
Multiple Imputation Approaches Applied to the Missing Value Problem in Bottom-Up Proteomics

Abstract
:1. Introduction
2. Results
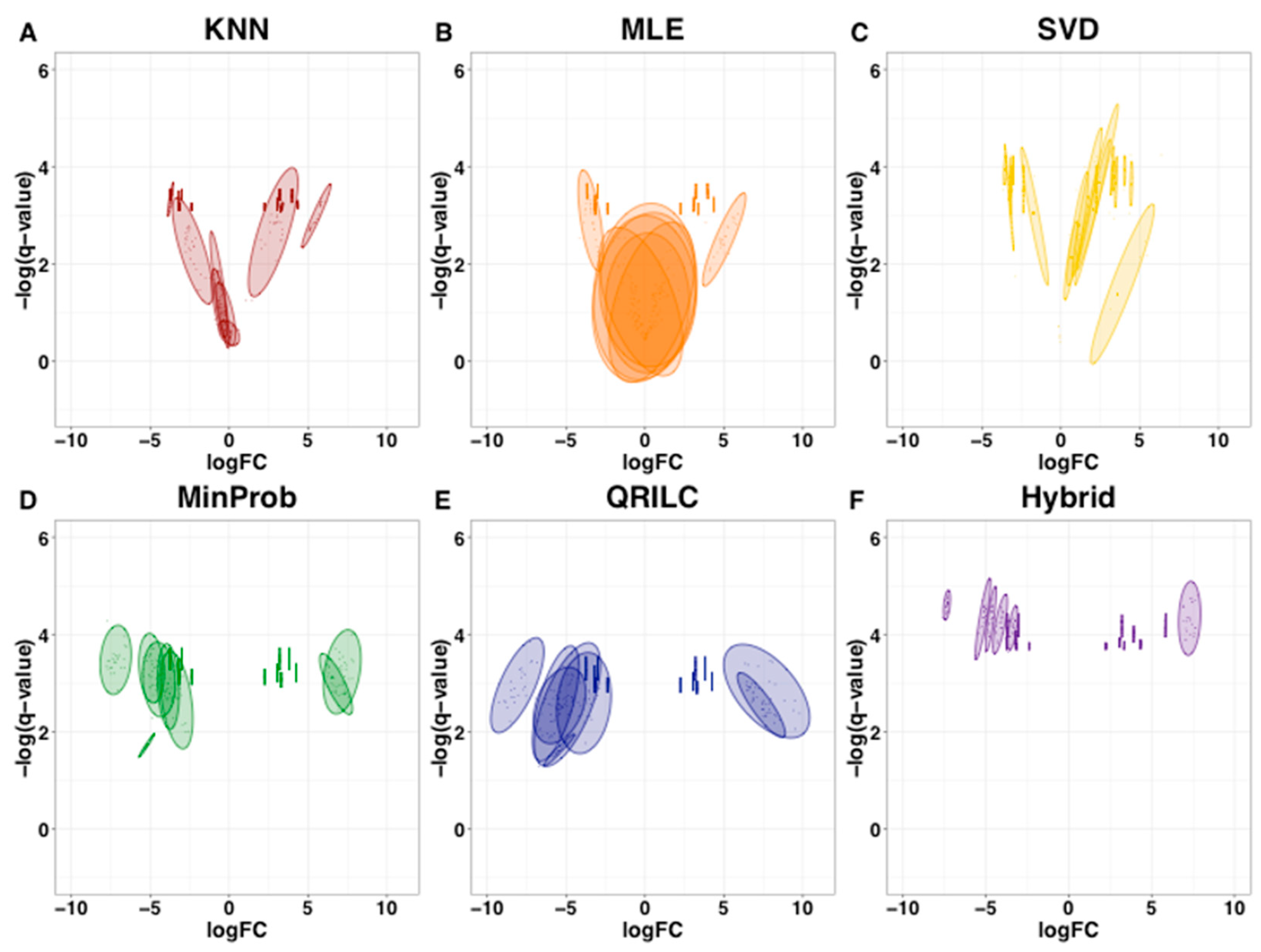
2.1. Imputation with Simulated Dataset
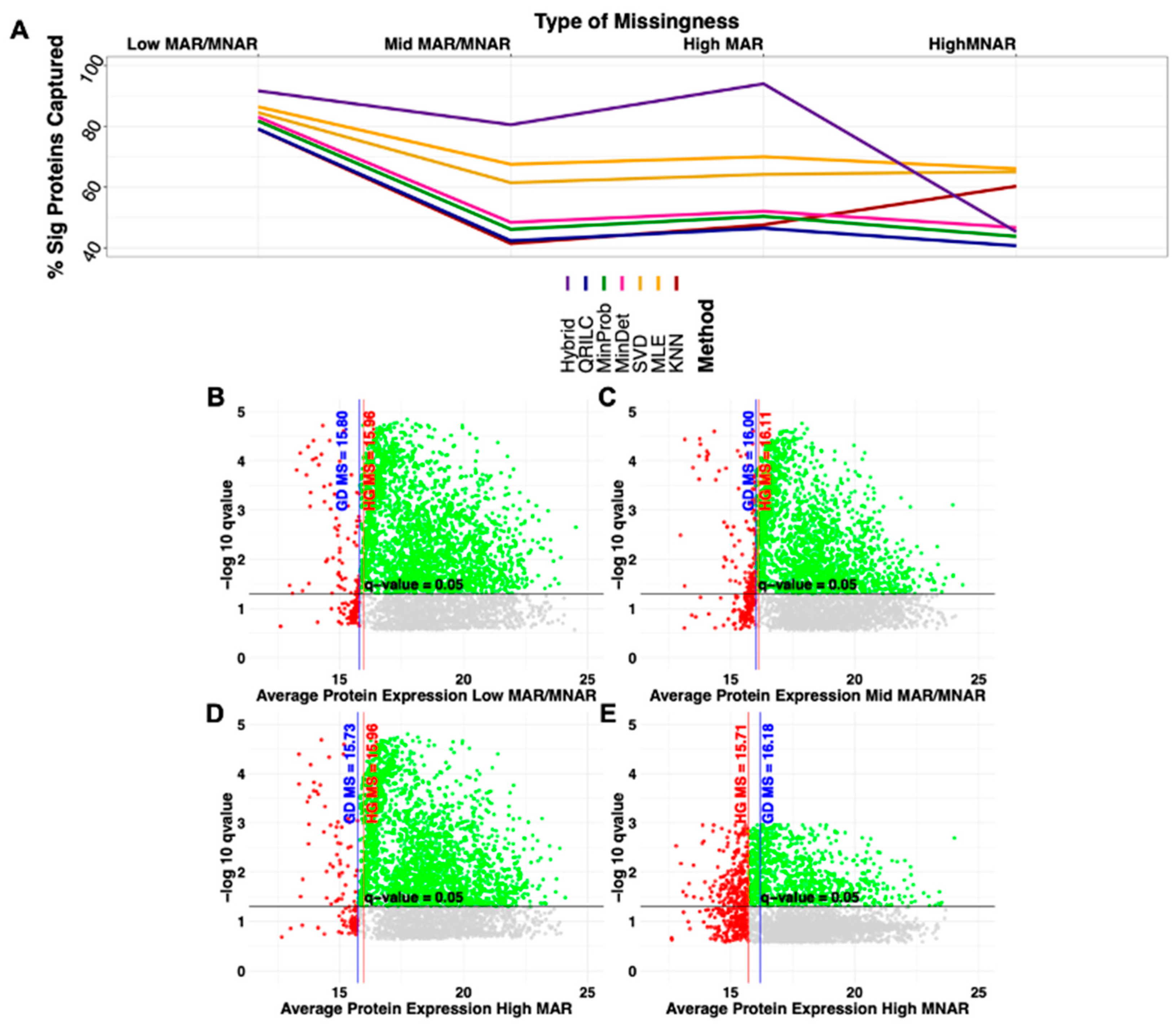
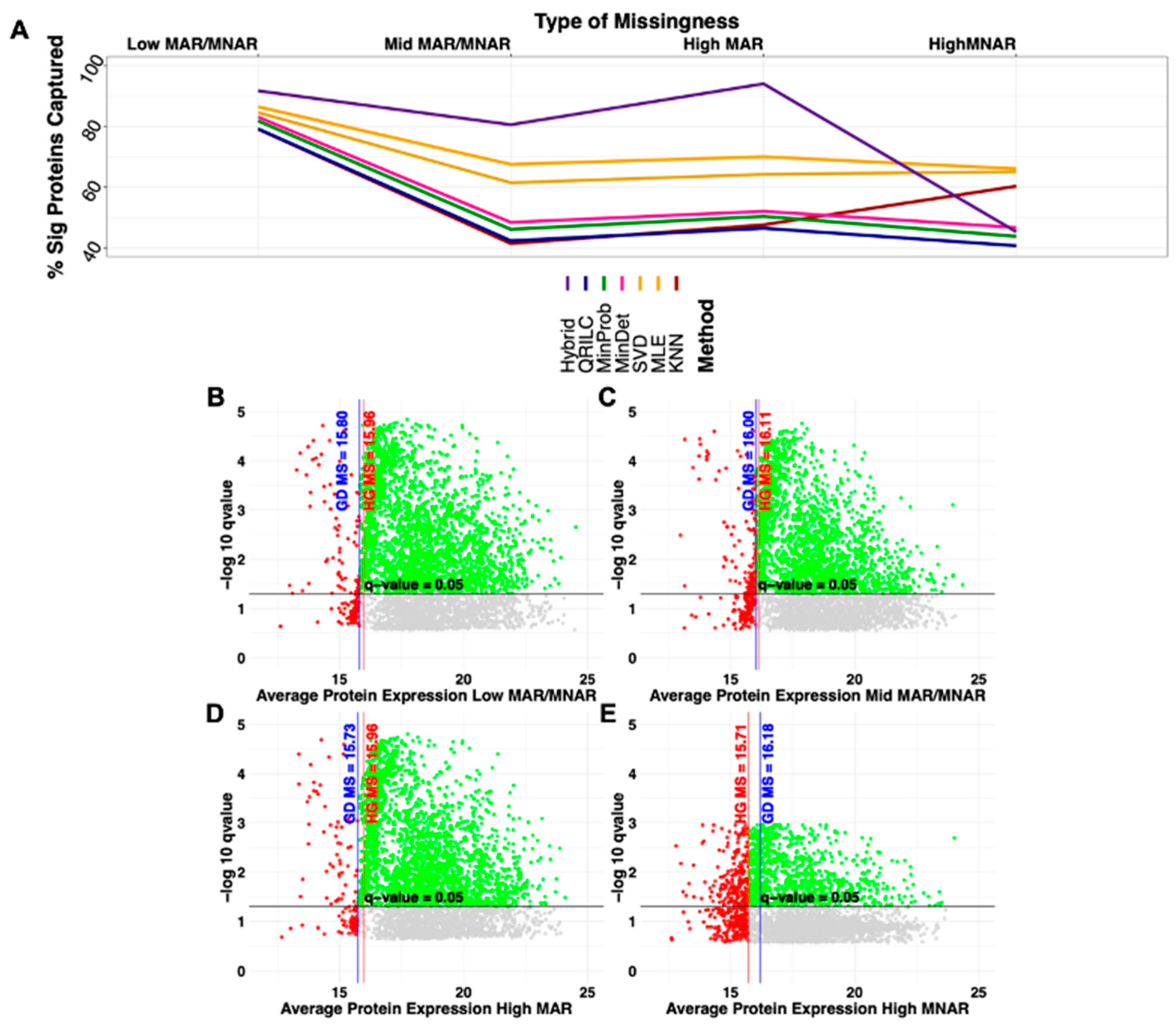
2.2. Type of Amputation and Increasing Missingness Influences on Significance
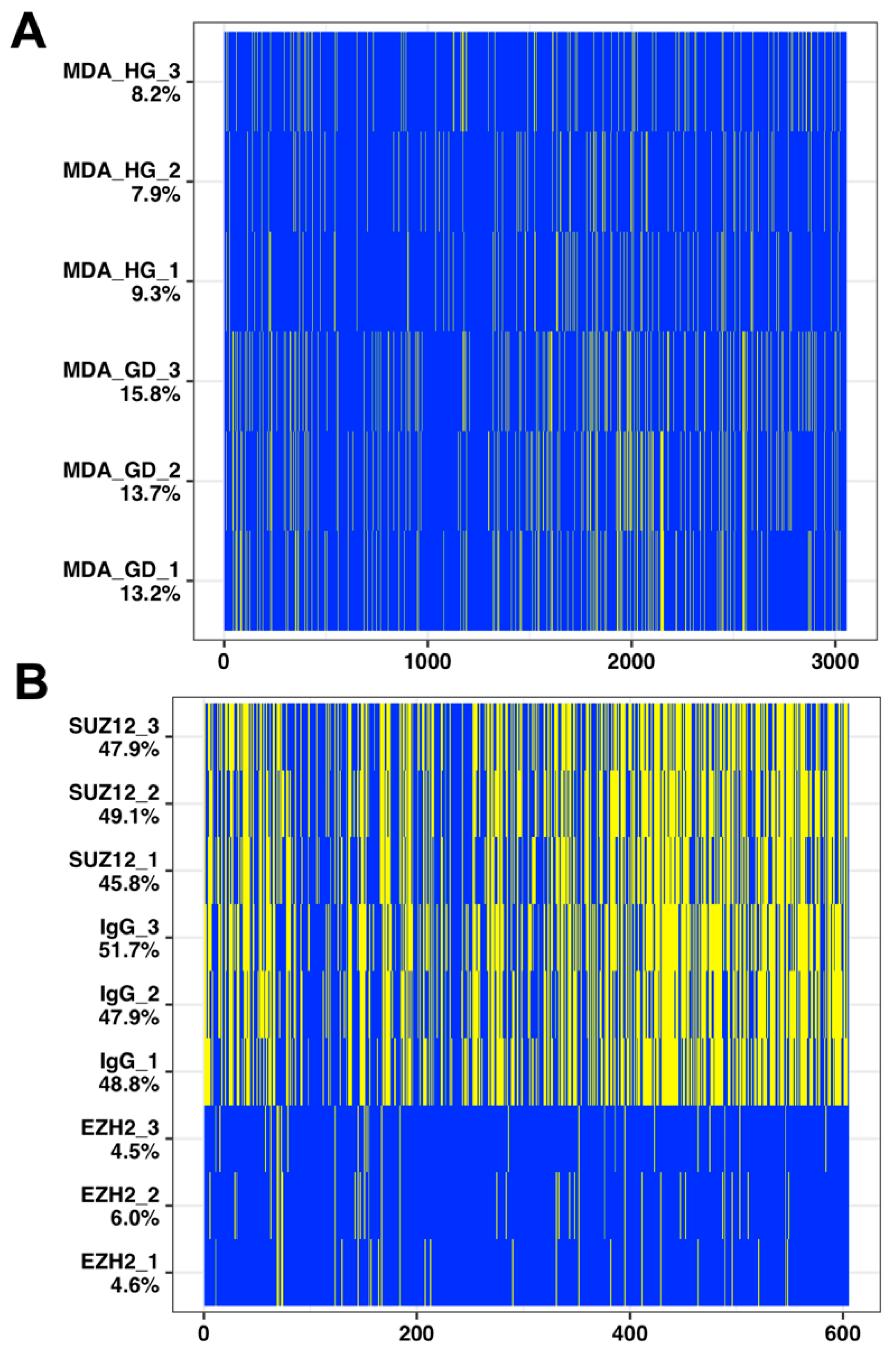
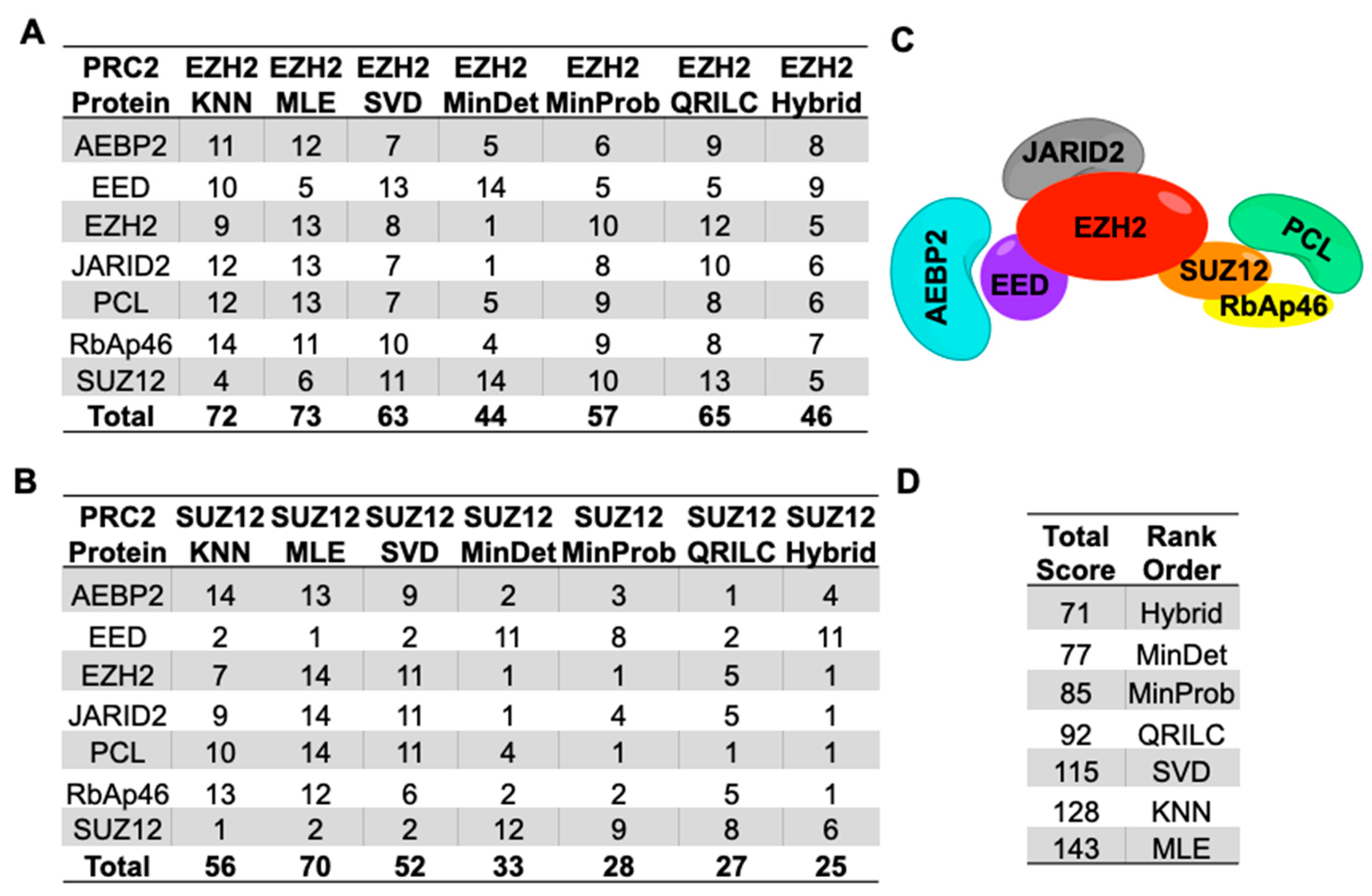
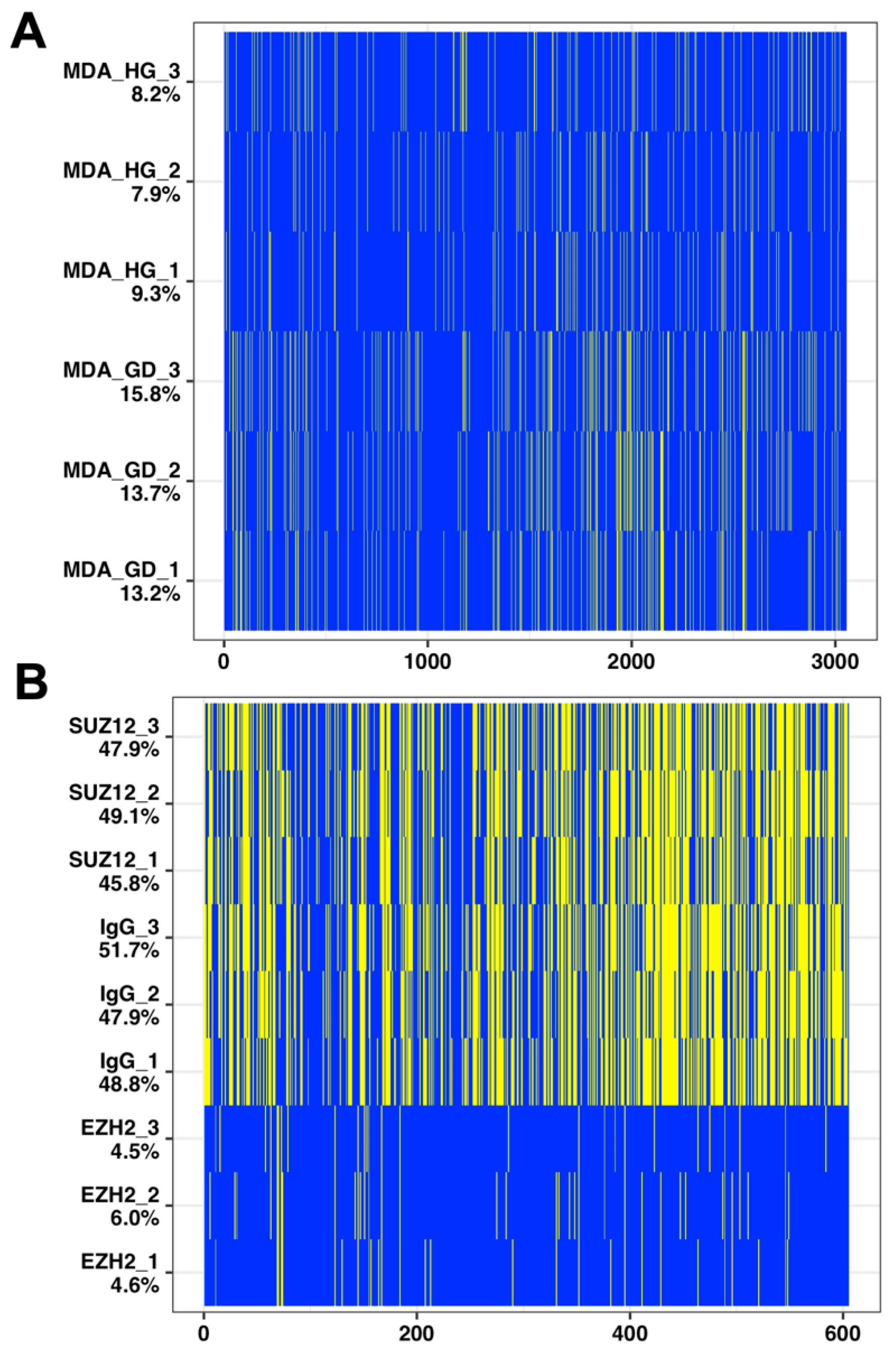
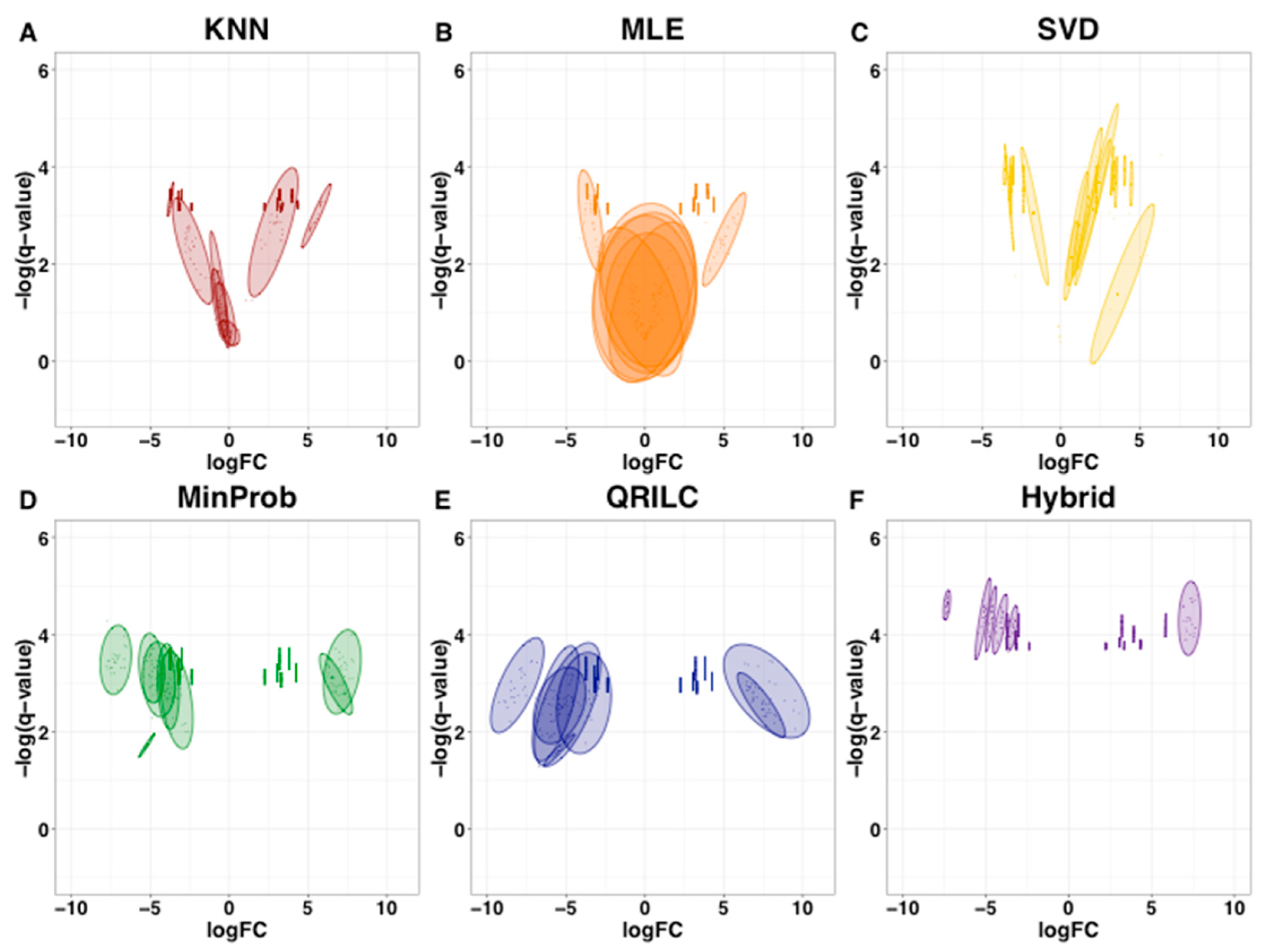
2.3. Imputation with Small Dataset of Similar Proteomic Profiles
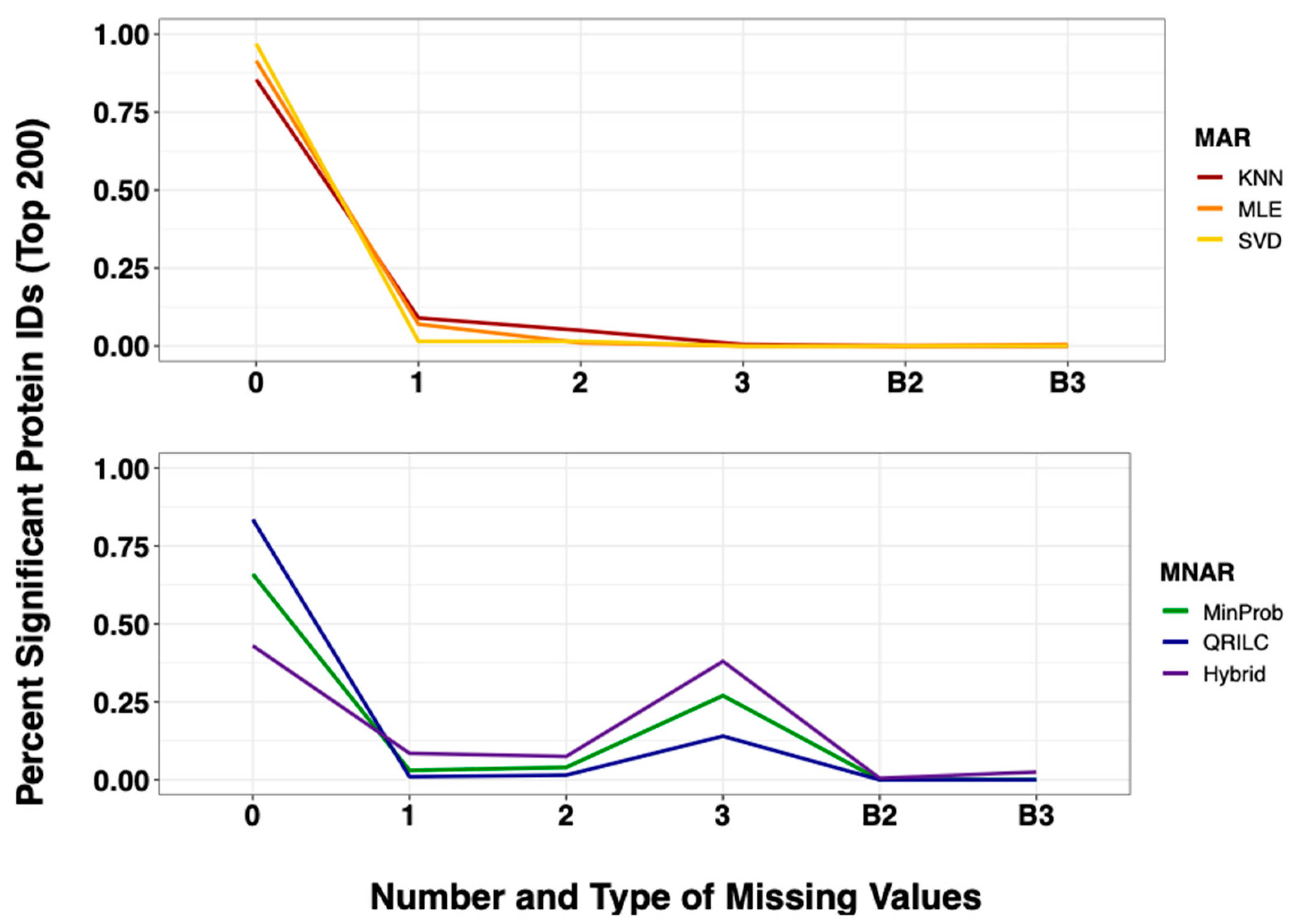
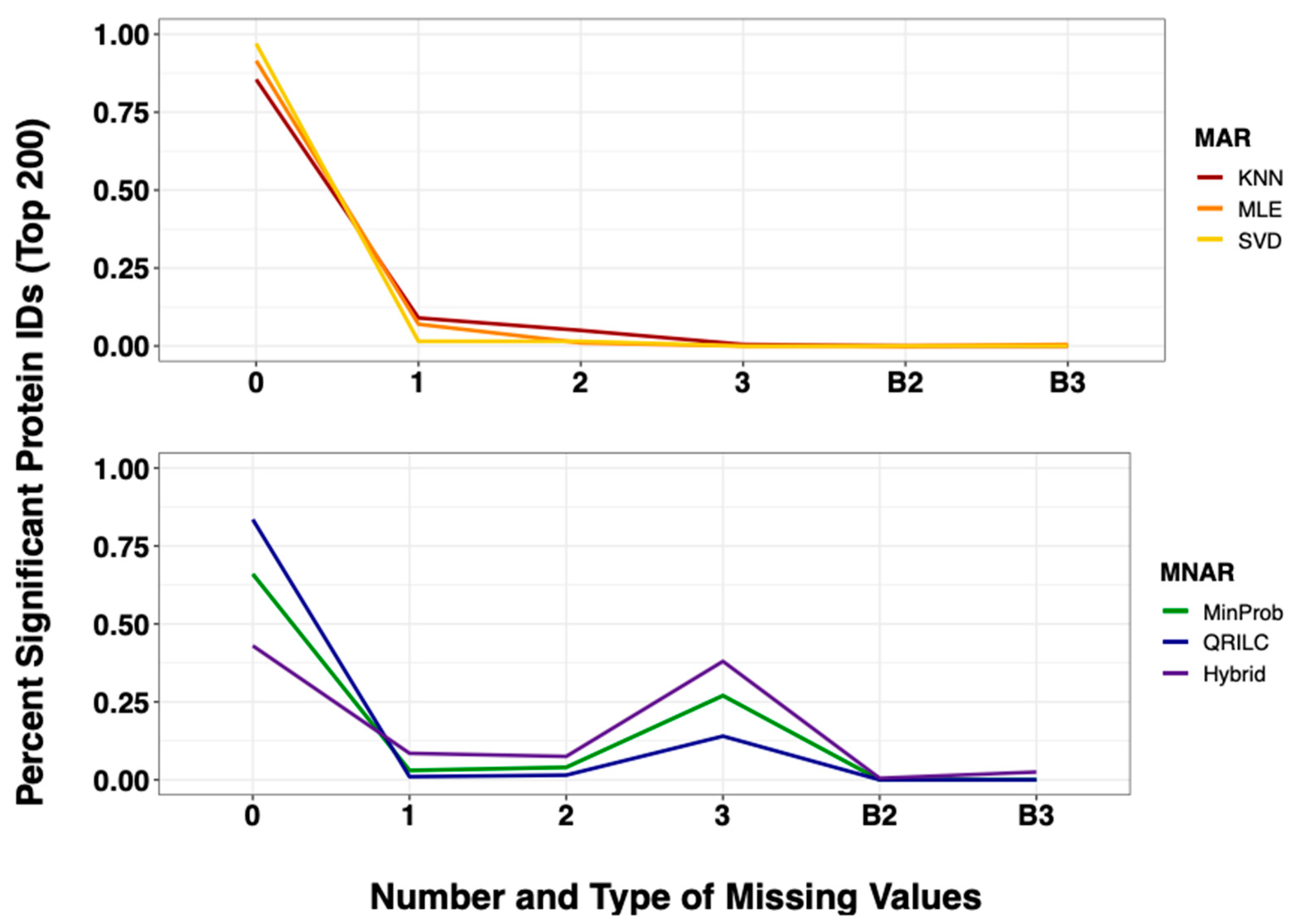
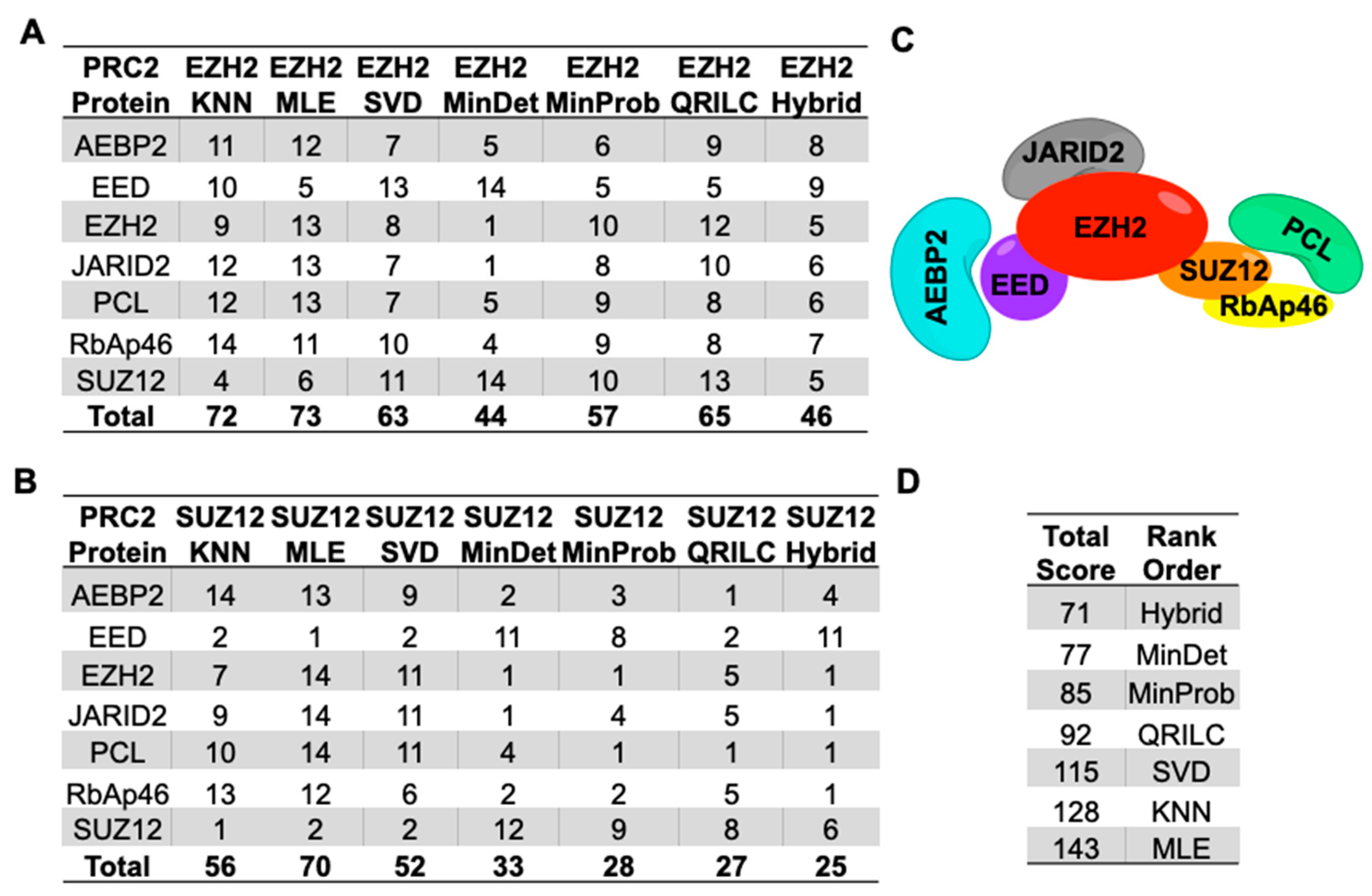
2.4. Imputation Influences with Increasing Number and Type of Missing Values
3. Discussion
4. Materials and Methods
4.1. Proteomic Datasets
4.1.1. Glucose Deprivation
4.1.2. Pluripotent Cell Differentiation
4.2. Database Searching and Label-Free Quantitation (LFQ)
4.3. Generation of Simulated and Amputed Datasets
4.4. Data Processing, Imputation and Differential Expression Analysis of Simulated Datasets
4.5. Data Processing, Imputation, Differential Expression/Enrichment Analysis and Top Protein Lists with Proteomic Data
4.6. Relative Ranking of Analyses
5. Conclusions
- Single MAR or MNAR strategies are acceptable approaches in proteomics only when the nature of missingness is known to the researcher;
- When an entire protein observation is missing from a treatment or group (three missing values in this case), a single MAR or MNAR imputation strategy is not recommended as the downstream statistics demonstrate the majority of significant identifications contain no missing values. This observation suggests that either the methods have a bias to choose complete cases, or the algorithms are imputing values too close to the observed to be considered significant;
- The statistics with single MAR or MNAR strategies (not the SFI-hybrid) are negatively impacted by increasing number and type of missingness, characterized by large standard deviations, logFC sign fluctuations and an overall trend toward non-significance as seen by the loss in the number of significant proteins from the ground truth and known protein complex interactors;
- To avoid unnecessarily excluding data as in a complete case analysis, a combinatorial MAR/MNAR approach, such as SFI-hybrid, that imputes missing values separately for each treatment group most accurately and reproducibly models bottom-up proteomics data regardless of the missing value type (with the exception of high MNAR as explained in the discussion section).
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DDA | data-dependent acquisition |
| MAR | missing at random |
| MNAR | missing not at random |
| LOD | limit of detection |
| MCAR | missing completely at random |
| kNN | k nearest neighbors |
| MLE | maximum likelihood estimation |
| SVD | singular value decomposition |
| IP | immunoprecipitation |
| IgG | immunoglobulin |
| MinDet | deterministic minimum |
| MinProb | probabilistic minimum |
| QRILC | quantile regression imputation of left-censored data |
| DEP | differentially expressed/enriched proteins |
| MI | multiple imputation |
| MI-MFA | multiple imputation in multi-factor analysis |
| PRIDE | PRoteomics IDEntifications |
| NT2 | NTERA2 cells |
| PRC2 | polycomb repressive complex 2 |
| FDR | false discovery rate |
| LFQ | label-free quantitation |
| GD | glucose deprivation |
| HG | high glucose |
| SFI | select filter imputation |
| IP-MS/MS | immunoprecipitation tandem mass spectrometry |
References
- Scheltema, R.A.; Hauschild, J.P.; Lange, O.; Hornburg, D.; Denisov, E.; Damoc, E.; Kuehn, A.; Makarov, A.; Mann, M. The Q Exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol. Cell Proteom. 2014, 13, 3698–3708. [Google Scholar] [CrossRef] [Green Version]
- Lubeck, M. Pasef on a Timstof Pro Defines New Performance Standards for Shotgun Proteomics with Dramatic Improvements in ms/ms Data Acquisition Rates and Sensitivity. Bruker Daltonik GmbH Tech. Rep. 2017. Available online: https://www.bruker.com/en/products-and-solutions/mass-spectrometry/timstof/timstof-pro.html (accessed on 28 August 2021).
- Papaioannou, D.; Petri, A.; Dovey, O.M.; Terreri, S.; Wang, E.; Collins, F.A.; Woodward, L.A.; Walker, A.E.; Nicolet, D.; Pepe, F.; et al. The long non-coding RNA HOXB-AS3 regulates ribosomal RNA transcription in NPM1-mutated acute myeloid leukemia. Nat. Commun. 2019, 10, 5351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.L.; Li, S.; Mertins, P.; Cao, S.; Gunawardena, H.P.; Ruggles, K.V.; Mani, D.R.; Clauser, K.R.; Tanioka, M.; Usary, J.; et al. Proteogenomic integration reveals therapeutic targets in breast cancer xenografts. Nat. Commun. 2017, 8, 14864. [Google Scholar] [CrossRef] [PubMed]
- Karpievitch, Y.V.; Dabney, A.R.; Smith, R.D. Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinform. 2012, 13 (Suppl. 16), S5. [Google Scholar] [CrossRef] [Green Version]
- Lazar, C.; Gatto, L.; Ferro, M.; Bruley, C.; Burger, T. Accounting for the Multiple Natures of Missing Values in Label-Free Quantitative Proteomics Data Sets to Compare Imputation Strategies. J. Proteome Res. 2016, 15, 1116–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valikangas, T.; Suomi, T.; Elo, L.L. A comprehensive evaluation of popular proteomics software workflows for label-free proteome quantification and imputation. Brief Bioinform. 2018, 19, 1344–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb-Robertson, B.J.; Wiberg, H.K.; Matzke, M.M.; Brown, J.N.; Wang, J.; McDermott, J.E.; Smith, R.D.; Rodland, K.D.; Metz, T.O.; Pounds, J.G.; et al. Review, evaluation, and discussion of the challenges of missing value imputation for mass spectrometry-based label-free global proteomics. J. Proteome Res. 2015, 14, 1993–2001. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.; Wang, J.; Su, M.; Jia, E.; Chen, S.; Chen, T.; Ni, Y. Missing Value Imputation Approach for Mass Spectrometry-based Metabolomics Data. Sci. Rep. 2018, 8, 663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.J.; Gunawardena, H.P.; Paulo, J.A.; Chen, X.; Ibrahim, J.G.; Gygi, S.P.; Qaqish, B.F. The effects of nonignorable missing data on label-free mass spectrometry proteomics experiments. Ann. Appl. Stat. 2018, 12, 2075–2095. [Google Scholar] [CrossRef]
- Wang, J.; Li, L.; Chen, T.; Ma, J.; Zhu, Y.; Zhuang, J.; Chang, C. In-depth method assessments of differentially expressed protein detection for shotgun proteomics data with missing values. Sci. Rep. 2017, 7, 3367. [Google Scholar] [CrossRef]
- Karpievitch, Y.; Stanley, J.; Taverner, T.; Huang, J.; Adkins, J.N.; Ansong, C.; Heffron, F.; Metz, T.O.; Qian, W.J.; Yoon, H.; et al. A statistical framework for protein quantitation in bottom-up MS-based proteomics. Bioinformatics 2009, 25, 2028–2034. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Fisher, K.; Meng, W.; Fang, B.; Welsh, E.; Haura, E.B.; Koomen, J.M.; Eschrich, S.A.; Fridley, B.L.; Chen, Y.A. GMSimpute: A generalized two-step Lasso approach to impute missing values in label-free mass spectrum analysis. Bioinformatics 2020, 36, 257–263. [Google Scholar] [CrossRef]
- Rubin, D.B. Inference and Missing Data. Biometrika 1976, 63, 581–592. [Google Scholar] [CrossRef]
- Troyanskaya, O.; Cantor, M.; Sherlock, G.; Brown, P.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing value estimation methods for DNA microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar] [CrossRef] [Green Version]
- Schafer, J.L.; Graham, J.W. Missing Data: Our View of the State of the Art. Psychol. Methods 2002, 7, 147–177. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, J.G.; Chen, M.-H.; Lipsitz, S.R.; Herring, A.H. Missing-Data Methods for Generalized Linear Models: A Comparative Review. J. Am. Stat. Assoc. 2005, 100, 332–346. [Google Scholar] [CrossRef]
- Almeida, J.S.; Stanislaus, R.; Krug, E.; Arthur, J.M. Normalization and analysis of residual variation in two-dimensional gel electrophoresis for quantitative differential proteomics. Proteomics 2005, 5, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Meleth, S.; Deshane, J.; Kim, H. The case for well-conducted experiments to validate statistical protocols for 2D gels: Different pre-processing = different lists of significant proteins. BMC Biotechnol. 2005, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chich, J.F.; David, O.; Villers, F.; Schaeffer, B.; Lutomski, D.; Huet, S. Statistics for proteomics: Experimental design and 2-DE differential analysis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 849, 261–272. [Google Scholar] [CrossRef]
- Fu, J.; Luo, Y.; Mou, M.; Zhang, H.; Tang, J.; Wang, Y.; Zhu, F. Advances in Current Diabetes Proteomics: From the Perspectives of Label-free Quantification and Biomarker Selection. Curr. Drug Targets 2020, 21, 34–54. [Google Scholar] [CrossRef]
- Zhao, L.; Cong, X.; Zhai, L.; Hu, H.; Xu, J.Y.; Zhao, W.; Zhu, M.; Tan, M.; Ye, B.C. Comparative evaluation of label-free quantification strategies. J. Proteom. 2020, 215, 103669. [Google Scholar] [CrossRef]
- Karpievitch, Y.V.; Taverner, T.; Adkins, J.N.; Callister, S.J.; Anderson, G.A.; Smith, R.D.; Dabney, A.R. Normalization of peak intensities in bottom-up MS-based proteomics using singular value decomposition. Bioinformatics 2009, 25, 2573–2580. [Google Scholar] [CrossRef]
- Wei, R.; Wang, J.; Jia, E.; Chen, T.; Ni, Y.; Jia, W. GSimp: A Gibbs sampler based left-censored missing value imputation approach for metabolomics studies. PLoS Comput. Biol. 2018, 14, e1005973. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Luan, H.; Ji, F.; Chen, Y.; Cai, Z. StatTarget: A streamlined tool for signal drift correction and interpretations of quantitative mass spectrometry-based omics data. Anal. Chim. Acta 2018, 1036, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, S.; Combes, F.; Lazar, C.; Giai Gianetto, Q.; Gatto, L.; Dorffer, A.; Hesse, A.M.; Coute, Y.; Ferro, M.; Bruley, C.; et al. DAPAR & ProStaR: Software to perform statistical analyses in quantitative discovery proteomics. Bioinformatics 2017, 33, 135–136. [Google Scholar] [PubMed] [Green Version]
- Wieczorek, S.; Combes, F.; Borges, H.; Burger, T. Protein-Level Statistical Analysis of Quantitative Label-Free Proteomics Data with ProStaR. Methods Mol. Biol. 2019, 1959, 225–246. [Google Scholar] [PubMed]
- Chang, C.; Xu, K.; Guo, C.; Wang, J.; Yan, Q.; Zhang, J.; He, F.; Zhu, Y. PANDA-view: An easy-to-use tool for statistical analysis and visualization of quantitative proteomics data. Bioinformatics 2018, 34, 3594–3596. [Google Scholar] [CrossRef] [Green Version]
- Gatto, L.; Lilley, K.S. MSnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 2012, 28, 288–289. [Google Scholar] [CrossRef] [Green Version]
- Gatto, L.; Gibb, S.; Rainer, J. MSnbase, Efficient and Elegant R-Based Processing and Visualization of Raw Mass Spectrometry Data. J. Proteome Res. 2021, 20, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Crowell, A.M.; Greene, C.S.; Loros, J.J.; Dunlap, J.C. Learning and Imputation for Mass-spec Bias Reduction (LIMBR). Bioinformatics 2019, 35, 1518–1526. [Google Scholar] [CrossRef]
- Liu, M.; Dongre, A. Proper imputation of missing values in proteomics datasets for differential expression analysis. Brief Bioinform. 2021, 22, bbaa112. [Google Scholar] [CrossRef]
- Voillet, V.; Besse, P.; Liaubet, L.; San Cristobal, M.; Gonzalez, I. Handling missing rows in multi-omics data integration: Multiple imputation in multiple factor analysis framework. BMC Bioinform. 2016, 17, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Levy, D.; Willinger, C.; Adourian, A.; Larson, M.G. Multiple imputation and analysis for high-dimensional incomplete proteomics data. Stat. Med. 2016, 35, 1315–1326. [Google Scholar] [CrossRef] [Green Version]
- The, M.; Kall, L. Integrated Identification and Quantification Error Probabilities for Shotgun Proteomics. Mol. Cell Proteom. 2019, 18, 561–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar, C. ImputeLCMD: A Collection of Methods for Left-Censored Missing Data Imputation. In R Package, Version 2.0. 2015. Available online: https://cran.r-project.org/web/packages/imputeLCMD/imputeLCMD.pdf (accessed on 28 August 2021).
- Lee, H.Y.; Kim, E.G.; Jung, H.R.; Jung, J.W.; Kim, H.B.; Cho, J.W.; Kim, K.M.; Yi, E.C. Refinements of LC-MS/MS Spectral Counting Statistics Improve Quantification of Low Abundance Proteins. Sci. Rep. 2019, 9, 13653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliviero, G.; Brien, G.L.; Waston, A.; Streubel, G.; Jerman, E.; Andrews, D.; Doyle, B.; Munawar, N.; Wynne, K.; Crean, J.; et al. Dynamic Protein Interactions of the Polycomb Repressive Complex 2 during Differentiation of Pluripotent Cells. Mol. Cell Proteom. 2016, 15, 3450–3460. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G.K.; Ritchie, M.; Thorne, N.; Shi, W.; Hu, Y. Limma: Linear Models for Microarray and RNA-Seq Data User’s Guide. 2002. Available online: https://www.bioconductor.org/packages/devel/bioc/vignettes/limma/inst/doc/usersguide.pdf (accessed on 28 August 2021).
- Kalyanasundaram, A.; Li, N.; Gardner, M.L.; Artiga, E.J.; Hansen, B.J.; Webb, A.; Freitas, M.A.; Pietrzak, M.; Whitson, B.A.; Mokadam, N.A.; et al. Fibroblast-Specific Proteotranscriptomes Reveal Distinct Fibrotic Signatures of Human Sinoatrial Node in Nonfailing and Failing Hearts. Circulation 2021, 144, 126–143. [Google Scholar] [CrossRef]
- Soliman, S.H.A.; Stark, A.E.; Gardner, M.L.; Harshman, S.W.; Breece, C.C.; Amari, F.; Orlacchio, A.; Chen, M.; Tessari, A.; Martin, J.A.; et al. Tagging enhances histochemical and biochemical detection of Ran Binding Protein 9 in vivo and reveals its interaction with Nucleolin. Sci. Rep. 2020, 10, 7138. [Google Scholar] [CrossRef] [PubMed]
- Dorayappan, K.D.P.; Gardner, M.L.; Hisey, C.L.; Zingarelli, R.A.; Smith, B.Q.; Lightfoot, M.D.S.; Gogna, R.; Flannery, M.M.; Hays, J.; Hansford, D.J.; et al. A microfluidic chip enables isolation of exosomes and establishment of their protein profiles and associated signaling pathways in ovarian cancer. Cancer Res. 2019, 79, 3503–3513. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.R.; Huang, S. Comparison of false discovery rate methods in identifying genes with differential expression. Genomics 2005, 86, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Manes, N.P.; Dong, L.; Zhou, W.; Du, X.; Reghu, N.; Kool, A.C.; Choi, D.; Bailey, C.L.; Petricoin, E.F., 3rd; Liotta, L.A.; et al. Discovery of mouse spleen signaling responses to anthrax using label-free quantitative phosphoproteomics via mass spectrometry. Mol. Cell Proteom. 2011, 10, M110.000927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branson, O.E.; Freitas, M.A. Tag-Count Analysis of Large-Scale Proteomic Data. J. Proteome Res. 2016, 15, 4742–4746. [Google Scholar] [CrossRef] [PubMed]
- Branson, O.E.; Freitas, M.A. A multi-model statistical approach for proteomic spectral count quantitation. J. Proteomics 2016, 144, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Simulated Dataset | Amputed Data | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| High MAR (0.8 MAR: 0.2 MNAR) | |||||||||||
| Method | % MV GD | % MV HG | % MV Total | Sig IDs | % MV GD | % MV HG | % MV Total | Sig IDs | % Sig IDs | Sig Lost | Sig Gain |
| kNN | 12.2 | 6.2 | 9.2 | 2570 | MAR 6.5 MNAR 1.5 TOTAL 8.0 | MAR 7.0 MNAR 1.6 TOTAL 8.6 | MAR 6.8 MNAR 1.5 TOTAL 8.3 | 1223 | 47.6 | 1368 | 21 |
| MLE | 1794 | 1256 | 70.0 | 583 | 45 | ||||||
| SVD | 2494 | 1601 | 64.2 | 916 | 23 | ||||||
| MinDet | 2647 | 1378 | 52.1 | 1276 | 7 | ||||||
| MinProb | 2527 | 1273 | 50.4 | 1267 | 13 | ||||||
| QRILC | 2509 | 1166 | 46.5 | 1347 | 4 | ||||||
| SFI-Hybrid | 2537 | 2386 | 94.1 | 254 | 103 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gardner, M.L.; Freitas, M.A. Multiple Imputation Approaches Applied to the Missing Value Problem in Bottom-Up Proteomics. Int. J. Mol. Sci. 2021, 22, 9650. https://doi.org/10.3390/ijms22179650
Gardner ML, Freitas MA. Multiple Imputation Approaches Applied to the Missing Value Problem in Bottom-Up Proteomics. International Journal of Molecular Sciences. 2021; 22(17):9650. https://doi.org/10.3390/ijms22179650
Chicago/Turabian StyleGardner, Miranda L., and Michael A. Freitas. 2021. "Multiple Imputation Approaches Applied to the Missing Value Problem in Bottom-Up Proteomics" International Journal of Molecular Sciences 22, no. 17: 9650. https://doi.org/10.3390/ijms22179650
APA StyleGardner, M. L., & Freitas, M. A. (2021). Multiple Imputation Approaches Applied to the Missing Value Problem in Bottom-Up Proteomics. International Journal of Molecular Sciences, 22(17), 9650. https://doi.org/10.3390/ijms22179650