
Discriminating Clonotypes of Influenza A Virus Genes by Nanopore Sequencing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
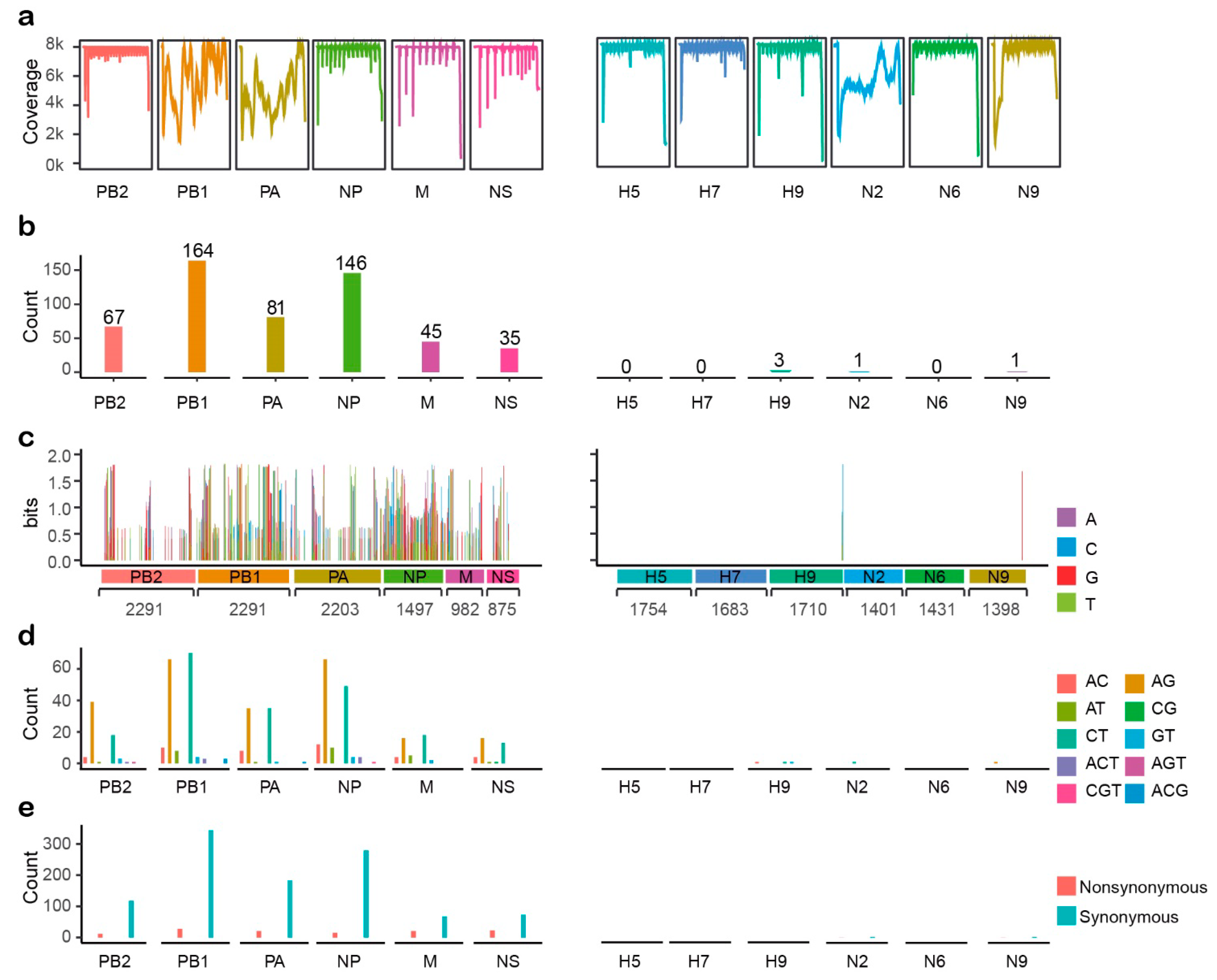
2.1. Assembly and Statistical Analyses of Illumina Data
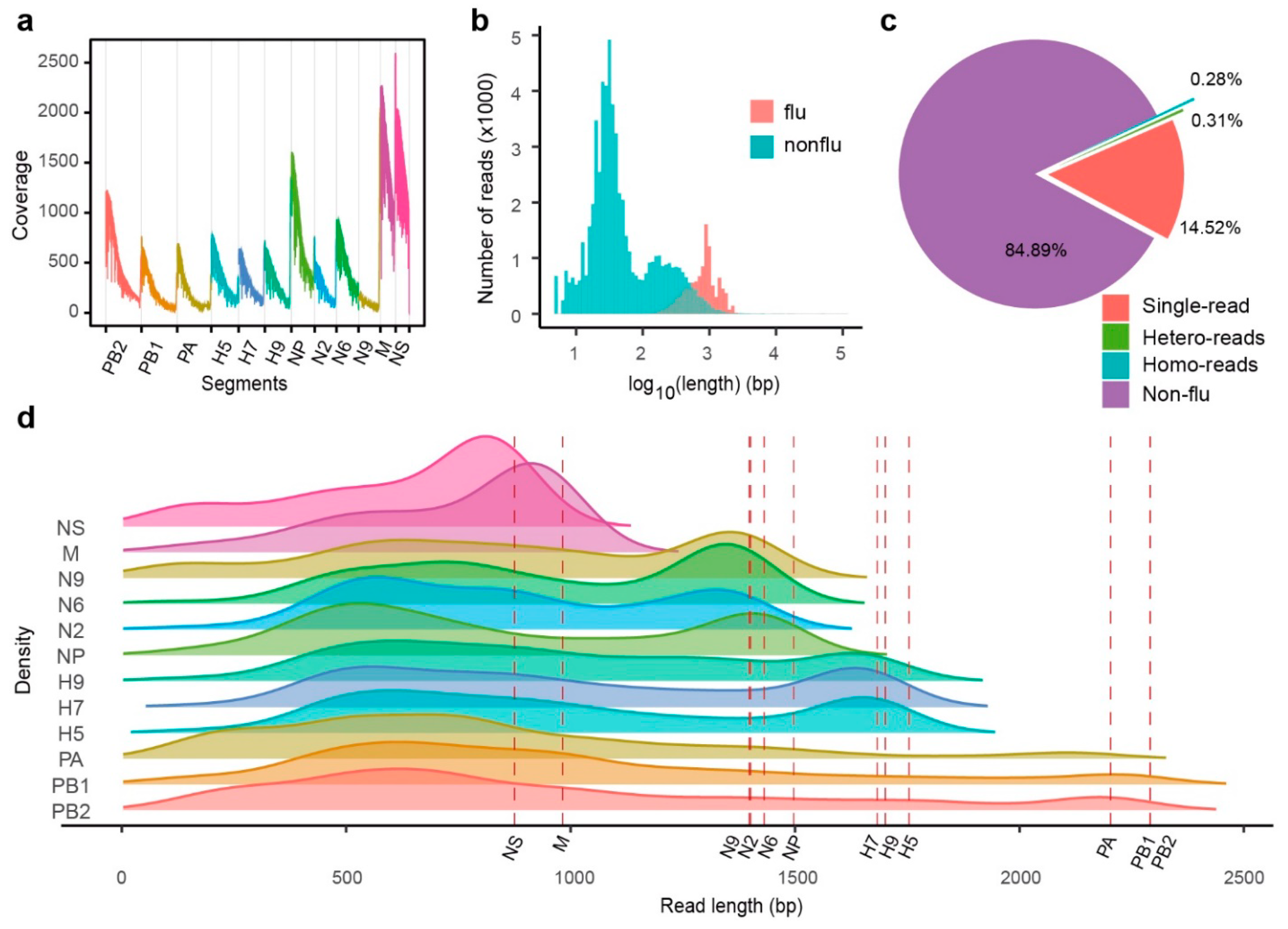
2.2. MinION RNA Sequencing of IAV Genes
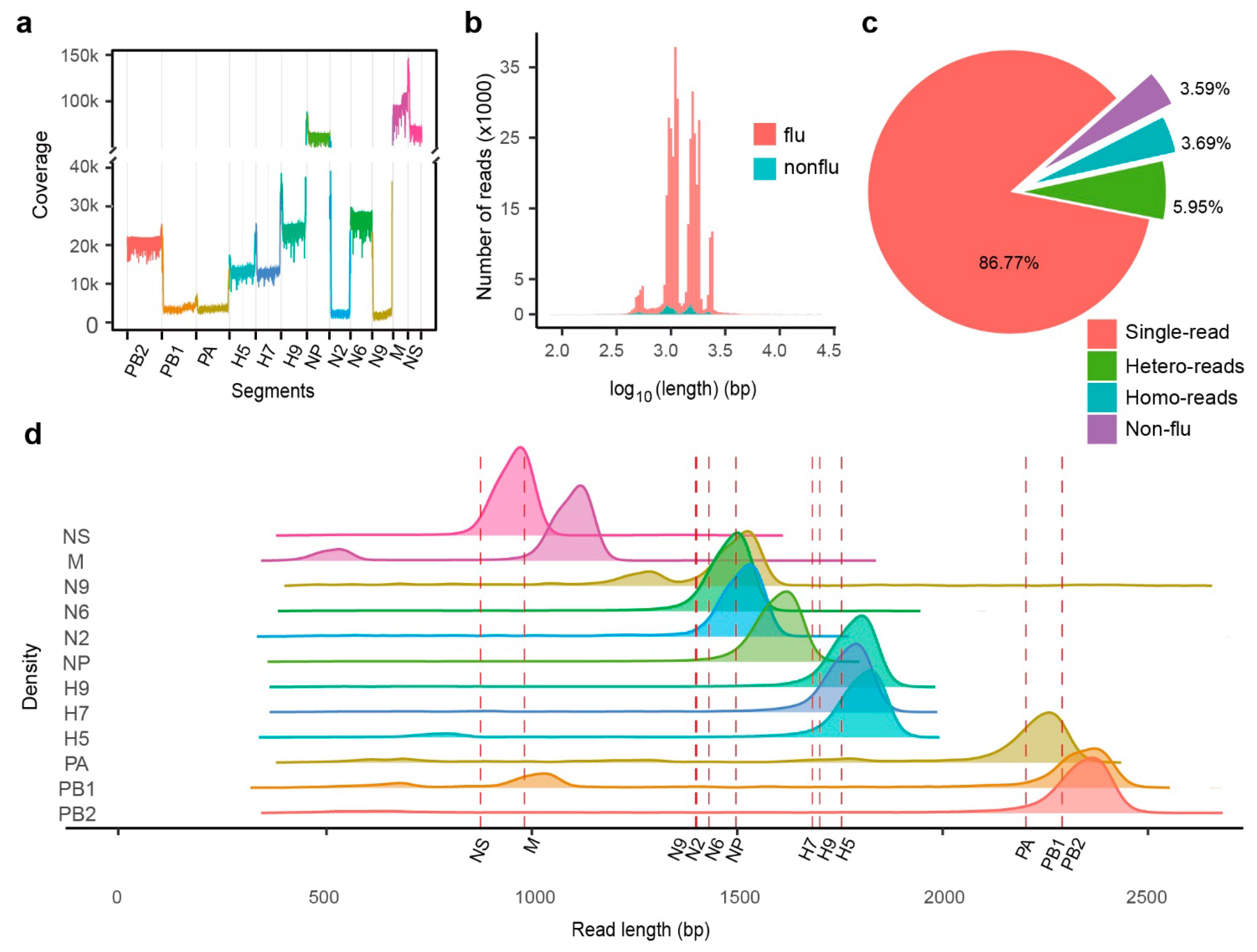
2.3. MinION cDNA Sequencing of IAV Genes
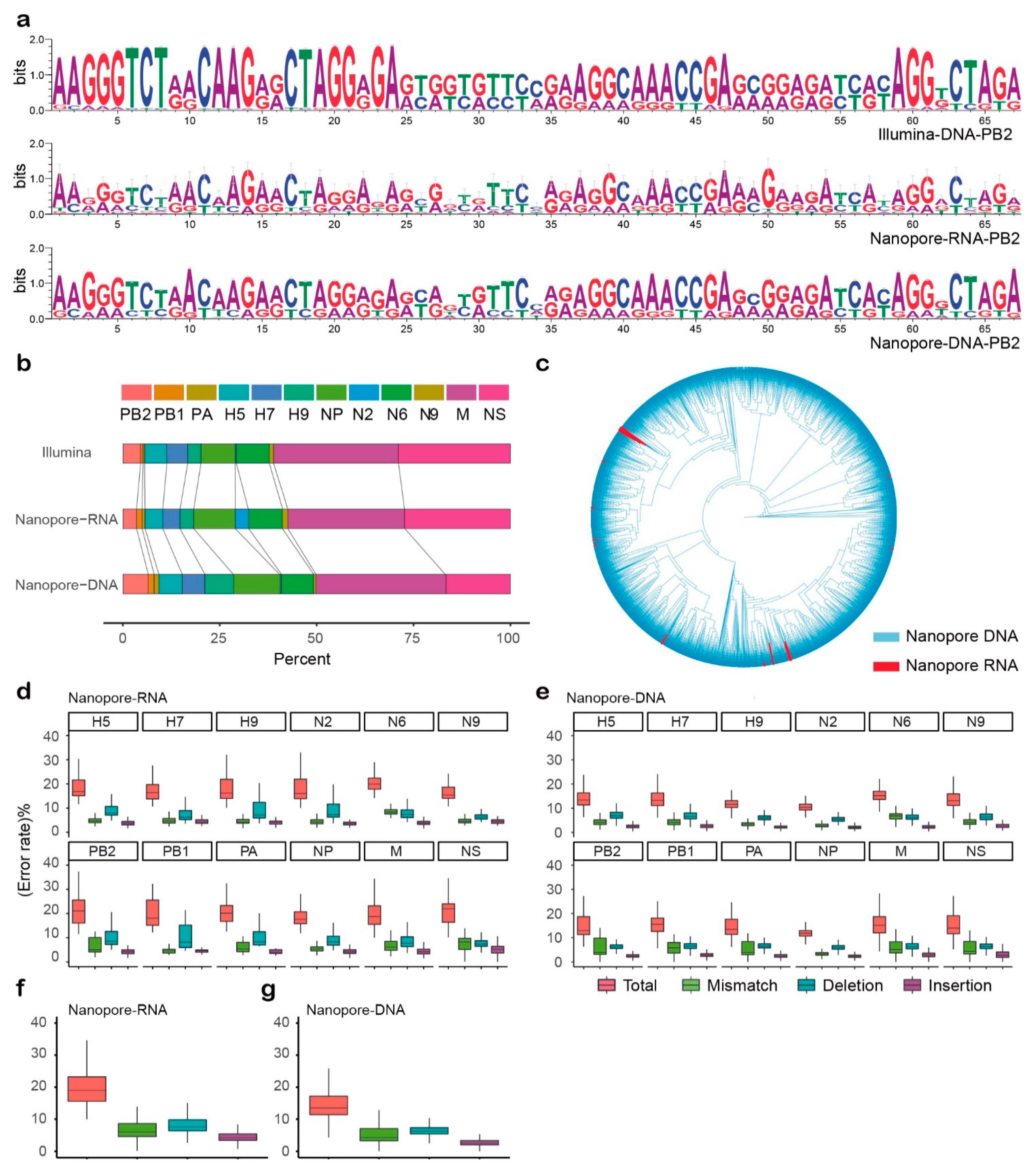
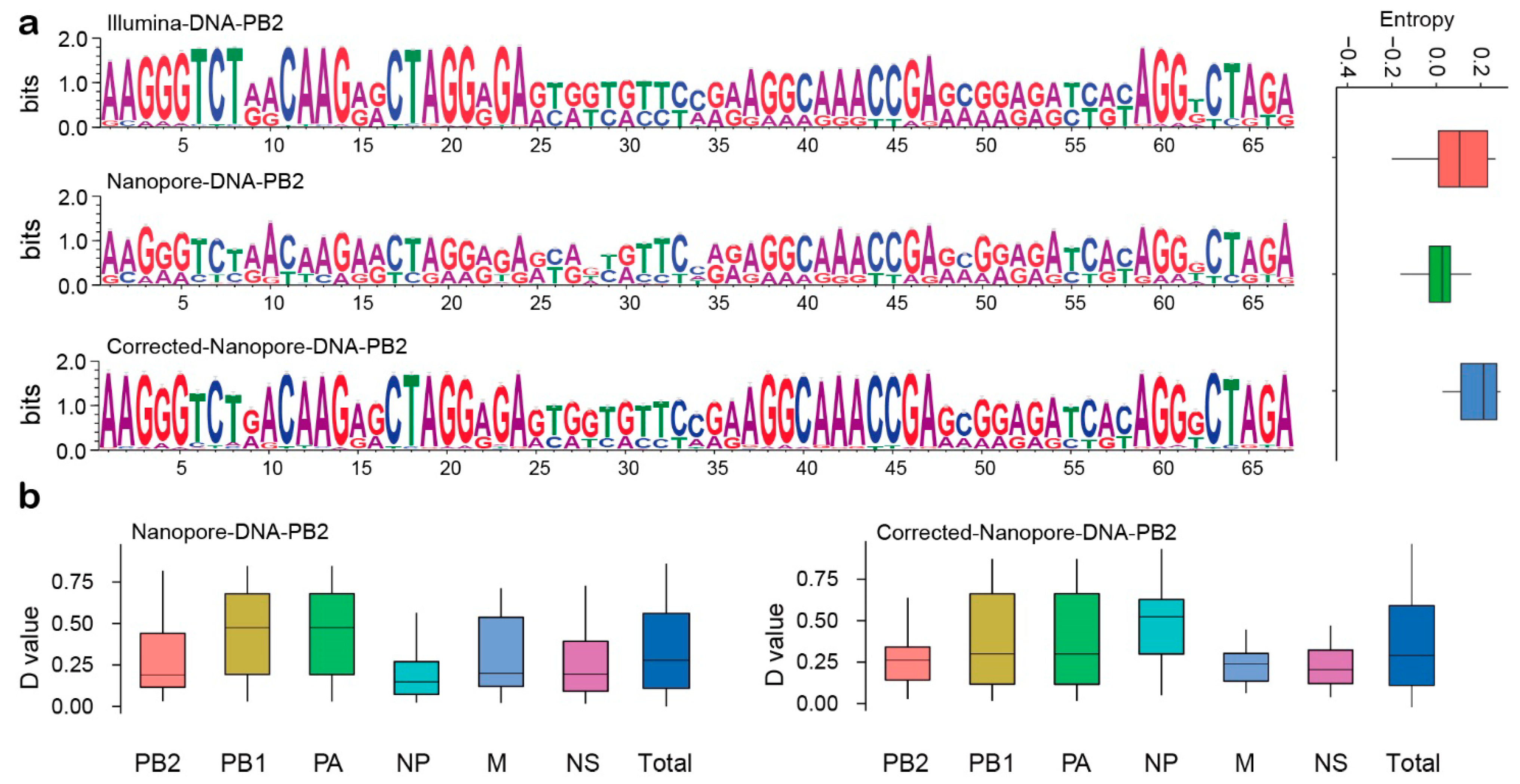
2.4. Comparison of Three Sequencing Strategies
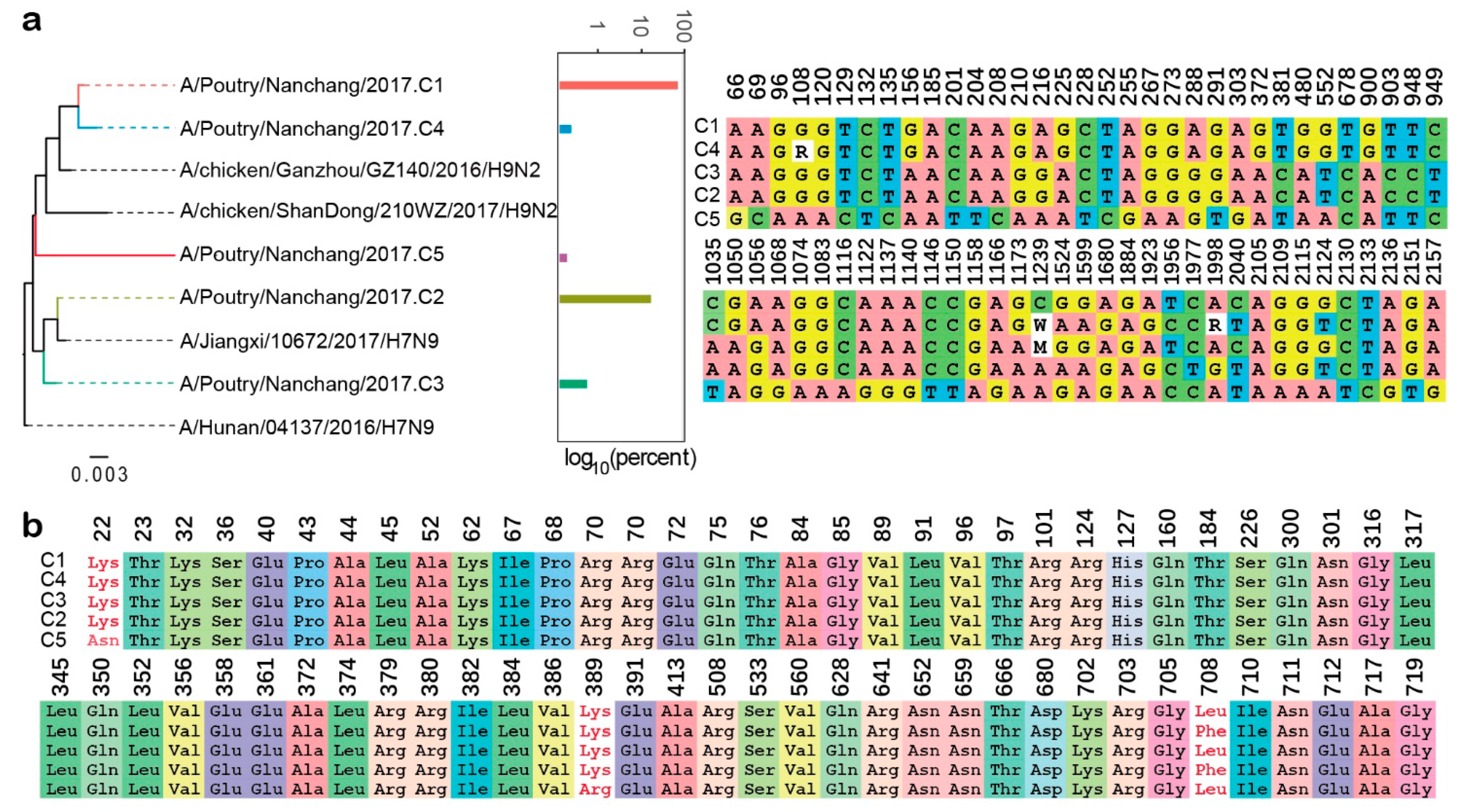
2.5. Clonotype Discrimination of IAV Internal Genes
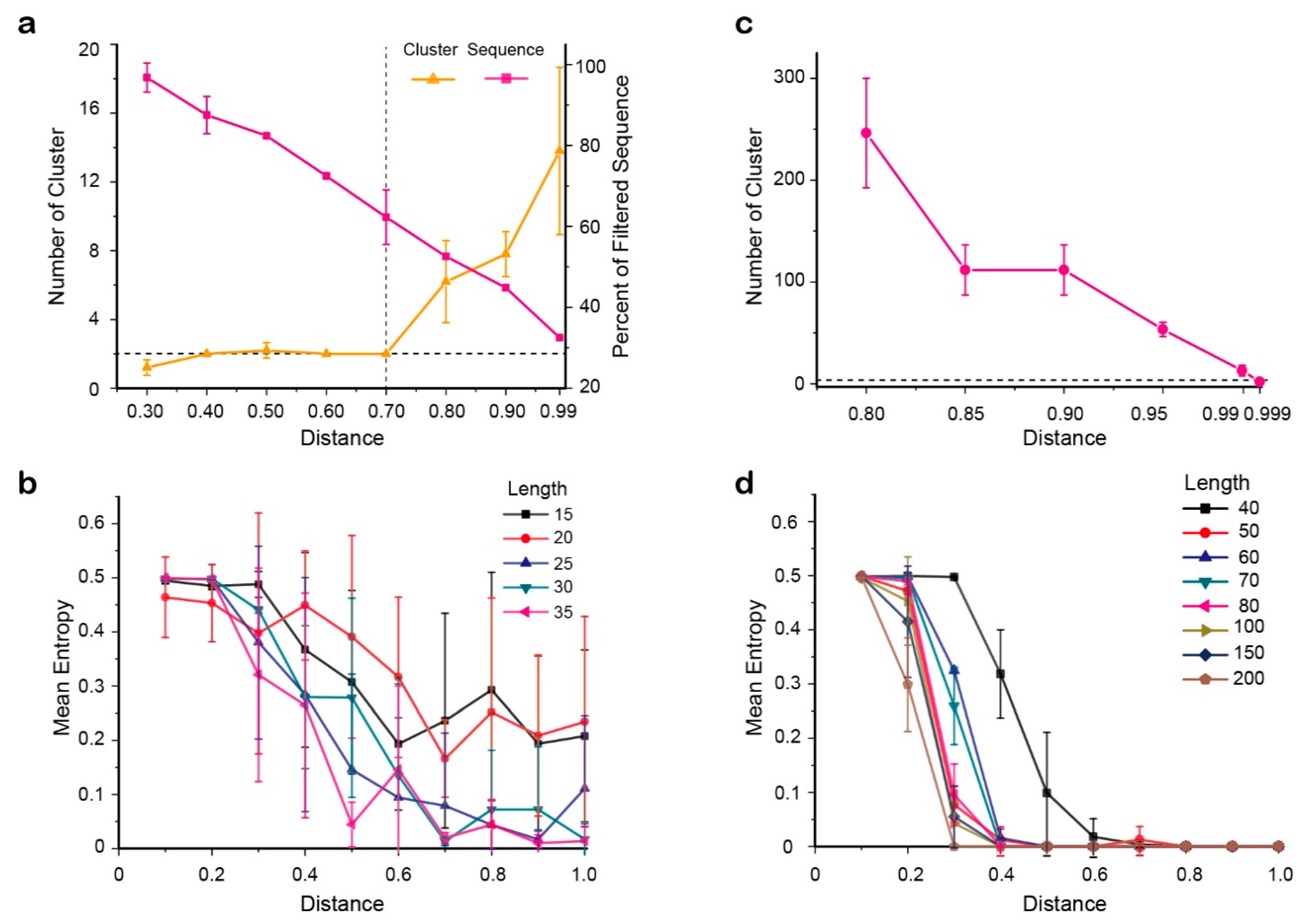
2.6. Determination of Parameters for Clonotype Discrimination Presentation
2.7. Comparison of Pre-Corrected and Modified Clonotype Sequences
3. Discussion
4. Material and Methods
4.1. Sample Screening and Preparation
4.2. Illumina Library Preparation and Sequencing
4.3. Illumina Data Analysis
4.4. MinION RNA Library Preparation and Sequencing
4.5. MinION cDNA Library Preparation and Sequencing
4.6. Analysis of MinION Data
4.7. Comparison of Direct RNA and cDNA Sequencing
4.8. Clonotype Analysis of Viral Genes
4.9. Phylogenetic Analysis
4.10. Determination of Cluster Threshold by Simulation
- (1)
- The location of the variant site used to call the clonotype is random (simulate the steps in Supplementary Figure S4a).
- (2)
- A sequence of 2000 bases in length was selected as the seed sequence of the original clonotype. A Perl script was used to generate a set of mutated sequences, of which the length was 2 kb, and imported random indels and mismatches according to the error rates of MinION results in previous analyses (simulate the steps in Supplementary Figure S4b). The clonotype dataset of these sequences was obtained by the same site variation calling script described above. For the selection of the dataset size, we generated simulation datasets for which the size varied (1000, 2000, 5000, 10,000, and 20,000), and calculate the each quartile, mean value of the matrix of the different clonotype datasets. The data size when the matrix eigenvalues tend to be stable was selected as the size of all data sets in the simulation experiment.
- (3)
- Another related dataset was created to identify the resolution scope of this procedure. This new clonotype sequence dataset originated from a sequence that was similar to the starting sequence but with n random nucleotide mutations. These two clonotype datasets were then merged for the determination of the two CD-HIT cluster threshold values. In this dataset, sequences with gaps of >10% of the length were removed.
- (4)
- The two cluster threshold values were independently determined by fixing the other one. To specify the first threshold value, the second distance value was fixed. The first distance value was set from 0.30–0.999 (Supplementary Figure S4c,d). The process of determining the second threshold is similar to the operation of the first threshold, and the second distance value is between 0.80 and 0.999 (including 0.80, 0.85, 0.90, 0.95, 0.99, and 0.999). (Here, the sequence identity threshold of CD-HIT (option -c) = 1 − distance value.)
4.11. Evaluation of the Effect and Bias of the Method
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bush, R.M.; Fitch, W.M.; Bender, C.A.; Cox, N.J. Positive selection on the H3 hemagglutinin gene of human influenza virus A. Mol. Biol. Evol. 1999, 16, 1457–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, J.W. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. USA 1993, 90, 4171–4175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daszak, P.; Cunningham, A.A.; Hyatt, A.D. Emerging infectious diseases of wildlife—Threats to biodiversity and human health. Science 2000, 287, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Härtl, G. Up to 650,000 People Die of Respiratory Diseases Linked to Seasonal Flu each Year. 2017. Available online: https://www.who.int/mediacentre/news/releases/2017/seasonal-flu/zh/ (accessed on 13 December 2017).
- Gao, G.F. Influenza and the Live Poultry Trade. Science 2014, 344, 235. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Deng, G.; Shi, J.; Wang, S.; Zhang, Q.; Kong, H.; Gu, C.; Guan, Y.; Suzuki, Y.; Li, Y.; et al. Genetics, Receptor Binding, Replication, and Mammalian Transmission of H4 Avian Influenza Viruses Isolated from Live Poultry Markets in China. J. Virol. 2016, 90, 1455–1469. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wu, J.; Zeng, X.; Huang, G.; Zou, L.; Song, Y.; Gopinath, D.; Zhang, X.; Kang, M.; Hui-Ling, Y.; et al. Isolation of H5N6, H7N9 and H9N2 avian influenza A viruses from air sampled at live poultry markets in China, 2014 and 2015. Eurosurveillance 2016, 21, 30331. [Google Scholar] [CrossRef] [Green Version]
- Bi, Y.; Li, J.; Li, S.; Fu, G.; Jin, T.; Zhang, C.; Yang, Y.; Ma, Z.; Tian, W.; Li, J.; et al. Dominant subtype switch in avian influenza viruses during 2016–2019 in China. Nat. Commun. 2020, 11, 5909. [Google Scholar] [CrossRef]
- Cui, L.; Liu, D.; Shi, W.; Pan, J.; Qi, X.; Li, X.; Guo, X.; Zhou, M.; Li, W.; Li, J.; et al. Dynamic reassortments and genetic heterogeneity of the human-infecting influenza A (H7N9) virus. Nat. Commun. 2014, 5, 3142. [Google Scholar] [CrossRef] [Green Version]
- Nowak, M.A. Evolutionary Dynamics; Harvard University Press: Harvard, MA, USA, 2006. [Google Scholar]
- Gao, R.; Cao, B.; Hu, Y.; Feng, Z.; Wang, D.; Hu, W.; Chen, J.; Jie, Z.; Qiu, H.; Xu, K.; et al. Human Infection with a Novel Avian-Origin Influenza A (H7N9) Virus. N. Engl. J. Med. 2013, 368, 1888–1897. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Shi, W.; Shi, Y.; Wang, D.; Xiao, H.; Li, W.; Bi, Y.; Wu, Y.; Li, X.; Yan, J.; et al. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: Phylogenetic, structural, and coalescent analyses. Lancet 2013, 381, 1926–1932. [Google Scholar] [CrossRef]
- Ni, M.; Chen, C.; Qian, J.; Xiao, H.-X.; Shi, W.-F.; Luo, Y.; Wang, H.-Y.; Li, Z.; Wu, J.; Xu, P.-S.; et al. Intra-host dynamics of Ebola virus during 2014. Nat. Microbiol. 2016, 1, 16151. [Google Scholar]
- Grubaugh, N.; Ladner, J.T.; Kraemer, M.U.G.; Dudas, G.; Tan, A.L.; Gangavarapu, K.; Wiley, M.; White, S.; Thézé, J.; Magnani, D.; et al. Genomic epidemiology reveals multiple introductions of Zika virus into the United States. Nat. Cell Biol. 2017, 546, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Metsky, H.C.; Matranga, C.B.; Wohl, S.; Schaffner, S.F.; Freije, C.A.; Winnicki, S.M.; West, K.; Quigley, J.E.; Baniecki, M.L.; Gladden-Young, A.; et al. Zika virus evolution and spread in the Americas. Nature 2017, 546, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soverini, S. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Fuselli, S.; Baptista, R.D.P.; Panziera, A.; Magi, A.; Guglielmi, S.; Tonin, R.; Benazzo, A.; Bauzer, L.; Mazzoni, C.J.; Bertorelle, G. A new hybrid approach for MHC genotyping: High-throughput NGS and long read MinION nanopore sequencing, with application to the non-model vertebrate Alpine chamois (Rupicapra rupicapra). Heredity 2018, 121, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Mondal, T.K.; Rawal, H.C.; Chowrasia, S.; Varshney, D.; Panda, A.K.; Mazumdar, A.; Kaur, H.; Gaikwad, K.; Sharma, T.R.; Singh, N.K. Draft genome sequence of first monocot-halophytic species Oryza coarctata reveals stress-specific genes. Sci. Rep. 2018, 8, 1–13. [Google Scholar]
- Shin, H.; Lee, E.; Shin, J.; Ko, S.-R.; Oh, H.-S.; Ahn, C.-Y.; Oh, H.-M.; Cho, B.-K.; Cho, S. Elucidation of the bacterial communities associated with the harmful microalgae Alexandrium tamarense and Cochlodinium polykrikoides using nanopore sequencing. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.W.; Rambo-Martin, B.L.; Wilson, M.M.; Ridenour, C.A.; Shepard, S.S.; Stark, T.J.; Neuhaus, E.B.; Dugan, V.G.; Wentworth, D.E.; Barnes, J.R. Direct RNA Sequencing of the Coding Complete Influenza A Virus Genome. Sci. Rep. 2018, 8, 14408. [Google Scholar] [CrossRef] [Green Version]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nat. Cell Biol. 2016, 530, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Moore, N.; Deng, Y.-M.; Eccles, D.; Hall, R. MinION nanopore sequencing of an influenza genome. Front. Microbiol. 2015, 6, 766. [Google Scholar] [CrossRef] [Green Version]
- Faria, N.R.; Quick, J.; Claro, I.; Thézé, J.; De Jesus, J.G.; Giovanetti, M.; Kraemer, M.U.G.; Hill, S.C.; Black, A.; Da Costa, A.C.; et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 2017, 546, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Kilianski, A.; Haas, J.L.; Corriveau, E.J.; Liem, A.T.; Willis, K.L.; Kadavy, D.R.; Rosenzweig, C.N.; Minot, S.S. Bacterial and viral identification and differentiation by amplicon sequencing on the MinION nanopore sequencer. GigaScience 2015, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boža, V.; Brejova, B.; Vinař, T. DeepNano: Deep recurrent neural networks for base calling in MinION nanopore reads. PLoS ONE 2017, 12, e0178751. [Google Scholar] [CrossRef] [PubMed]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef]
- Viehweger, A.; Krautwurst, S.; Lamkiewicz, K.; Madhugiri, R.; Ziebuhr, J.; Hölzer, M.; Marz, M. Nanopore direct RNA sequencing reveals modification in full-length coronavirus genomes. bioRxiv 2018, 483693. [Google Scholar]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Hernandez, D.; Francois, P.; Farinelli, L.L.D.; Østerås, M.; Schrenzel, J. De novo bacterial genome sequencing: Millions of very short reads assembled on a desktop computer. Genome Res. 2008, 18, 802–809. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, Y.; Chen, Q.; Wang, Q.; Chen, J.; Jin, T.; Wong, G.; Quan, C.; Liu, J.; Wu, J.; Yin, R.; et al. Genesis, Evolution and Prevalence of H5N6 Avian Influenza Viruses in China. Cell Host Microbe 2016, 20, 810–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sović, I.; Šikić, M.; Wilm, A.; Fenlon, S.; Chen, S.; Nagarajan, N. Fast and sensitive mapping of nanopore sequencing reads with GraphMap. Nat. Commun. 2016, 7, 11307. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Bolotov, P.; Dernovoy, D.; Kiryutin, B.; Zaslavsky, L.; Tatusova, T.; Ostell, J.; Lipman, D. The Influenza Virus Resource at the National Center for Biotechnology Information. J. Virol. 2008, 82, 596–601. [Google Scholar] [CrossRef] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinform. 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; López, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; IEEE: New Orleans, LA, USA, 2010; pp. 1–8. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Y.; Liu, H.; Yan, Y.; Liu, W.; Liu, D.; Li, J. Discriminating Clonotypes of Influenza A Virus Genes by Nanopore Sequencing. Int. J. Mol. Sci. 2021, 22, 10069. https://doi.org/10.3390/ijms221810069
Cao Y, Liu H, Yan Y, Liu W, Liu D, Li J. Discriminating Clonotypes of Influenza A Virus Genes by Nanopore Sequencing. International Journal of Molecular Sciences. 2021; 22(18):10069. https://doi.org/10.3390/ijms221810069
Chicago/Turabian StyleCao, Ying, Haizhou Liu, Yi Yan, Wenjun Liu, Di Liu, and Jing Li. 2021. "Discriminating Clonotypes of Influenza A Virus Genes by Nanopore Sequencing" International Journal of Molecular Sciences 22, no. 18: 10069. https://doi.org/10.3390/ijms221810069
APA StyleCao, Y., Liu, H., Yan, Y., Liu, W., Liu, D., & Li, J. (2021). Discriminating Clonotypes of Influenza A Virus Genes by Nanopore Sequencing. International Journal of Molecular Sciences, 22(18), 10069. https://doi.org/10.3390/ijms221810069