kinase by Akabane virus is required for apoptosis. Res. Vet. Sci. 2016, 107, 147–151. [Google Scholar] [CrossRef]
- Lin, C.; Zimmer, S.G.; Lu, Z.; Holland, R.J.; Dong, Q.; Chambers, T.M. The involvement of a stress-activated pathway in equine influenza virus-mediated apoptosis. Virology 2001, 287, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Shu, W.; Dai, W.; Gao, B.; Xiong, S. Reactive oxygen species-mediated c-Jun NH2-terminal kinase activation contributes to hepatitis B virus X protein-induced autophagy via regulation of the beclin-1/Bcl-2 interaction. J. Virol. 2017, 91, e00001-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, S.R.; Piya, S.; Lu, Z.; Xia, Y.; Alonso, M.M.; White, E.J.; Wei, J.; Gomez-Manzano, C.; Jiang, H.; Fueyo, J. C-Jun N-terminal kinases are required for oncolytic adenovirus-mediated autophagy. Oncogene 2015, 34, 5295–5301. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, M.A.; Malathi, K. RNase L induces autophagy via c-Jun N-terminal kinase and double-stranded RNA-dependent protein kinase signaling pathways. J. Biol. Chem. 2012, 287, 43651–43664. [Google Scholar] [CrossRef] [Green Version]
- Wileman, T. Aggresomes and autophagy generate sites for virus replication. Science 2006, 312, 875–878. [Google Scholar] [CrossRef]
- Hu, W.; Hofstetter, W.; Guo, W.; Li, H.; Pataer, A.; Peng, H.H.; Guo, Z.S.; Bartlett, D.L.; Lin, A.; Swisher, S.G.; et al. JNK-deficiency enhanced oncolytic vaccinia virus replication and blocked activation of double-stranded RNA-dependent protein kinase. Cancer Gene Ther. 2008, 15, 616–624. [Google Scholar] [CrossRef] [Green Version]
- Rahaus, M.; Desloges, N.; Wolff, M.H. Replication of varicella-zoster virus is influenced by the levels of JNK/SAPK and p38/MAPK activation. J. Gen. Virol. 2004, 85, 3529–3540. [Google Scholar] [CrossRef]
- Kurapati, S.; Sadaoka, T.; Rajbhandari, L.; Jagdish, B.; Shukla, P.; Ali, M.A.; Kim, Y.J.; Lee, G.; Cohen, J.I.; Venkatesan, A. Role of the JNK pathway in varicella-zoster virus lytic infection and reactivation. J. Virol. 2017, 91, e00640-17. [Google Scholar] [CrossRef] [Green Version]
- Marudhupandiyan, S.; Balamurugan, K. Intrinsic JNK-MAPK pathway involvement requires daf-16-mediated immune response during Shigella flexneri infection in C. elegans. Immunol. Res. 2017, 65, 609–621. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Qu, F.; Xiang, Z.; Xiao, S.; Wang, F.; Li, J.; Zhang, Y.; Zhang, Y.; Qin, Y.; Yu, Z. c-Jun N-terminal kinase (JNK) is involved in immune defense against bacterial infection in Crassostrea hongkongensis. Dev. Comp. Immunol. 2017, 67, 77–85. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, L.; Zhang, M.; Li, R.; Li, Y.; Hu, X.; Wang, S.; Bao, Z. Characterization of three mitogen-activated protein kinases (MAPK) genes reveals involvement of ERK and JNK, not p38 in defense against bacterial infection in Yesso scallop Patinopecten yessoensis. Fish Shellfish Immunol. 2016, 54, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Hambleton, J.; Weinstein, S.L.; Lem, L.; DeFranco, A.L. Activation of c-Jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophages. Proc. Natl. Acad. Sci. USA 1996, 93, 2774–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Peng, H. Lipopolysaccharides attenuates growth of HS cells through the JNK pathway. Cytotechnology 2016, 68, 2389–2394. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Luo, J.; Chen, D.; Xu, H.; Shi, H.; Jing, X.; Zang, W. CD36 regulates lipopolysaccharide-induced signaling pathways and mediates the internalization of Escherichia coli in cooperation with TLR4 in goat mammary gland epithelial cells. Sci. Rep. 2016, 6, 23132. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Young, D.W.; Gusovsky, F.; Chow, J.C. Cellular events mediated by lipopolysaccharide-stimulated toll-like receptor 4. MD-2 is required for activation of mitogen-activated protein kinases and Elk-1. J. Biol. Chem. 2000, 275, 20861–20866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, C.T.; Kim, E.H.; Luong, T.T.; Pyo, S.; Rhee, D.K. TLR4 mediates pneumolysin-induced ATF3 expression through the JNK/p38 pathway in Streptococcus pneumoniae-infected RAW 264.7 cells. Mol. Cells 2015, 38, 58–64. [Google Scholar] [PubMed] [Green Version]
- Smith, W.E.; Kane, A.V.; Campbell, S.T.; Acheson, D.W.; Cochran, B.H.; Thorpe, C.M. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 2003, 71, 1497–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, J.; Alaoui-El-Azher, M.; Chow, M.; Chambers, T.C.; Baker, H.; Jin, S. c-Jun NH2-terminal kinase-mediated signaling is essential for Pseudomonas aeruginosa ExoS-induced apoptosis. Infect. Immun. 2003, 71, 3361–3370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurarie, L.; Rosenshine, I. Subversion of MAPK signaling by pathogenic bacteria. MAP Kinase 2015, 4, 6–11. [Google Scholar]
- Jones, R.M.; Wu, H.; Wentworth, C.; Luo, L.; Collier-Hyams, L.; Neish, A.S. Salmonella AvrA coordinates suppression of host immune and apoptotic defenses via JNK pathway blockade. Cell Host Microbe 2008, 3, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trosky, J.E.; Mukherjee, S.; Burdette, D.L.; Roberts, M.; McCarter, L.; Siegel, R.M.; Orth, K. Inhibition of MAPK signaling pathways by VopA from Vibrio parahaemolyticus. J. Biol. Chem. 2004, 279, 51953–51957. [Google Scholar] [CrossRef] [Green Version]
- Baruch, K.; Gur-Arie, L.; Nadler, C.; Koby, S.; Yerushalmi, G.; Ben-Neriah, Y.; Yogev, O.; Shaulian, E.; Guttman, C.; Zarivach, R.; et al. Metalloprotease type III effectors that specifically cleave JNK and NF-κB. EMBO J. 2011, 30, 221–231. [Google Scholar] [CrossRef]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar] [CrossRef]
- Pellizzari, R.; Guidi-Rontani, C.; Vitale, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFN gamma-induced release of NO and TNF alpha. FEBS Lett. 1999, 462, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Vitale, G.; Pellizzari, R.; Recchi, C.; Napolitani, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 1998, 248, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.M.; Jeon, B.Y.; Lee, H.M.; Jin, H.S.; Yuk, J.M.; Song, C.H.; Lee, S.H.; Lee, Z.W.; Cho, S.N.; Kim, J.M.; et al. Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog. 2010, 6, e1001230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, N.; Liu, Z.S.; Xue, W.; Bai, Z.F.; Wang, Q.Y.; Dai, J.; Liu, X.; Huang, Y.J.; Cai, H.; Zhan, X.Y.; et al. NLRP3 Phosphorylation is an essential priming event for inflammasome activation. Mol. Cell 2017, 68, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Hara, H.; Tsuchiya, K.; Kawamura, I.; Fang, R.; Hernandez-Cuellar, E.; Shen, Y.; Mizuguchi, J.; Schweighoffer, E.; Tybulewicz, V.; Mitsuyama, M. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 2013, 14, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Huang, Q.; Ye, C.; Wu, R.; Lei, G.; Jiang, J.; Chen, T.; Peng, Y.; Fang, R. Syk and JNK signaling pathways are involved in inflammasome activation in macrophages infected with Streptococcus pneumoniae. Biochem. Biophys. Res. Commun. 2018, 507, 217–222. [Google Scholar] [CrossRef]
- Okada, M.; Matsuzawa, A.; Yoshimura, A.; Ichijo, H. The lysosome rupture-activated TAK1-JNK pathway regulates NLRP3 inflammasome activation. J. Biol. Chem. 2014, 289, 32926–32936. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Yang, D.; Han, F.; Tan, J.; Zhang, L.; Xiao, J.; Zhang, Y.; Liu, Q. The bacterial T6SS effector EvpP prevents NLRP3 inflammasome activation by inhibiting the Ca2+-dependent MAPK-Jnk pathway. Cell Host Microbe 2017, 21, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Achterman, R.R.; Moyes, D.L.; Thavaraj, S.; Smith, A.R.; Blair, K.M.; White, T.C.; Naglik, J.R. Dermatophytes activate skin keratinocytes via mitogen-activated protein kinase signaling and induce immune responses. Infect. Immun. 2015, 83, 1705–1714. [Google Scholar] [CrossRef] [Green Version]
- Geissler, A.; Haun, F.; Frank, D.O.; Wieland, K.; Simon, M.M.; Idzko, M.; Davis, R.J.; Maurer, U.; Borner, C. Apoptosis induced by the fungal pathogen gliotoxin requires a triple phosphorylation of Bim by JNK. Cell Death Differ. 2013, 20, 1317–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galley, Y.; Hagens, G.; Glaser, I.; Davis, W.; Eichhorn, M.; Dobbelaere, D. Jun NH2-terminal kinase is constitutively activated in T cells transformed by the intracellular parasite Theileria parva. Proc. Natl. Acad. Sci. USA 1997, 94, 5119–5124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valère, A.; Garnotel, R.; Villena, I.; Guenounou, M.; Pinon, J.M.; Aubert, D. Activation of the cellular mitogen-activated protein kinase pathways ERK, P38 and JNK during Toxoplasma gondii invasion. Parasite 2003, 10, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suman, S.; Rachakonda, G.; Mandape, S.N.; Sakhare, S.S.; Villalta, F.; Pratap, S.; Lima, M.F.; Nde, P.N. Phospho-proteomic analysis of primary human colon epithelial cells during the early Trypanosoma cruzi infection phase. PLoS Negl. Trop. Dis. 2018, 12, e0006792. [Google Scholar] [CrossRef]
- Anand, S.S.; Babu, P.P. c-Jun N terminal kinases (JNK) are activated in the brain during the pathology of experimental cerebral malaria. Neurosci. Lett. 2011, 488, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Gong, P.; Tai, L.; Li, X.; Wang, X.; Zhao, C.; Zhang, X.; Yang, Z.; Yang, J.; Li, J.; et al. Extracellular vesicles secreted by Neospora caninum are recognized by toll-like receptor 2 and modulate host cell innate immunity through the MAPK signaling pathway. Front. Immunol. 2018, 9, 1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukhumavasi, W.; Warren, A.L.; Del-Rio, L.; Denkers, E.Y. Absence of mitogen-activated protein kinase family member c-Jun N-terminal kinase-2 enhances resistance to Toxoplasma gondii. Exp. Parasitol. 2010, 126, 415–420. [Google Scholar] [CrossRef] [Green Version]
- Ramphul, U.N.; Garver, L.S.; Molina-Cruz, A.; Canepa, G.E.; Barillas-Mury, C. Plasmodium falciparum evades mosquito immunity by disrupting JNK-mediated apoptosis of invaded midgut cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Kaneto, H.; Kawamori, D.; Nakatani, Y.; Gorogawa, S.; Matsuoka, T.A. Oxidative stress and the JNK pathway as a potential therapeutic target for diabetes. Drug News Perspect. 2004, 17, 447–453. [Google Scholar] [CrossRef]
- Wu, Q.; Wu, W.; Jacevic, V.; Franca, T.C.C.; Wang, X.; Kuca, K. Selective inhibitors for JNK signalling: A potential targeted therapy in cancer. J. Enzym. Inhib. Med. Chem. 2020, 35, 574–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Lin, W. The JNK signaling pathway as a potential new target for depression. Chin. Sci. Bull. 2018, 63, 1998–2009. (In Chinese) [Google Scholar] [CrossRef]
- Bogoyevitch, M.A. Therapeutic promise of JNK ATP-noncompetitive inhibitors. Trends Mol. Med. 2005, 11, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; You, Z.; Wang, Q.; Zhou, Z.J.; Qiu, Y.; Luo, R.; Ge, X.Y. The epidemic of 2019-novel-coronavirus (2019-nCoV) pneumonia and insights for emerging infectious diseases in the future. Microbes Infect. 2020, 22, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Zhang, M.; Zhang, Y.; Xu, Z. JNK pathway: Diseases and therapeutic potential. Acta Pharmacol. Sin. 2007, 28, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Niu, X.; Qian, Z.; Qian, J.; Xuan, B. The c-Jun N-terminal kinase inhibitor SP600125 inhibits human cytomegalovirus replication. J. Med. Virol. 2015, 87, 2135–2144. [Google Scholar] [CrossRef]
- He, Z.; Chen, X.; Fu, M.; Tang, J.; Li, X.; Cao, H.; Wang, Y.; Zheng, S.J. Inhibition of fowl adenovirus serotype 4 replication in Leghorn male hepatoma cells by SP600125 via blocking JNK MAPK pathway. Vet. Microbiol. 2019, 228, 45–52. [Google Scholar] [CrossRef]
- Zhu, L.; Yuan, C.; Huang, L.; Ding, X.; Wang, J.; Zhang, D.; Zhu, G. The activation of p38MAPK and JNK pathways in bovine herpesvirus 1 infected MDBK cells. Vet. Res. 2016, 47, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shai, B.; Schmukler, E.; Yaniv, R.; Ziv, N.; Horn, G.; Bumbarov, V.; Yadin, H.; Smorodinsky, N.I.; Bacharach, E.; Pinkas-Kramarski, R.; et al. Epizootic hemorrhagic disease virus induces and benefits from cell stress, autophagy, and apoptosis. J. Virol. 2013, 87, 13397–13408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, A.C.; Soares-Martins, J.A.; Leite, F.G.; Da-Cruz, A.F.; Torres, A.A.; Souto-Padrón, T.; Kroon, E.G.; Ferreira, P.C.; Bonjardim, C.A. SP600125 inhibits Orthopoxviruses replication in a JNK1/2 -independent manner: Implication as a potential antipoxviral. Antivir. Res. 2012, 93, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Dimitrakopoulos, O.; Liopeta, K.; Dimitracopoulos, G.; Paliogianni, F. Replication of Brucella melitensis inside primary human monocytes depends on mitogen activated protein kinase signaling. Microbes Infect. 2013, 15, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Kobayashi, M.; Eshita, Y.; Shirato, K.; Kimura, T.; Ako, Y.; Miyoshi, H.; Takasaki, T.; Kurane, I.; Kariwa, H.; et al. Involvement of the JNK-like protein of the Aedes albopictus mosquito cell line, C6/36, in phagocytosis, endocytosis and infection of West Nile virus. Insect Mol. Biol. 2003, 12, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ishida, H.; Yamane, D.; Yi, M.; Swinney, D.C.; Foung, S.; Lemon, S.M. Contrasting roles of mitogen-activated protein kinases in cellular entry and replication of hepatitis C virus: MKNK1 facilitates cell entry. J. Virol. 2013, 87, 4214–4224. [Google Scholar] [CrossRef] [Green Version]
- Kortesoja, M.; Trofin, R.E.; Hanski, L. A platform for studying the transfer of Chlamydia pneumoniae infection between respiratory epithelium and phagocytes. J. Microbiol. Methods 2020, 171, 105857. [Google Scholar] [CrossRef]
- Huang, M.; Xu, A.; Wu, X.; Zhang, Y.; Guo, Y.; Guo, F.; Pan, Z.; Kong, L. Japanese encephalitis virus induces apoptosis by the IRE1/JNK pathway of ER stress response in BHK-21 cells. Arch. Virol. 2016, 161, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Si, X.; Luo, H.; Morgan, A.; Zhang, J.; Wong, J.; Yuan, J.; Esfandiarei, M.; Gao, G.; Cheung, C.; McManus, B.M. Stress-Activated Protein Kinases Are Involved in Coxsackievirus B3 Viral Progeny Release. J. Virol. 2005, 79, 13875–13881. [Google Scholar] [CrossRef] [Green Version]
- Marozin, S.; Altomonte, J.; Apfel, S.; Dinh, P.X.; De-Toni, E.N.; Rizzani, A.; Nüssler, A.; Kato, N.; Schmid, R.M.; Pattnaik, A.K.; et al. Posttranslational modification of vesicular stomatitis virus glycoprotein, but not JNK inhibition, is the antiviral mechanism of SP600125. J. Virol. 2012, 86, 4844–4855. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.; Huo, Y.; Dong, Y.; Fan, L.; Yang, H.; Wang, L.; Ning, Y.; Hu, H. Activation of c-Jun NH(2)-terminal kinase is required for porcine reproductive and respiratory syndrome virus-induced apoptosis but not for virus replication. Virus Res. 2012, 166, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Caly, L.; Li, H.M.; Bogoyevitch, M.A.; Jans, D.A. c-Jun N-terminal kinase activity is required for efficient respiratory syncytial virus production. Biochem. Biophys. Res. Commun. 2017, 483, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Messoussi, A.; Feneyrolles, C.; Bros, A.; Deroide, A.; Daydé-Cazals, B.; Chevé, G.; Van-Hijfte, N.; Fauvel, B.; Bougrin, K.; Yasri, A. Recent progress in the design, study, and development of c-Jun N-terminal kinase inhibitors as anticancer agents. Chem. Biol. 2014, 21, 1433–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogoyevitch, M.A.; Arthur, P.G. Inhibitors of c-Jun N-terminal kinases: JuNK no more? Biochim. Biophys. Acta 2008, 1784, 76–93. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhu, Z.; Wang, J.; Liu, J. JNK and p38 mitogen-activated protein kinase pathways contribute to porcine circovirus type 2 infection. J. Virol. 2009, 83, 6039–6047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, T.I.; Bachenheimer, S.L. Activation of cJUN N-terminal kinase by herpes simplex virus type 1 enhances viral replication. J. Virol. 1999, 73, 8415–8426. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Inhibitor | Inhibitor | Effects on Pathogens | Pathogens | References |
|---|---|---|---|---|
| ATP-competitive inhibitors | SP600125 | Reduce pathogen replication | Viruses: Kaposi’s sarcoma-associated herpesvirus, human cytomegalovirus, rotavirus, fowl adenovirus serotype 4, bovine herpesvirus 1, epizootic hemorrhagic disease virus, Orthopoxviruses. Bacteria: Brucella melitensis Fungus: Candida albicans | [21,41,44,109,110,111,112,113,114] |
| Inhibit pathogen entry | Viruses: West Nile virus, hepatitis C Virus Bacteria: Chlamydia pneumonia | [115,116,117] | ||
| Enhance pathogen replication | Viruses: Oncolytic vaccinia virus | [66] | ||
| No significant effects | Viruses: Japanese encephalitis virus, coxsackievirus B3, vesicular stomatitis virus, porcine reproductive, respiratory syndrome virus, and poliovirus. | [49,118,119,120,121] | ||
| AS601245 | Reduce pathogen replication | Viruses: hepatitis C Virus | [116] | |
| JNK-IN-8 | Reduce pathogen replication | Viruses: respiratory syncytial virus | [122] | |
| Small peptide inhibitors | JNK peptide inhibitor 1 | Reduce pathogen replication | Viruses: porcine circovirus type 2 | [125] |
| JIP-1 | Reduce pathogen replication | Viruses: herpes simplex virus type 1 | [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Ye, C.; Wan, C.; Li, G.; Peng, L.; Peng, Y.; Fang, R. The Roles of c-Jun N-Terminal Kinase (JNK) in Infectious Diseases. Int. J. Mol. Sci. 2021, 22, 9640. https://doi.org/10.3390/ijms22179640
Chen J, Ye C, Wan C, Li G, Peng L, Peng Y, Fang R. The Roles of c-Jun N-Terminal Kinase (JNK) in Infectious Diseases. International Journal of Molecular Sciences. 2021; 22(17):9640. https://doi.org/10.3390/ijms22179640
Chicago/Turabian StyleChen, Jing, Chao Ye, Chao Wan, Gang Li, Lianci Peng, Yuanyi Peng, and Rendong Fang. 2021. "The Roles of c-Jun N-Terminal Kinase (JNK) in Infectious Diseases" International Journal of Molecular Sciences 22, no. 17: 9640. https://doi.org/10.3390/ijms22179640
APA StyleChen, J., Ye, C., Wan, C., Li, G., Peng, L., Peng, Y., & Fang, R. (2021). The Roles of c-Jun N-Terminal Kinase (JNK) in Infectious Diseases. International Journal of Molecular Sciences, 22(17), 9640. https://doi.org/10.3390/ijms22179640