
Elucidating the Neuroprotective Role of PPARs in Parkinson’s Disease: A Neoteric and Prospective Target


 ,
, 
 , ,
, , 
Abstract
:1. Introduction
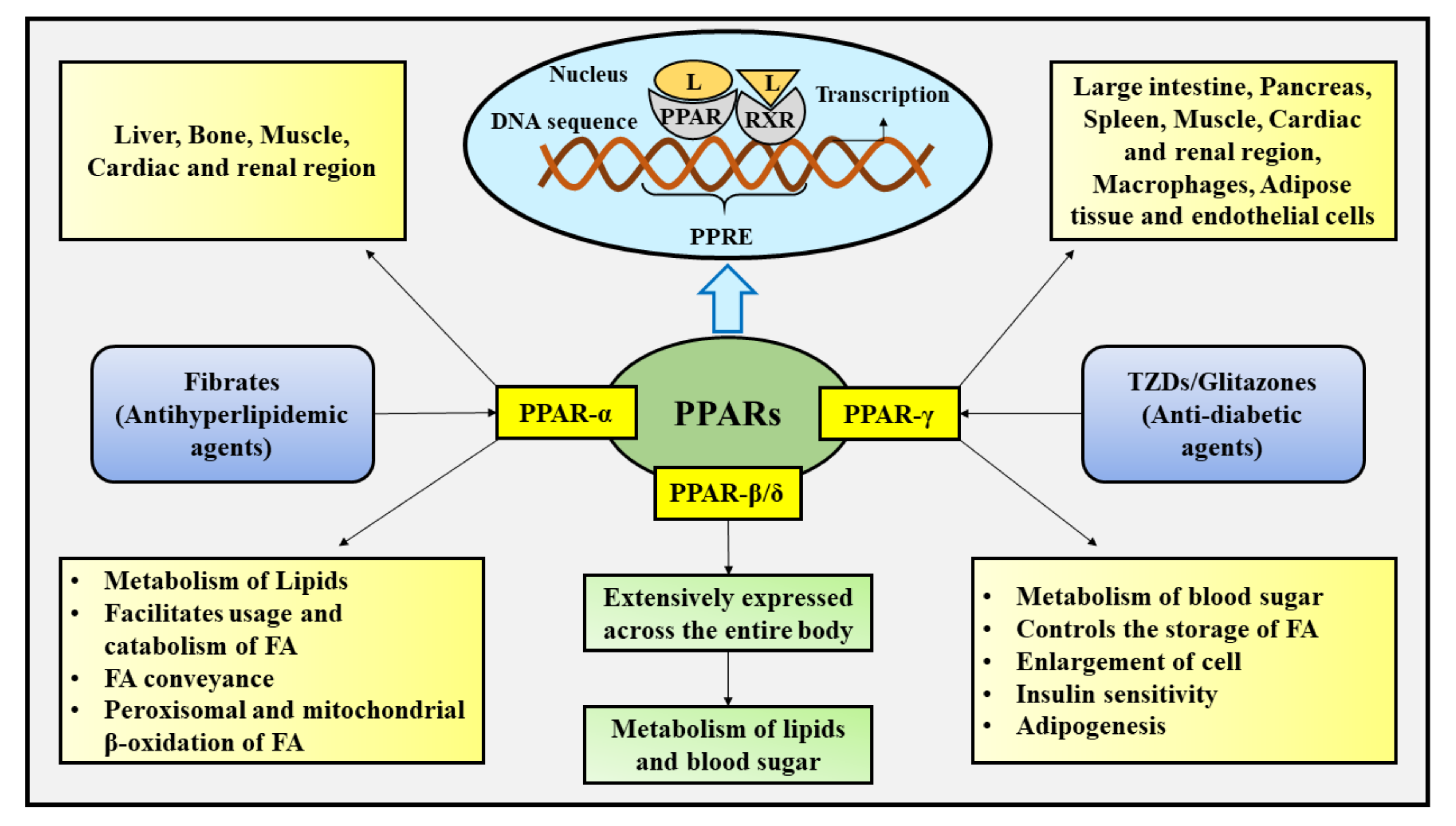
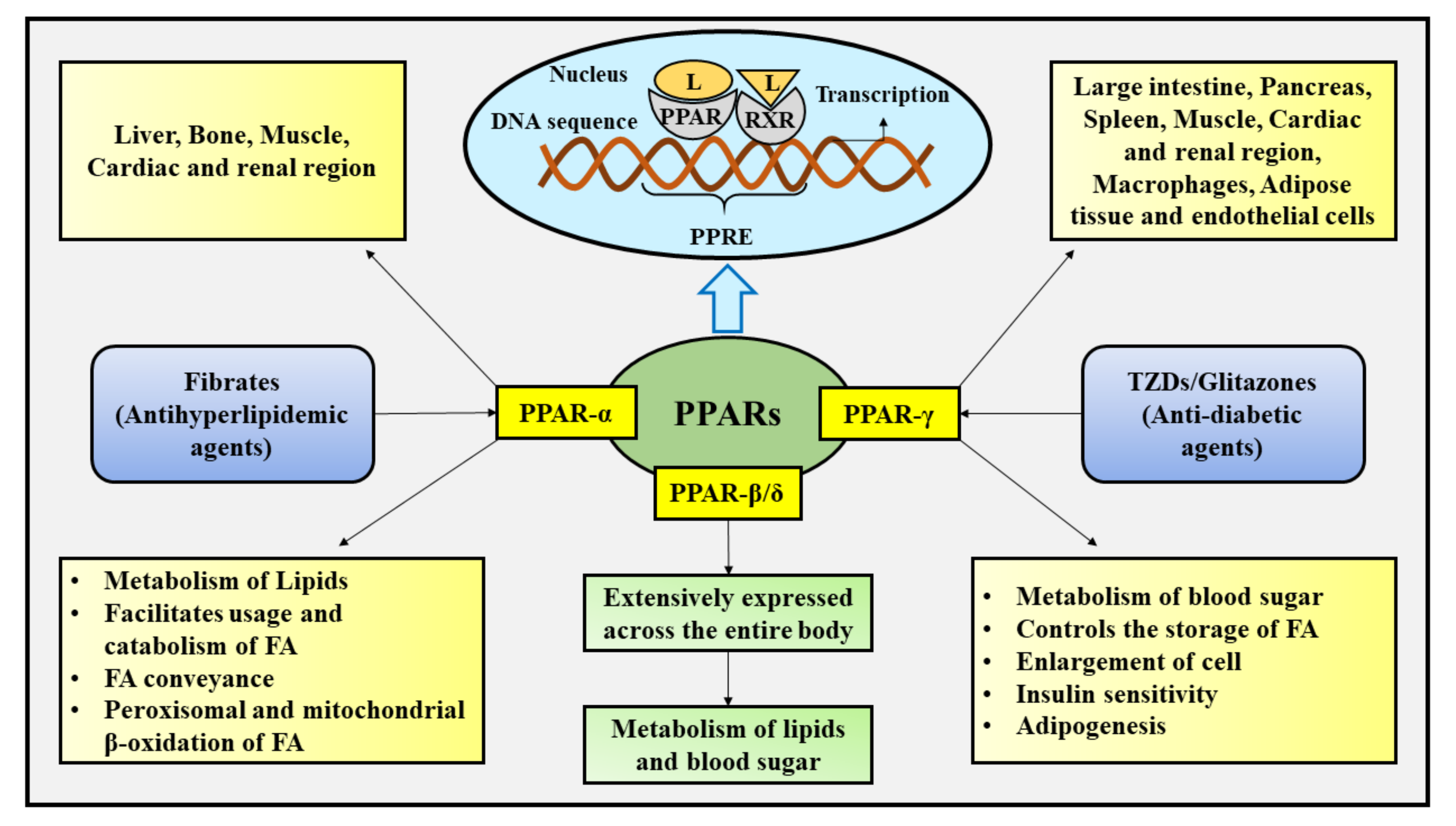
2. Cellular Influences of PPARs
3. Parkinson’s Disease
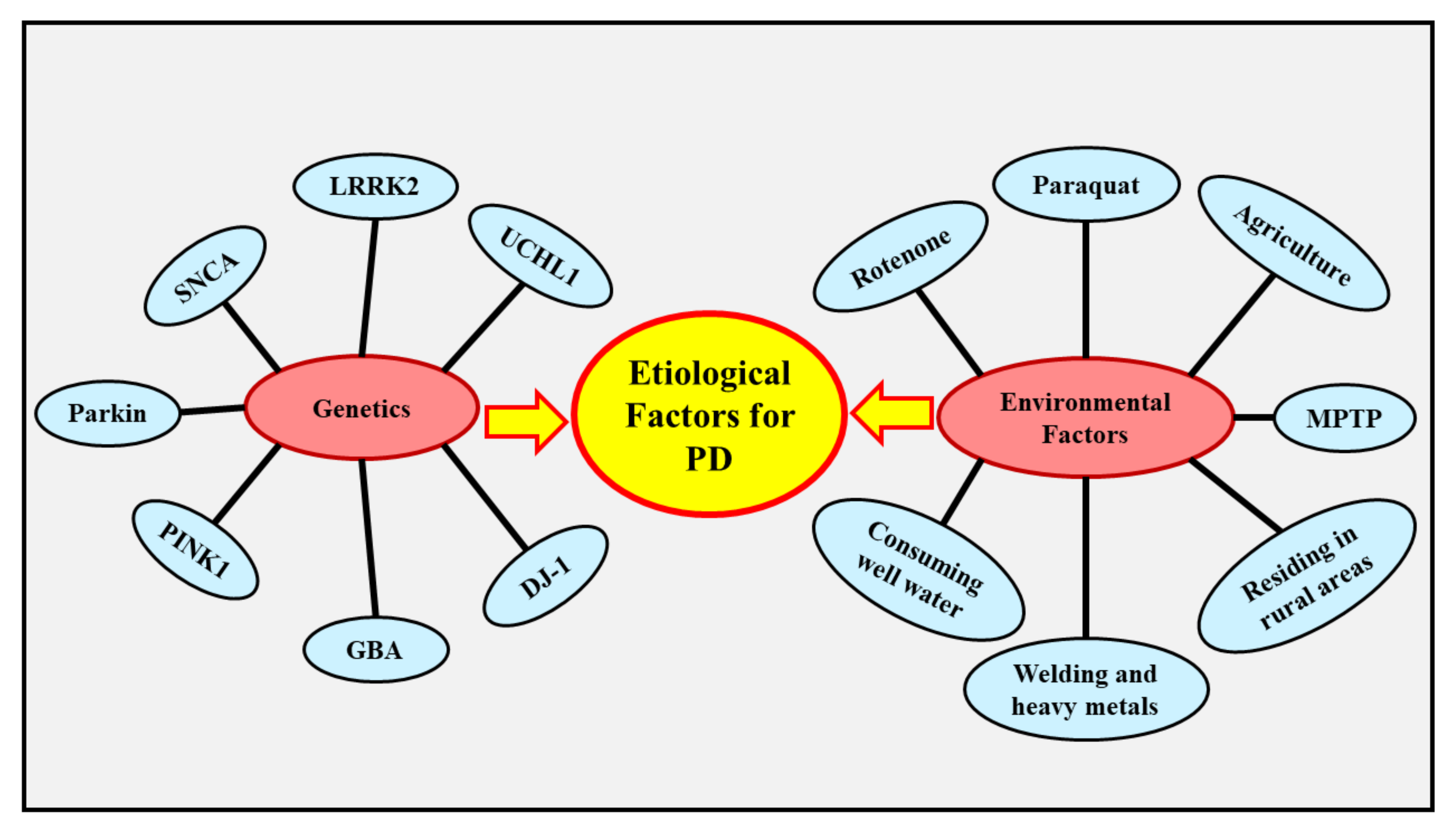
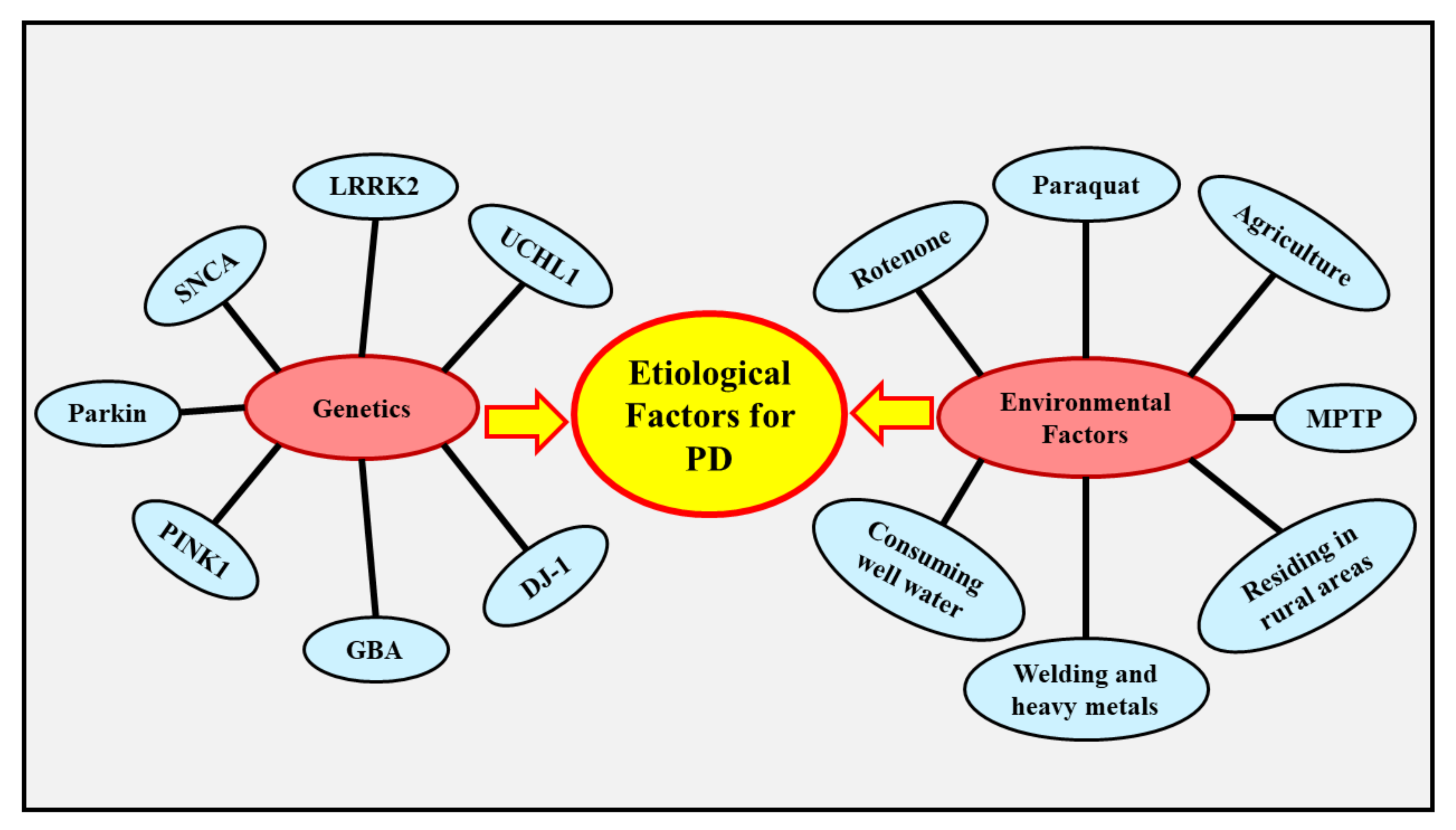
4. Etiology of PD
4.1. Genetics
4.2. Environmental Factors
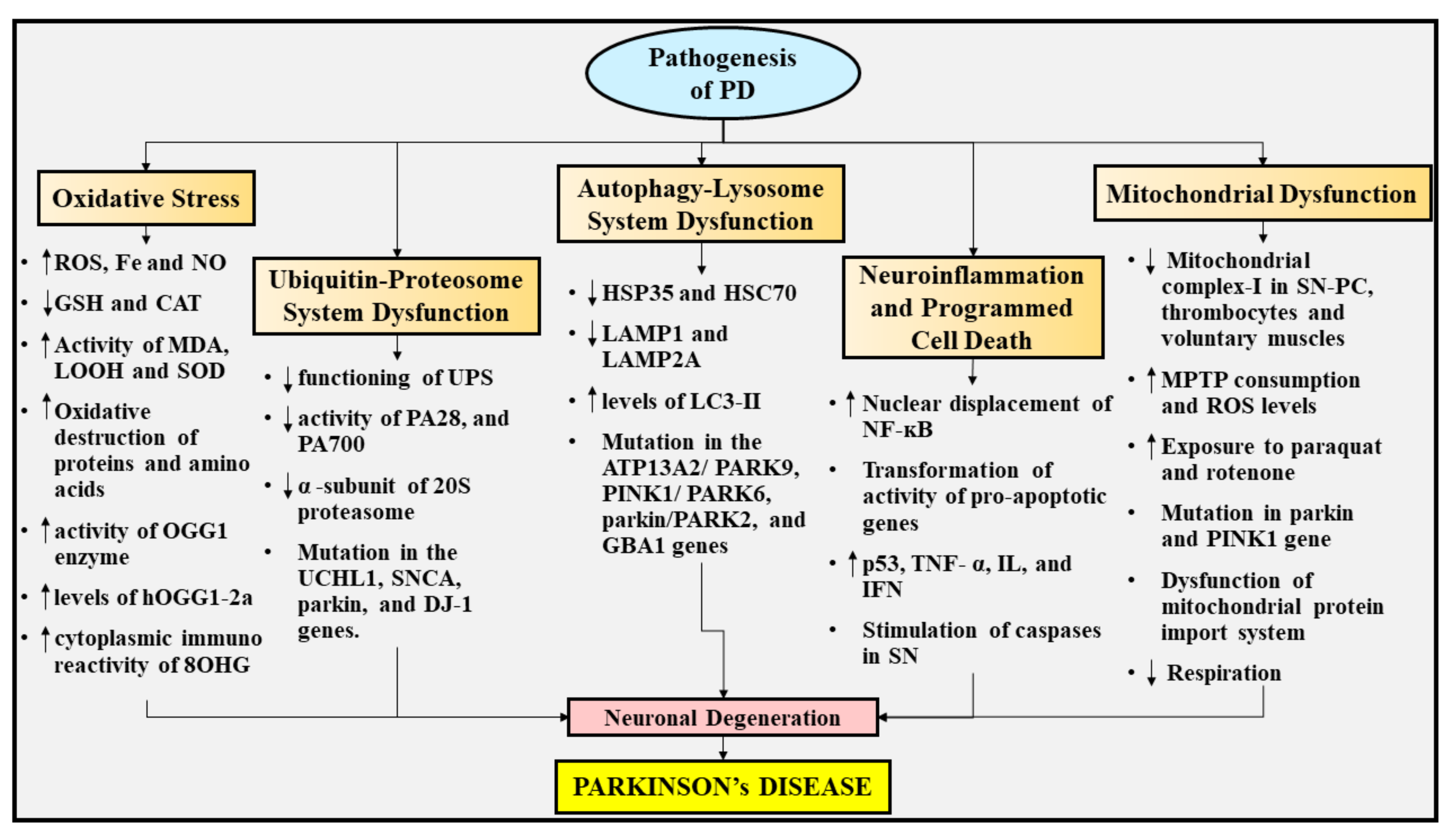
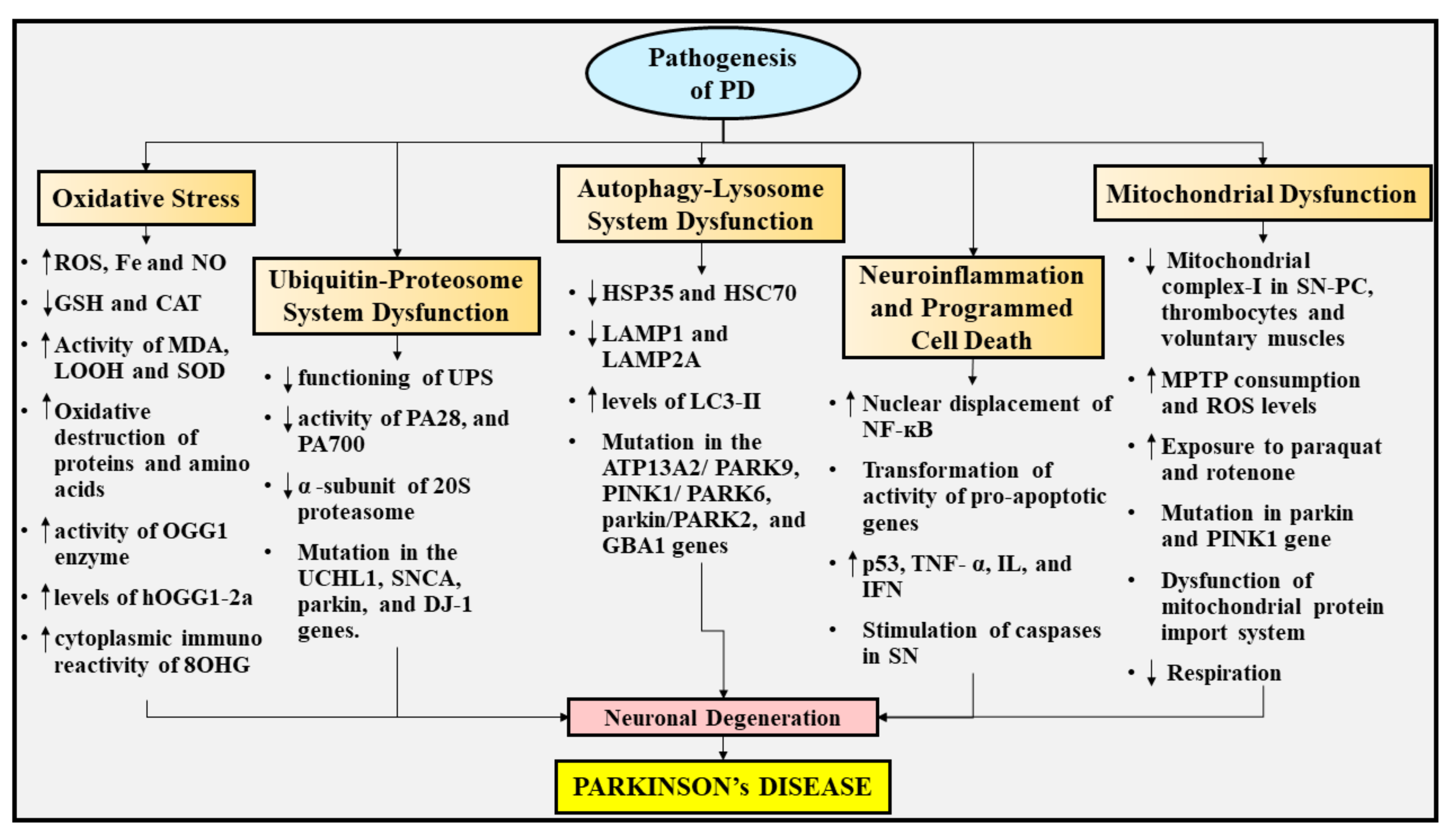
5. Pathogenesis of PD
5.1. Oxidative Stress
5.2. UPS Dysfunction
5.3. Autophagy-Lysosome System Dysfunction
5.4. Neuroinflammation and Programmed Cell Death
5.5. Mitochondrial Dysfunction
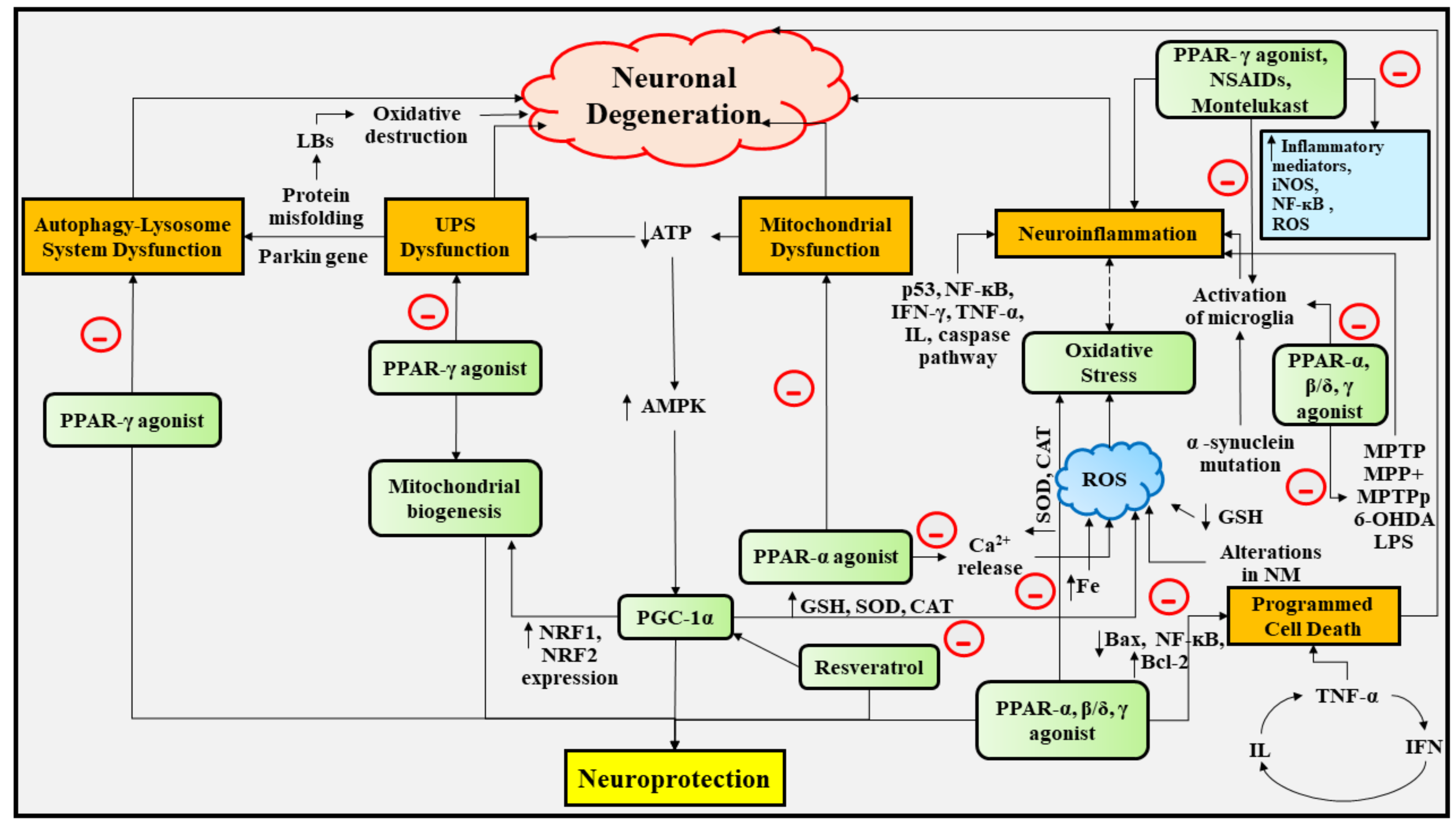
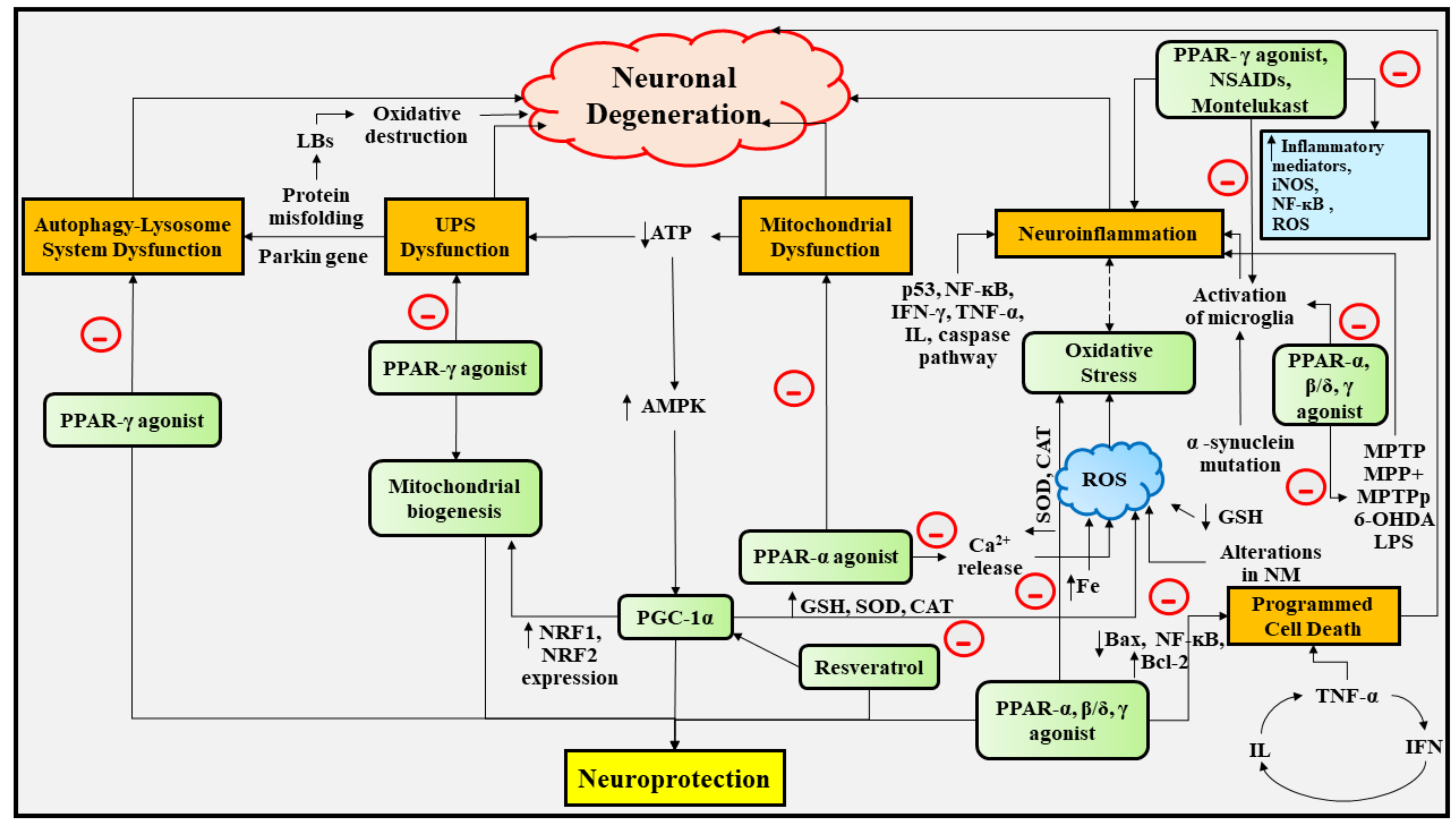
6. Experimental Studies Portraying the Deep Insights into the Neuroprotective Role of PPAR Agonists in PD
6.1. Therapeutic Implications of PPAR-γ Agonists in PD
6.2. Therapeutic Implications of PPAR-β/δ Agonists in PD
6.3. Therapeutic Implications of PPAR-α, and PPAR-α/γ Agonists in PD
6.4. Therapeutic Implications of NSAIDs, Leukotriene Receptor Antagonist, and Vitamin E in PD
6.5. Therapeutic Implications of PGC-1α in PD
6.6. Therapeutic Implications of Smoking, Caffeine, and Alcohol Consumption in PD
6.7. Therapeutic Implications of Physical Exercise in PD
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Capriotti, T.; Terzakis, K. Parkinson Disease. Home Healthc. Now 2016, 34, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M. Parkinson’s Disease and Parkinsonism. Am J Med. 2019, 132, 802–807. [Google Scholar] [CrossRef]
- Tysnes, O.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Picillo, M.; Nicoletti, A.; Fetoni, V.; Garavaglia, B.; Barone, P.; Pellecchia, M. The relevance of gender in Parkinson’s disease: A review. J. Neurol. 2017, 264, 1583–1607. [Google Scholar] [CrossRef]
- Vázquez-Vélez, G.; Zoghbi, H. Parkinson’s Disease Genetics and Pathophysiology. Annu. Rev. Neurosci. 2021, 44, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, A.; Dahiya, N. Management of Parkinson׳s disease: Current and future pharmacotherapy. Eur. J. Pharmacol. 2015, 750, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Mal, S.; Dwivedi, A.; Kumar, V.; Kumar, N.; Kumar, B. Role of Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) in Different Disease States: Recent Updates. Curr. Med. Chem. 2021, 28, 3193–3215. [Google Scholar] [CrossRef]
- Deb, R.; Joshi, N.; Nagotu, S. Peroxisomes of the Brain: Distribution, Functions, and Associated Diseases. Neurotox Res. 2021, 39, 986–1006. [Google Scholar] [CrossRef] [PubMed]
- Ciccocioppo, R.; Ubaldi, M. Nuclear peroxisome proliferator activated receptor-gamma (PPARγ) as a therapeutic target to treat neurodegeneration and dependence elicited by drugs of abuse. Neural Regen. Res. 2021, 16, 984–985. [Google Scholar] [CrossRef] [PubMed]
- Avarachan, J.; Augustine, A.; Shinde, P.M.; Gunasekaran, V. A Mechanistic approach of Peroxisome Proliferator-Activated Receptors and its subtypes on Clinical and preclinical model of Neurodegenerative disorders. Res. J. Pharm. Technol. 2021, 14, 3967–3975. [Google Scholar] [CrossRef]
- Upadhyay, A.; Amanullah, A.; Joshi, V.; Dhiman, R.; Prajapati, V.; Poluri, K.; Mishra, A. Ibuprofen-based advanced therapeutics: Breaking the inflammatory link in cancer, neurodegeneration, and diseases. Drug Metab. Rev. 2021, 53, 100–121. [Google Scholar] [CrossRef] [PubMed]
- Hain, E.; Sparenberg, M.; Rasińska, J.; Klein, C.; Akyüz, L.; Steiner, B. Indomethacin promotes survival of new neurons in the adult murine hippocampus accompanied by anti-inflammatory effects following MPTP-induced dopamine depletion. J. Neuroinflamm. 2018, 15, 162. [Google Scholar] [CrossRef] [Green Version]
- Michael, J.; Zirknitzer, J.; Unger, M.S.; Poupardin, R.; Rieß, T.; Paiement, N.; Zerbe, H.; Hutter-Paier, B.; Reitsamer, H.; Aigner, L. The Leukotriene Receptor Antagonist Montelukast Attenuates Neuroinflammation and Affects Cognition in Transgenic 5 × FAD Mice. Int. J. Mol. Sci. 2021, 22, 2782. [Google Scholar] [CrossRef] [PubMed]
- Mahalakshmi, B.; Maurya, N.; Lee, S.-D.; Bharath Kumar, V. Possible Neuroprotective Mechanisms of Physical Exercise in Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 5895. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236. [Google Scholar] [CrossRef]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar]
- Kostadinova, R.; Wahli, W.; Michalik, L. PPARs in diseases: Control mechanisms of inflammation. Curr. Med. Chem. 2005, 12, 2995–3009. [Google Scholar] [CrossRef] [PubMed]
- Corona, J.C.; Duchen, M.R. PPARγ as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic. Biol. Med. 2016, 100, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Corona, J.C.; Duchen, M.R. PPARγ and PGC-1α as therapeutic targets in Parkinson’s. Neurochem. Res. 2015, 40, 308–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-C.; Wu, J.-S.; Tsai, H.-D.; Huang, C.-Y.; Chen, J.-J.; Sun, G.Y.; Lin, T.-N. Peroxisome proliferator-activated receptor gamma (PPAR-γ) and neurodegenerative disorders. Mol. Neurobiol. 2012, 46, 114–124. [Google Scholar] [CrossRef]
- Puigserver, P. Tissue-specific regulation of metabolic pathways through the transcriptional coactivator PGC1-α. Int. J. Obes. 2005, 29, S5–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Neerven, S.; Mey, J. RAR/RXR and PPAR/RXR signaling in spinal cord injury. PPAR Res. 2007, 2007, 29275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Zhu, Y.; Reddy, J.K. Peroxisome proliferator-activated receptors, coactivators, and downstream targets. Cell Biochem. Biophys. 2000, 32, 187–204. [Google Scholar] [CrossRef]
- Ziouzenkova, O.; Plutzky, J. Retinoid metabolism and nuclear receptor responses: New insights into coordinated regulation of the PPAR–RXR complex. FEBS Lett. 2008, 582, 32–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straus, D.S.; Glass, C.K. Anti-inflammatory actions of PPAR ligands: New insights on cellular and molecular mechanisms. Trends Immunol. 2007, 28, 551–558. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Lenhard, J.M.; Oliver, B.B.; Ringold, G.M.; Kliewer, S.A. Peroxisome proliferator-activated receptors α and γ are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 1997, 272, 3406–3410. [Google Scholar] [CrossRef] [Green Version]
- Jaradat, M.S.; Wongsud, B.; Phornchirasilp, S.; Rangwala, S.M.; Shams, G.; Sutton, M.; Romstedt, K.J.; Noonan, D.J.; Feller, D.R. Activation of peroxisome proliferator-activated receptor isoforms and inhibition of prostaglandin H2 synthases by ibuprofen, naproxen, and indomethacin. Biochem. Pharmacol. 2001, 62, 1587–1595. [Google Scholar] [CrossRef]
- Marion-Letellier, R.; Savoye, G.; Ghosh, S. Fatty acids, eicosanoids and PPAR gamma. Eur. J. Pharmacol. 2016, 785, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Mottillo, E.P.; Bloch, A.E.; Leff, T.; Granneman, J.G. Lipolytic products activate peroxisome proliferator-activated receptor (PPAR) α and δ in brown adipocytes to match fatty acid oxidation with supply. J. Biol. Chem. 2012, 287, 25038–25048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177. [Google Scholar] [CrossRef]
- Janani, C.; Kumari, B.R. PPAR gamma gene–a review. Diabetes Metab. Syndr. Clin. Res. Rev. 2015, 9, 46–50. [Google Scholar] [CrossRef]
- Chung, J.H.; Seo, A.Y.; Chung, S.W.; Kim, M.K.; Leeuwenburgh, C.; Yu, B.P.; Chung, H.Y. Molecular mechanism of PPAR in the regulation of age-related inflammation. Ageing Res. Rev. 2008, 7, 126–136. [Google Scholar] [CrossRef]
- Pascual, G.; Glass, C.K. Nuclear receptors versus inflammation: Mechanisms of transrepression. Trends Endocrinol. Metab. 2006, 17, 321–327. [Google Scholar] [CrossRef]
- Brown, G.C.; Murphy, M.P.; Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogacka, I.; Xie, H.; Bray, G.A.; Smith, S.R. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes 2005, 54, 1392–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Patel, N.; Rahn, D.; McAllister, J.; Sadeghi, S.; Horwitz, G.; Berry, D.; Wang, K.X.; Swerdlow, R.H. The thiazolidinedione pioglitazone alters mitochondrial function in human neuron-like cells. Mol. Pharmacol. 2007, 71, 1695–1702. [Google Scholar] [CrossRef] [Green Version]
- Russo, C.D.; Gavrilyuk, V.; Weinberg, G.; Almeida, A.; Bolanos, J.P.; Palmer, J.; Pelligrino, D.; Galea, E.; Feinstein, D.L. Peroxisome proliferator-activated receptor γ thiazolidinedione agonists increase glucose metabolism in astrocytes. J. Biol. Chem. 2003, 278, 5828–5836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geldenhuys, W.J.; Leeper, T.C.; Carroll, R.T. mitoNEET as a novel drug target for mitochondrial dysfunction. Drug Discov. Today 2014, 19, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Paddock, M.L.; Wiley, S.E.; Axelrod, H.L.; Cohen, A.E.; Roy, M.; Abresch, E.C.; Capraro, D.; Murphy, A.N.; Nechushtai, R.; Dixon, J.E. MitoNEET is a uniquely folded 2Fe–2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc. Natl. Acad. Sci. USA 2007, 104, 14342–14347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strum, J.C.; Shehee, R.; Virley, D.; Richardson, J.; Mattie, M.; Selley, P.; Ghosh, S.; Nock, C.; Saunders, A.; Roses, A. Rosiglitazone induces mitochondrial biogenesis in mouse brain. J. Alzheimer’s Dis. 2007, 11, 45–51. [Google Scholar] [CrossRef]
- Wang, Y.L.; Frauwirth, K.A.; Rangwala, S.M.; Lazar, M.A.; Thompson, C.B. Thiazolidinedione activation of peroxisome proliferator-activated receptor γ can enhance mitochondrial potential and promote cell survival. J. Biol. Chem. 2002, 277, 31781–31788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, J.K.; Hashimoto, T. Peroxisomal β-oxidation and peroxisome proliferator–activated receptor α: An adaptive metabolic system. Annu. Rev. Nutr. 2001, 21, 193–230. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Chong, L.-W.; Evans, R.M. PPAR-γ regulates osteoclastogenesis in mice. Nat. Med. 2007, 13, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, D. Effects of rosiglitazone on Kruppul-like factor 6 (KLF6) signaling in the livers of rats with nonalcoholic fatty liver fibrosis. Zhonghua Gan Zang Bing Za Zhi/Zhonghua Ganzangbing Zazhi/Chin. J. Hepatol. 2007, 15, 649–653. [Google Scholar]
- Lee, J.; Reding, M. Effects of thiazolidinediones on stroke recovery: A case-matched controlled study. Neurochem. Res. 2007, 32, 635–638. [Google Scholar] [CrossRef]
- Gou, Q.; Gong, X.; Jin, J.; Shi, J.; Hou, Y. Peroxisome proliferator-activated receptors (PPARs) are potential drug targets for cancer therapy. Oncotarget 2017, 8, 60704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Roman, J. Peroxisome proliferator-activated receptor γ: A novel target for cancer therapeutics? Anti-Cancer Drugs 2007, 18, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Khuchua, Z.; Glukhov, A.I.; Strauss, A.W.; Javadov, S. Elucidating the beneficial role of PPAR agonists in cardiac diseases. Int. J. Mol. Sci. 2018, 19, 3464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, B.; Staels, B. PPAR agonists: Multimodal drugs for the treatment of type-2 diabetes. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 687–710. [Google Scholar] [CrossRef]
- Heneka, M.T.; Landreth, G.E. PPARs in the brain. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 1031–1045. [Google Scholar] [CrossRef] [PubMed]
- Tureyen, K.; Kapadia, R.; Bowen, K.K.; Satriotomo, I.; Liang, J.; Feinstein, D.L.; Vemuganti, R. Peroxisome proliferator-activated receptor-γ agonists induce neuroprotection following transient focal ischemia in normotensive, normoglycemic as well as hypertensive and type-2 diabetic rodents. J. Neurochem. 2007, 101, 41–56. [Google Scholar] [CrossRef]
- Luo, Y.; Yin, W.; Signore, A.P.; Zhang, F.; Hong, Z.; Wang, S.; Graham, S.H.; Chen, J. Neuroprotection against focal ischemic brain injury by the peroxisome proliferator-activated receptor-γ agonist rosiglitazone. J. Neurochem. 2006, 97, 435–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuenzalida, K.; Quintanilla, R.; Ramos, P.; Piderit, D.; Fuentealba, R.A.; Martinez, G.; Inestrosa, N.C.; Bronfman, M. Peroxisome proliferator-activated receptor γ up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J. Biol. Chem. 2007, 282, 37006–37015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.-W.; Yi, J.-H.; Miranpuri, G.; Satriotomo, I.; Bowen, K.; Resnick, D.K.; Vemuganti, R. Thiazolidinedione class of peroxisome proliferator-activated receptor γ agonists prevents neuronal damage, motor dysfunction, myelin loss, neuropathic pain, and inflammation after spinal cord injury in adult rats. J. Pharmacol. Exp. Ther. 2007, 320, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- McTigue, D.M.; Tripathi, R.; Wei, P.; Lash, A.T. The PPAR gamma agonist Pioglitazone improves anatomical and locomotor recovery after rodent spinal cord injury. Exp. Neurol. 2007, 205, 396–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, I.E.; Serneels, L.; Spittaels, K.; Merchiers, P.; Dominguez, D.; De Strooper, B. Peroxisome proliferator-activated receptor γ induces a clearance mechanism for the amyloid-β peptide. J. Neurosci. 2004, 24, 10908–10917. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, R.K.; Beal, M.F. PPAR: A therapeutic target in Parkinson’s disease. J. Neurochem. 2008, 106, 506–518. [Google Scholar] [CrossRef]
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajder, J.B. The novel role of PPAR alpha in the brain: Promising target in therapy of Alzheimer’s disease and other neurodegenerative disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiaei, M. Peroxisome Proliferator-Activated Receptor-in Amyotrophic Lateral Sclerosis and Huntington’s Disease. PPAR Res. 2008, 2008, 418765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bright, J.; Walline, C.; Kanakasabai, S.; Chakraborty, S. Targeting PPAR as a therapy to treat multiple sclerosis. Expert Opin. Ther. Targets 2008, 12, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Yankee, E. A review on Parkinson’s disease treatment. Neuroimmunol. Neuroinflamm. 2021, 8. [Google Scholar] [CrossRef]
- McGregor, M.M.; Nelson, A.B. Circuit mechanisms of Parkinson’s disease. Neuron 2019, 101, 1042–1056. [Google Scholar] [CrossRef] [Green Version]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Benito, M.; Granado, N.; García-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s disease with the alpha-synuclein protein. Front. Pharmacol. 2020, 11, 356. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Pu, J. Alpha-synuclein in Parkinson’s disease: From pathogenetic dysfunction to potential clinical application. Parkinson’s Dis. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Váradi, C. Clinical features of Parkinson’s disease: The evolution of critical symptoms. Biology 2020, 9, 103. [Google Scholar] [CrossRef]
- Hirsch, L.; Jette, N.; Frolkis, A.; Steeves, T.; Pringsheim, T. The incidence of Parkinson’s disease: A systematic review and meta-analysis. Neuroepidemiology 2016, 46, 292–300. [Google Scholar] [CrossRef]
- Driver, J.A.; Logroscino, G.; Gaziano, J.M.; Kurth, T. Incidence and remaining lifetime risk of Parkinson disease in advanced age. Neurology 2009, 72, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Cerri, S.; Mus, L.; Blandini, F. Parkinson’s disease in women and men: What’s the difference? J. Parkinson’s Dis. 2019, 9, 501–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maraganore, D.M.; Lesnick, T.G.; Elbaz, A.; Chartier-Harlin, M.C.; Gasser, T.; Krüger, R.; Hattori, N.; Mellick, G.D.; Quattrone, A.; Satoh, J.I. UCHL1 is a Parkinson’s disease susceptibility gene. Ann. Neurol. 2004, 55, 512–521. [Google Scholar] [CrossRef]
- Siddiqui, I.J.; Pervaiz, N.; Abbasi, A.A. The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Sci. Rep. 2016, 6, 1–11. [Google Scholar]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The role of LRRK2 in neurodegeneration of Parkinson disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.M.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Repici, M.; Giorgini, F. DJ-1 in Parkinson’s disease: Clinical insights and therapeutic perspectives. J. Clin. Med. 2019, 8, 1377. [Google Scholar] [CrossRef] [Green Version]
- O’Regan, G.; DeSouza, R.-M.; Balestrino, R.; Schapira, A.H. Glucocerebrosidase mutations in Parkinson disease. J. Parkinson’s Dis. 2017, 7, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, E.A.; Zhang, J.; Checkoway, H. Solvents and Parkinson disease: A systematic review of toxicological and epidemiological evidence. Toxicol. Appl. Pharmacol. 2013, 266, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Kenborg, L.; Rugbjerg, K.; Lee, P.-C.; Ravnskjær, L.; Christensen, J.; Ritz, B.; Lassen, C.F. Head injury and risk for Parkinson disease: Results from a Danish case-control study. Neurology 2015, 84, 1098–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.-Y.; Chou, M.-C.; Lin, C.-L.; Kao, C.-H. Increased risk of Parkinson disease in patients with carbon monoxide intoxication: A population-based cohort study. Medicine 2015, 94, e869. [Google Scholar] [CrossRef]
- Gopalakrishna, A.; Alexander, S.A. Understanding Parkinson disease: A complex and multifaceted illness. J. Neurosci. Nurs. 2015, 47, 320–326. [Google Scholar] [CrossRef]
- VanItallie, T.B. Parkinson disease: Primacy of age as a risk factor for mitochondrial dysfunction. Metabolism 2008, 57, S50–S55. [Google Scholar] [CrossRef] [PubMed]
- De Rijk, M.; Breteler, M.; Graveland, G.; Ott, A.; Grobbee, D.; Van der Meche, F.; Hofman, A. Prevalence of Parkinson’s disease in the elderly: The Rotterdam Study. Neurology 1995, 45, 2143–2146. [Google Scholar] [CrossRef]
- Bower, J.H.; Maraganore, D.M.; McDonnell, S.K.; Rocca, W.A. Incidence and distribution of parkinsonism in Olmsted County, Minnesota, 1976–1990. Neurology 1999, 52, 1214. [Google Scholar] [CrossRef]
- Sauerbier, A.; Aris, A.; Lim, E.W.; Bhattacharya, K.; Ray Chaudhuri, K. Impact of ethnicity on the natural history of Parkinson disease. Med. J. Aust. 2018, 208, 410–414. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef] [PubMed]
- C Lee, Y.-T.; D Hsu, S.-T. Familial mutations and post-translational modifications of UCH-L1 in Parkinson’s disease and neurodegenerative disorders. Curr. Protein Pept. Sci. 2017, 18, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Solano, S.M.; Miller, D.W.; Augood, S.J.; Young, A.B.; Penney Jr, J.B. Expression of α-synuclein, parkin, and ubiquitin carboxy-terminal hydrolase L1 mRNA in human brain: Genes associated with familial Parkinson’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child. Neurol. Soc. 2000, 47, 201–210. [Google Scholar] [CrossRef]
- Jackson, P.; Thompson, R. The demonstration of new human brain-specific proteins by high-resolution two-dimensional polyacrylamide gel electrophoresis. J. Neurol. Sci. 1981, 49, 429–438. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Olanow, C.W. Proteolytic stress: A unifying concept for the etiopathogenesis of Parkinson’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child. Neurol. Soc. 2003, 53, S73–S86. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M. Axon degeneration mechanisms: Commonality amid diversity. Nat. Rev. Neurosci. 2005, 6, 889–898. [Google Scholar] [CrossRef]
- Saigoh, K.; Wang, Y.-L.; Suh, J.-G.; Yamanishi, T.; Sakai, Y.; Kiyosawa, H.; Harada, T.; Ichihara, N.; Wakana, S.; Kikuchi, T. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat. Genet. 1999, 23, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Ardley, H.C.; Scott, G.B.; Rose, S.A.; Tan, N.G.; Robinson, P.A. UCH-L1 aggresome formation in response to proteasome impairment indicates a role in inclusion formation in Parkinson’s disease. J. Neurochem. 2004, 90, 379–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recchia, A.; Debetto, P.; Negro, A.; Guidolin, D.; Skaper, S.D.; Giusti, P. α-Synuclein and Parkinson’s disease. FASEB J. 2004, 18, 617–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuytemans, K.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, W.W.; Pei, Z.; Jiang, H.; Moore, D.J.; Liang, Y.; West, A.B.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 18676–18681. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef]
- Gandhi, P.N.; Chen, S.G.; Wilson-Delfosse, A.L. Leucine-rich repeat kinase 2 (LRRK2): A key player in the pathogenesis of Parkinson’s disease. J. Neurosci. Res. 2009, 87, 1283–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, E.K.; Skipper, L.M. Pathogenic mutations in Parkinson disease. Hum. Mutat. 2007, 28, 641–653. [Google Scholar] [CrossRef]
- Hilker, R.; Pilatus, U.; Eggers, C.; Hagenah, J.; Roggendorf, J.; Baudrexel, S.; Klein, J.C.; Neumaier, B.; Fink, G.R.; Steinmetz, H. The bioenergetic status relates to dopamine neuron loss in familial PD with PINK1 mutations. PLoS ONE 2012, 7, e51308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.-Y.; Whitworth, A.J.; Schapira, A.H. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Maple-Grødem, J.; Dalen, I.; Tysnes, O.-B.; Macleod, A.D.; Forsgren, L.; Counsell, C.E.; Alves, G. Association of GBA Genotype With Motor and Functional Decline in Patients With Newly Diagnosed Parkinson Disease. Neurology 2021, 96, e1036–e1044. [Google Scholar] [PubMed]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hallett, P.; Isacson, O. Progressive decline of glucocerebrosidase in aging and P arkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 433–438. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardi, S.P.; Cheng, S.H.; Shihabuddin, L.S. Gaucher-related synucleinopathies: The examination of sporadic neurodegeneration from a rare (disease) angle. Prog. Neurobiol. 2015, 125, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Bajpai, P.; Sangar, M.C.; Singh, S.; Tang, W.; Bansal, S.; Chowdhury, G.; Cheng, Q.; Fang, J.-K.; Martin, M.V.; Guengerich, F.P. Metabolism of 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine by mitochondrion-targeted cytochrome P450 2D6: Implications in Parkinson disease. J. Biol. Chem. 2013, 288, 4436–4451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustapha, M.; Taib, C.N.M. MPTP-induced mouse model of Parkinson’s disease: A promising direction for therapeutic strategies. Bosn. J. Basic Med. Sci. 2021, 21, 422. [Google Scholar] [PubMed]
- Behl, T.; Kaur, G.; Sehgal, A.; Bhardwaj, S.; Singh, S.; Buhas, C.; Judea-Pusta, C.; Uivarosan, D.; Munteanu, M.A.; Bungau, S. Multifaceted Role of Matrix Metalloproteinases in Neurodegenerative Diseases: Pathophysiological and Therapeutic Perspectives. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, A.; Clavel, J.; Rathouz, P.J.; Moisan, F.; Galanaud, J.P.; Delemotte, B.; Alpérovitch, A.; Tzourio, C. Professional exposure to pesticides and Parkinson disease. Ann. Neurol. 2009, 66, 494–504. [Google Scholar] [CrossRef]
- Xiong, N.; Long, X.; Xiong, J.; Jia, M.; Chen, C.; Huang, J.; Ghoorah, D.; Kong, X.; Lin, Z.; Wang, T. Mitochondrial complex I inhibitor rotenone-induced toxicity and its potential mechanisms in Parkinson’s disease models. Crit. Rev. Toxicol. 2012, 42, 613–632. [Google Scholar] [CrossRef] [PubMed]
- Di Monte, D.; Sandy, M.S.; Ekström, G.; Smith, M.T. Comparative studies on the mechanisms of paraquat and 1-methyl-4-phenylpyridine (MPP+) cytotoxicity. Biochem. Biophys. Res. Commun. 1986, 137, 303–309. [Google Scholar] [CrossRef]
- Bungau, S.; Behl, T.; Aleya, L.; Bourgeade, P.; Aloui-Sosse, B.; Purza, A.L.; Abid, A.; Samuel, A.D. Expatiating the impact of anthropogenic aspects and climatic factors on long-term soil monitoring and management. Environ. Sci. Pollut. Res. 2021, 28, 30528–30550. [Google Scholar] [CrossRef]
- Federoff, H.J.; Burke, R.E.; Fahn, S.; Fiskum, G. Parkinson’s disease (the life cycle of the dopamine neuron). Ann. N. Y. Acad. Sci. 2003, 991, 1–360. [Google Scholar]
- Wang, J.-Y.; Zhuang, Q.-Q.; Zhu, L.-B.; Zhu, H.; Li, T.; Li, R.; Chen, S.-F.; Huang, C.-P.; Zhang, X.; Zhu, J.-H. Meta-analysis of brain iron levels of Parkinson’s disease patients determined by postmortem and MRI measurements. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Li, X.; Li, X.; Liu, Q.; Cheng, Y. Oxidative stress in Parkinson’s disease: A systematic review and meta-analysis. Front. Mol. Neurosci. 2018, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Fukae, J.; Takanashi, M.; Kubo, S.-i.; Nishioka, K.-i.; Nakabeppu, Y.; Mori, H.; Mizuno, Y.; Hattori, N. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson’s disease and related neurodegenerative disorders. Acta Neuropathol. 2005, 109, 256–262. [Google Scholar] [CrossRef]
- Zhang, J.; Perry, G.; Smith, M.A.; Robertson, D.; Olson, S.J.; Graham, D.G.; Montine, T.J. Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am. J. Pathol. 1999, 154, 1423–1429. [Google Scholar] [CrossRef]
- Percário, S.; Da Silva Barbosa, A.; Varela, E.L.P.; Gomes, A.R.Q.; Ferreira, M.E.S.; De Nazaré Araújo Moreira, T.; Dolabela, M.F. Oxidative stress in parkinson’s disease: Potential benefits of antioxidant supplementation. Oxidative Med. Cell. Longev. 2020, 2020, 2360872. [Google Scholar] [CrossRef] [PubMed]
- Subedi, L.; Gaire, B.P.; Kim, S.-Y.; Parveen, A. Nitric Oxide as a Target for Phytochemicals in Anti-Neuroinflammatory Prevention Therapy. Int. J. Mol. Sci. 2021, 22, 4771. [Google Scholar] [CrossRef]
- Yao, D.; Gu, Z.; Nakamura, T.; Shi, Z.Q.; Ma, Y.; Gaston, B.; Palmer, L.A.; Rockenstein, E.M.; Zhang, Z.; Masliah, E.; et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10810–10814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavya, R.; Saluja, R.; Singh, S.; Dikshit, M. Nitric oxide synthase regulation and diversity: Implications in Parkinson’s disease. Nitric Oxide 2006, 15, 280–294. [Google Scholar] [CrossRef]
- Yeung, P.K.K.; Lai, A.K.W.; Son, H.J.; Zhang, X.; Hwang, O.; Chung, S.S.M.; Chung, S.K. Aldose reductase deficiency leads to oxidative stress-induced dopaminergic neuronal loss and autophagic abnormality in an animal model of Parkinson’s disease. Neurobiol Aging. 2017, 50, 119–133. [Google Scholar] [CrossRef]
- Rathnayake, D.; Chang, T.; Udagama, P. Selected serum cytokines and nitric oxide as potential multi-marker biosignature panels for Parkinson disease of varying durations: A case-control study. BMC Neurol. 2019, 19, 56. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- McKinnon, C.; Tabrizi, S.J. The ubiquitin-proteasome system in neurodegeneration. Antioxid. Redox Signal. 2014, 21, 2302–2321. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.P.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- Ullrich, C.; Mlekusch, R.; Kuschnig, A.; Marksteiner, J.; Humpel, C. Ubiquitin enzymes, ubiquitin and proteasome activity in blood mononuclear cells of MCI, Alzheimer and Parkinson patients. Curr. Alzheimer Res. 2010, 7, 549–555. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Belizaire, R.; Jenner, P.; Olanow, C.W.; Isacson, O. Selective loss of 20S proteasome α-subunits in the substantia nigra pars compacta in Parkinson’s disease. Neurosci. Lett. 2002, 326, 155–158. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Rare genetic mutations shed light on the pathogenesis of Parkinson disease. J. Clin. Investig. 2003, 111, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Miwa, H.; Kubo, T.; Suzuki, A.; Nishi, K.; Kondo, T. Retrograde dopaminergic neuron degeneration following intrastriatal proteasome inhibition. Neurosci. Lett. 2005, 380, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Alteration of autophagosomal proteins (LC3, GABARAP and GATE-16) in Lewy body disease. Neurobiol. Dis. 2011, 43, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Xu, Y. Mutations in the ATP13A2 gene and Parkinsonism: A preliminary review. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, E.M.; Salvi, S.; Ialongo, T.; Marongiu, R.; Elia, A.E.; Caputo, V.; Romito, L.; Albanese, A.; Dallapiccola, B.; Bentivoglio, A.R. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann. Neurol. 2004, 56, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Ryan, E.; Seehra, G.; Sharma, P.; Sidransky, E. GBA1-associated parkinsonism: New insights and therapeutic opportunities. Curr. Opin. Neurol. 2019, 32, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.; Michel, P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar] [PubMed]
- Hunot, S.; Brugg, B.; Ricard, D.; Michel, P.P.; Muriel, M.-P.; Ruberg, M.; Faucheux, B.A.; Agid, Y.; Hirsch, E.C. Nuclear translocation of NF-κB is increased in dopaminergic neurons of patients with Parkinson disease. Proc. Natl. Acad. Sci. USA 1997, 94, 7531–7536. [Google Scholar] [CrossRef] [Green Version]
- Mogi, M.; Harada, M.; Kondo, T.; Mizuno, Y.; Narabayashi, H.; Riedere, P.; Nagatsu, T. bcl-2 protein is increased in the brain from parkinsonian patients. Neurosci. Lett. 1996, 215, 137–139. [Google Scholar] [CrossRef]
- Hartmann, A.; Hunot, S.; Michel, P.P.; Muriel, M.-P.; Vyas, S.; Faucheux, B.A.; Mouatt-Prigent, A.; Turmel, H.; Srinivasan, A.; Ruberg, M. Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2875–2880. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, A.; Mouatt-Prigent, A.; Vila, M.; Abbas, N.; Perier, C.; Faucheux, B.A.; Vyas, S.; Hirsch, E.C. Increased expression and redistribution of the antiapoptotic molecule Bcl-xL in Parkinson’s disease. Neurobiol. Dis. 2002, 10, 28–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogi, M.; Kondo, T.; Mizuno, Y.; Nagatsu, T. p53 protein, interferon-γ, and NF-κB levels are elevated in the parkinsonian brain. Neurosci. Lett. 2007, 414, 94–97. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S. Aggregated α-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.E.; Paek, S.H. Mitochondrial dysfunction in Parkinson’s disease. Exp. Neurobiol. 2015, 24, 103. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.; Cooper, J.; Dexter, D.; Clark, J.; Jenner, P.; Marsden, C. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Krige, D.; Carroll, M.T.; Cooper, J.M.; Marsden, C.D.; Schapira, A.H.; Group), R.K.Q.P.D.R. Platelet mitochondria function in Parkinson’s disease. Ann. Neurol. 1992, 32, 782–788. [Google Scholar] [CrossRef]
- Bindoff, L.; Birch-Machin, M.A.; Cartlidge, N.; Parker Jr, W.; Turnbull, D. Respiratory chain abnormalities in skeletal muscle from patients with Parkinson’s disease. J. Neurol. Sci. 1991, 104, 203–208. [Google Scholar] [CrossRef]
- Langston, J.; Forno, L.; Tetrud, J.; Reeves, A.; Kaplan, J.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine exposure. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child. Neurol. Soc. 1999, 46, 598–605. [Google Scholar] [CrossRef]
- Nicklas, W.J.; Vyas, I.; Heikkila, R.E. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1, 2, 5, 6-tetrahydropyridine. Life Sci. 1985, 36, 2503–2508. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [Green Version]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef] [Green Version]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, ra342–ra378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breidert, T.; Callebert, J.; Heneka, M.; Landreth, G.; Launay, J.; Hirsch, E. Protective action of the peroxisome proliferator-activated receptor-γ agonist pioglitazone in a mouse model of Parkinson’s disease. J. Neurochem. 2002, 82, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Dehmer, T.; Heneka, M.T.; Sastre, M.; Dichgans, J.; Schulz, J.B. Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with IκBα induction and block of NFκB and iNOS activation. J. Neurochem. 2004, 88, 494–501. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Darvesh, A.S.; Funk, M.O.; Van der Schyf, C.J.; Carroll, R.T. Identification of novel monoamine oxidase B inhibitors by structure-based virtual screening. Bioorg. Med. Chem. Lett. 2010, 20, 5295–5298. [Google Scholar] [CrossRef]
- Ridder, D.A.; Schwaninger, M. In search of the neuroprotective mechanism of thiazolidinediones in Parkinson’s disease. Exp. Neurol. 2012, 238, 133–137. [Google Scholar] [CrossRef]
- Carta, A.R.; Pisanu, A.; Carboni, E. Do PPAR-gamma agonists have a future in Parkinson’s disease therapy? Parkinson’s Dis. 2011, 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, C.R.; Joers, V.; Bondarenko, V.; Brunner, K.; Simmons, H.A.; Ziegler, T.E.; Kemnitz, J.W.; Johnson, J.A.; Emborg, M.E. The PPAR-γ agonist pioglitazone modulates inflammation and induces neuroprotection in parkinsonian monkeys. J. Neuroinflammation 2011, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, M.M.F.; Bassani, T.B.; Cóppola-Segovia, V.; Moura, E.L.R.; Zanata, S.M.; Andreatini, R.; Vital, M.A.B.F. PPAR-γ agonist pioglitazone reduces microglial proliferation and NF-κB activation in the substantia nigra in the 6-hydroxydopamine model of Parkinson’s disease. Pharmacol. Rep. 2019, 71, 556–564. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, W.; Li, G.; Chen, J.; Guan, X.; Chen, X.; Guan, Z. Neuroprotective effect and mechanism of thiazolidinedione on dopaminergic neurons in vivo and in vitro in Parkinson’s disease. PPAR Res. 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, R.L.; Dragicevic, N.; Seifert, K.; Choi, D.Y.; Liu, M.; Kim, H.C.; Cass, W.A.; Sullivan, P.G.; Bing, G. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J. Neurochem. 2007, 100, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.L.; Choi, D.-Y.; Ross, S.A.; Bing, G. Protective properties afforded by pioglitazone against intrastriatal LPS in Sprague–Dawley rats. Neurosci. Lett. 2008, 432, 198–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, B.; Liu, M.; Bing, G. Neuroprotection with pioglitazone against LPS insult on dopaminergic neurons may be associated with its inhibition of NF-κB and JNK activation and suppression of COX-2 activity. J. Neuroimmunol. 2007, 192, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Xing, B.; Xin, T.; Hunter, R.L.; Bing, G. Pioglitazone inhibition of lipopolysaccharide-induced nitric oxide synthase is associated with altered activity of p38 MAP kinase and PI3K/Akt. J. Neuroinflamm. 2008, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Carta, A.; Frau, L.; Pisanu, A.; Wardas, J.; Spiga, S.; Carboni, E. Rosiglitazone decreases peroxisome proliferator receptor-gamma levels in microglia and inhibits TNF-alpha production: New evidences on neuroprotection in a progressive Parkinson’s disease model. Neuroscience 2011, 194, 250–261. [Google Scholar] [CrossRef]
- Normando, E.M.; Davis, B.M.; De Groef, L.; Nizari, S.; Turner, L.A.; Ravindran, N.; Pahlitzsch, M.; Brenton, J.; Malaguarnera, G.; Guo, L. The retina as an early biomarker of neurodegeneration in a rotenone-induced model of Parkinson’s disease: Evidence for a neuroprotective effect of rosiglitazone in the eye and brain. Acta Neuropathol. Commun. 2016, 4, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, T.W.; Lee, J.Y.; Shim, W.S.; Kang, E.S.; Kim, S.K.; Ahn, C.W.; Lee, H.C.; Cha, B.S. Rosiglitazone protects human neuroblastoma SH-SY5Y cells against acetaldehyde-induced cytotoxicity. Biochem. Biophys. Res. Commun. 2006, 340, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.W.; Lee, J.Y.; Shim, W.S.; Kang, E.S.; Kim, S.K.; Ahn, C.W.; Lee, H.C.; Cha, B.S. Rosiglitazone protects human neuroblastoma SH-SY5Y cells against MPP+ induced cytotoxicity via inhibition of mitochondrial dysfunction and ROS production. J. Neurol. Sci. 2007, 253, 53–60. [Google Scholar] [CrossRef]
- Altinoz, M.A.; Elmaci, I.; Hacimuftuoglu, A.; Ozpinar, A.; Hacker, E.; Ozpinar, A. PPARδ and its ligand erucic acid may act anti-tumoral, neuroprotective, and myelin protective in neuroblastoma, glioblastoma, and Parkinson’s disease. Mol. Asp. Med. 2021, 78, 100871. [Google Scholar] [CrossRef] [PubMed]
- Das, N.R.; Gangwal, R.P.; Damre, M.V.; Sangamwar, A.T.; Sharma, S.S. A PPAR-β/δ agonist is neuroprotective and decreases cognitive impairment in a rodent model of Parkinson’s disease. Curr. Neurovasc. Res. 2014, 11, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Strosznajder, A.K.; Wójtowicz, S.; Jeżyna, M.J.; Sun, G.Y.; Strosznajder, J.B. Recent Insights on the Role of PPAR-β/δ in Neuroinflammation and Neurodegeneration, and Its Potential Target for Therapy. NeuroMol. Med. 2021, 23, 86–98. [Google Scholar] [CrossRef]
- Kreisler, A.; Gelé, P.; Wiart, J.-F.; Lhermitte, M.; Destée, A.; Bordet, R. Lipid-lowering drugs in the MPTP mouse model of Parkinson’s disease: Fenofibrate has a neuroprotective effect, whereas bezafibrate and HMG-CoA reductase inhibitors do not. Brain Res. 2007, 1135, 77–84. [Google Scholar] [CrossRef]
- Barbiero, J.K.; Santiago, R.; Tonin, F.S.; Boschen, S.; Da Silva, L.M.; De Paula Werner, M.F.; Da Cunha, C.; Lima, M.M.; Vital, M.A. PPAR-α agonist fenofibrate protects against the damaging effects of MPTP in a rat model of Parkinson’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 53, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Cho, J.-H.; Lee, S.; Lee, W.; Chang, S.-C.; Chung, H.Y.; Moon, H.R.; Lee, J. Neuroprotective effects of MHY908, a PPAR α/γ dual agonist, in a MPTP-induced Parkinson’s disease model. Brain Res. 2019, 1704, 47–58. [Google Scholar] [CrossRef]
- Casper, D.; Yaparpalvi, U.; Rempel, N.; Werner, P. Ibuprofen protects dopaminergic neurons against glutamate toxicity in vitro. Neurosci. Lett. 2000, 289, 201–204. [Google Scholar] [CrossRef]
- Singh, A.; Tripathi, P.; Singh, S. Neuroinflammatory responses in Parkinson’s disease: Relevance of Ibuprofen in therapeutics. Inflammopharmacology 2021, 29, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.; Fernandez-Villalba, E.; Izura, V.; Lucas-Ochoa, A.; Menezes-Filho, N.; Santana, R.; De Oliveira, M.; Araújo, F.; Estrada, C.; Silva, V. Combined 1-deoxynojirimycin and ibuprofen treatment decreases Microglial activation, phagocytosis and dopaminergic degeneration in MPTP-treated mice. J. Neuroimmune Pharmacol. 2021, 16, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Ko, J.H.; Jeon, H.; Park, S.-S.; Kim, S.-N. Co-Administration of Gagam-Sipjeondaebo-Tang and Ibuprofen Alleviates the Inflammatory Response in MPTP-Induced Parkinson’s Disease Mouse Model and RAW264. 7 Macrophages. Pathogens 2021, 10, 268. [Google Scholar] [CrossRef] [PubMed]
- Kurkowska-Jastrzębska, I.; Babiuch, M.; Joniec, I.; Przybyłkowski, A.; Członkowski, A.; Członkowska, A. Indomethacin protects against neurodegeneration caused by MPTP intoxication in mice. Int. Immunopharmacol. 2002, 2, 1213–1218. [Google Scholar] [CrossRef]
- Dassati, S.; Schweigreiter, R.; Buechner, S.; Waldner, A. Celecoxib promotes survival and upregulates the expression of neuroprotective marker genes in two different in vitro models of Parkinson’s disease. Neuropharmacology 2021, 194, 108378. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Kim, S.; Lee, J.M.; Oh, Y.-S.; Park, S.M.; Kim, S.R. Montelukast treatment protects nigral dopaminergic neurons against microglial activation in the 6-hydroxydopamine mouse model of Parkinson’s disease. Neuroreport 2017, 28, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, V.B.; Marathe, P.A. Effect of montelukast in experimental model of Parkinson’s disease. Neurosci. Lett. 2018, 682, 100–105. [Google Scholar] [CrossRef]
- Mansour, R.M.; Ahmed, M.A.; El-Sahar, A.E.; El Sayed, N.S. Montelukast attenuates rotenone-induced microglial activation/p38 MAPK expression in rats: Possible role of its antioxidant, anti-inflammatory and antiapoptotic effects. Toxicol. Appl. Pharmacol. 2018, 358, 76–85. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Landreth, G.E.; Hüll, M. Drug insight: Effects mediated by peroxisome proliferator-activated receptor-γ in CNS disorders. Nat. Clin. Pract. Neurol. 2007, 3, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Sastre, M.; Dewachter, I.; Rossner, S.; Bogdanovic, N.; Rosen, E.; Borghgraef, P.; Evert, B.O.; Dumitrescu-Ozimek, L.; Thal, D.R.; Landreth, G. Nonsteroidal anti-inflammatory drugs repress β-secretase gene promoter activity by the activation of PPARγ. Proc. Natl. Acad. Sci. USA 2006, 103, 443–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, E.; Di Matteo, V.; Benigno, A.; Pierucci, M.; Crescimanno, G.; Di Giovanni, G. Non-steroidal anti-inflammatory drugs in Parkinson’s disease. Exp. Neurol. 2007, 205, 295–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallin, J.; Svenningsson, P. Potential Effects of Leukotriene Receptor Antagonist Montelukast in Treatment of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 5606. [Google Scholar] [CrossRef]
- Chang, M.C.; Kwak, S.G.; Kwak, S. Effect of dietary vitamins C and E on the risk of Parkinson’s disease: A meta-analysis. Clin. Nutr. 2021, 40, 3922–3930. [Google Scholar] [CrossRef] [PubMed]
- Makkar, R.; Behl, T.; Bungau, S.; Zengin, G.; Mehta, V.; Kumar, A.; Uddin, M.S.; Ashraf, G.M.; Abdel-Daim, M.M.; Arora, S.; et al. Nutraceuticals in Neurological Disorders. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, inflammation, and oxidative stress: An integrative view in metabolism. Oxidative Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Piccinin, E.; Sardanelli, A.M.; Seibel, P.; Moschetta, A.; Cocco, T.; Villani, G. PGC-1s in the Spotlight with Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 3487. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahim, A.S.; Ko, L.-w.; Yen, S.-H. Reduced expression of peroxisome-proliferator activated receptor gamma coactivator-1α enhances α-synuclein oligomerization and down regulates AKT/GSK3β signaling pathway in human neuronal cells that inducibly express α-synuclein. Neurosci. Lett. 2010, 473, 120–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.-H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.-I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Seibler, P.; Graziotto, J.; Jeong, H.; Simunovic, F.; Klein, C.; Krainc, D. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 2011, 31, 5970–5976. [Google Scholar] [CrossRef] [PubMed]
- Mudo, G.; Mäkelä, J.; Di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Mälkiä, A.; Bonomo, A.; Kairisalo, M. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell. Mol. Life Sci. 2012, 69, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Grandinetti, A.; Morens, D.M.; Reed, D.; MacEachern, D. Prospective study of cigarette smoking and the risk of developing idiopathic Parkinson’s disease. Am. J. Epidemiol. 1994, 139, 1129–1138. [Google Scholar] [CrossRef]
- Hernán, M.A.; Zhang, S.M.; Rueda-DeCastro, A.M.; Colditz, G.A.; Speizer, F.E.; Ascherio, A. Cigarette smoking and the incidence of Parkinson’s disease in two prospective studies. Ann. Neurol. 2001, 50, 780–786. [Google Scholar] [CrossRef]
- Paganini-Hill, A. Risk factors for Parkinson’s disease: The leisure world cohort study. Neuroepidemiology 2001, 20, 118–124. [Google Scholar] [CrossRef]
- Hernán, M.A.; Takkouche, B.; Caamaño-Isorna, F.; Gestal-Otero, J.J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 2002, 52, 276–284. [Google Scholar] [CrossRef]
- Breckenridge, C.B.; Berry, C.; Chang, E.T.; Sielken Jr, R.L.; Mandel, J.S. Association between Parkinson’s disease and cigarette smoking, rural living, well-water consumption, farming and pesticide use: Systematic review and meta-analysis. PLoS ONE 2016, 11, e0151841. [Google Scholar]
- Ritz, B.; Ascherio, A.; Checkoway, H.; Marder, K.S.; Nelson, L.M.; Rocca, W.A.; Ross, G.W.; Strickland, D.; Van Den Eeden, S.K.; Gorell, J. Pooled analysis of tobacco use and risk of Parkinson disease. Arch. Neurol. 2007, 64, 990–997. [Google Scholar] [CrossRef] [Green Version]
- Morens, D.; Grandinetti, A.; Reed, D.; White, L.; Ross, G. Cigarette smoking and protection from Parkinson’s disease: False association or etiologic clue? Neurology 1995, 45, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Quik, M. Smoking, nicotine and Parkinson’s disease. Trends Neurosci. 2004, 27, 561–568. [Google Scholar] [CrossRef]
- Hong, D.-P.; Fink, A.L.; Uversky, V.N. Smoking and Parkinson’s disease: Does nicotine affect α-synuclein fibrillation? Biochim. Biophys. Acta Proteins Proteom. 2009, 1794, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Ritz, B.; Lee, P.-C.; Lassen, C.F.; Arah, O.A. Parkinson disease and smoking revisited: Ease of quitting is an early sign of the disease. Neurology 2014, 83, 1396–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, D.G.; Fujioka, S. Caffeine and Parkinson disease: A possible diagnostic and pathogenic breakthrough. Neurology 2018, 90, 205–206. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Chen, J.-F. Caffeine and Parkinson’s Disease: Multiple Benefits and Emerging Mechanisms. Front. Neurosci. 2020, 14, 1334. [Google Scholar] [CrossRef]
- Hong, C.T.; Chan, L.; Bai, C.-H. The effect of caffeine on the risk and progression of Parkinson’s disease: A meta-analysis. Nutrients 2020, 12, 1860. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.-H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F.; Xu, K.; Petzer, J.P.; Staal, R.; Xu, Y.-H.; Beilstein, M.; Sonsalla, P.K.; Castagnoli, K.; Castagnoli, N.; Schwarzschild, M.A. Neuroprotection by caffeine and A2A adenosine receptor inactivation in a model of Parkinson’s disease. J. Neurosci. 2001, 21, RC143. [Google Scholar] [CrossRef] [Green Version]
- Ascherio, A.; Zhang, S.M.; Hernán, M.A.; Kawachi, I.; Colditz, G.A.; Speizer, F.E.; Willett, W.C. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child. Neurol. Soc. 2001, 50, 56–63. [Google Scholar] [CrossRef]
- Benedetti, M.; Bower, J.; Maraganore, D.; McDonnell, S.; Peterson, B.; Ahlskog, J.; Schaid, D.; Rocca, W. Smoking, alcohol, and coffee consumption preceding Parkinson’s disease: A case-control study. Neurology 2000, 55, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Xu, Y.; Brown-Jermyn, D.; Chen, J.-F.; Ascherio, A.; Dluzen, D.E.; Schwarzschild, M.A. Estrogen prevents neuroprotection by caffeine in the mouse 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine model of Parkinson’s disease. J. Neurosci. 2006, 26, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Agúndez, J.A.G. Alcohol consumption and risk for Parkinson’s disease: A systematic review and meta-analysis. J. Neurol. 2019, 266, 1821–1834. [Google Scholar] [CrossRef]
- Zhang, D.; Jiang, H.; Xie, J. Alcohol intake and risk of Parkinson’s disease: A meta-analysis of observational studies. Mov. Disord. 2014, 29, 819–822. [Google Scholar] [CrossRef]
- Peng, B.; Yang, Q.; Joshi, R.B.; Liu, Y.; Akbar, M.; Song, B.J.; Zhou, S.; Wang, X. Role of Alcohol Drinking in Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 2316. [Google Scholar] [CrossRef] [Green Version]
- Zigmond, M.J.; Smeyne, R.J. Exercise: Is it a neuroprotective and if so, how does it work? Parkinsonism Relat Disord. 2014, 20, S123–S127. [Google Scholar] [CrossRef]
- Jadavji, N.M.; Kolb, B.; Metz, G.A. Enriched environment improves motor function in intact and unilateral dopamine-depleted rats. Neuroscience 2006, 140, 1127–1138. [Google Scholar] [CrossRef]
- Mabandla, M.; Kellaway, L.; St Clair Gibson, A.; Russell, V.A. Voluntary running provides neuroprotection in rats after 6-hydroxydopamine injection into the medial forebrain bundle. Metab. Brain Dis. 2004, 19, 43–50. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, S.J.; Gross, N.B.; Fricks, A.N.; Casiano, B.D.; Nguyen, T.B.; Marshall, J.F. Running wheel exercise enhances recovery from nigrostriatal dopamine injury without inducing neuroprotection. Neuroscience 2007, 144, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Steiner, B.; Winter, C.; Hosman, K.; Siebert, E.; Kempermann, G.; Petrus, D.S.; Kupsch, A. Enriched environment induces cellular plasticity in the adult substantia nigra and improves motor behavior function in the 6-OHDA rat model of Parkinson’s disease. Exp. Neurol. 2006, 199, 291–300. [Google Scholar] [CrossRef]
- Fisher, B.E.; Petzinger, G.M.; Nixon, K.; Hogg, E.; Bremmer, S.; Meshul, C.K.; Jakowec, M.W. Exercise-induced behavioral recovery and neuroplasticity in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse basal ganglia. J. Neurosci. Res. 2004, 77, 378–390. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PPAR Agent/ Ligand | Compound/ Toxin Utilized | Animal Model/ Cell Type | Outcomes | Ref. |
|---|---|---|---|---|
| 1. PPAR-γ agonist | ||||
| Pioglitazone (oral, 20 mg/kg) | MPTP (i.p, 15 mg/kg) | Mouse | Reduced MPTP-inebriation prompted microglia stimulation. Precluded forfeiture of DArgic nerve cells within the SN-PC. | [158] |
| Pioglitazone (oral) | MPTP | Mouse | Extended safeguardance towards MPTP-prompted nerve cell destruction in TH-immunoreactive SN nerve cells. Decreased stimulation of microglia, inflammatory mediators, nitro tyrosine activity in DArgic nerve cells, and the fraction of GFAP positive cells in the SN and striate nucleus. | [159] |
| MPTP | Rhesus monkeys | Preserved DA striatal fibers and SN nerve cells. | [163] | |
| Pioglitazone (oral, 30 mg/kg) | 6-OHDA | Male Wistar Rats | Rendered protection to DArgic nerve cells in the SN against neuronal destruction. Offered a significant reduction in NF-κB and microglial stimulation. | [164] |
| Pioglitazone | LPS (i.c.v) | Rat | Decreased LPS triggered microglial inflammatory processes, and oxidative damage. Upgraded mitochondrial operation, incompletely reinstated DA levels, and enhanced DArgic neuroprotection. | [166,167] |
| LPS | DArgic nerve cells-neuroglia co-culture | Safeguarded DA nerve cells via suppression of microglial activation, diminished phosphorylation of NF-κB and JNK, and reduced expression of COX-2. Diminished expression of iNOS, and synthesis of NO through distinguishably modulating the p38 MAPK and PI3K/Akt processes. | [168,169] | |
| Rosiglitazone (i.p, 10 mg/kg) | MPTP/MPTPp | Mice (C57BL/6J) | Decreased generation of TNF-α in the microglia cells, and arrested MPTPp-instigated nerve cell degeneration in the SN-PC. | [170] |
| Rosiglitazone (liposome-encapsulated form), (i.p, 1mL/kg) | Rotenone | Rat | Can safeguard retinal nerve cells from the abnormalities provoked by subjection to rotenone, and elevated neuroprotection in the retina and CNS. | [171] |
| Rosiglitazone | Acetaldehyde | Human neuroblastoma SH-SY5Y cells | Safeguarded DA nerve cells from acetaldehyde prompted programmed cell death via enhancing the activity of antioxidant enzymes, and by controlling the expression of Bax and Bcl-2. | [172] |
| MPP+ | Safeguards SH-SY5Y cells from MPP+ prompted cellular damage via the suppression of impairment in the functioning of mitochondria and ROS generation. Raised CAT, SOD, Bcl-2 expression, and diminished Bax expression. | [173] | ||
| 2. PPAR-β/δ agonist | ||||
| GW-501516 | Rotenone | Rat | Safeguards DArgic nerve cells from damage caused by deleterious substances and upgrades behavioral performance by diminishing ER-related stress. | [174] |
| GW-501516 and L-165041 | Staurosporine, and MPP+ | SH-SY5Y cells | Safeguarded SH-SY5Y cells from staurosporine and MPP+ elicited programmed cell death via suppressing the caspase-3 pathway activation. | [10] |
| GW0742 (30 and 100 μg/kg) | MPTP | Rat | Incomplete reinstatement of MPTP-damaged cognitive activities. Reduced oxidative destruction and splitting-up of DNA strands into fragments. | [175,176] |
| 3. PPAR-α agonist | ||||
| Fenofibrate (0.2% in diet) and Benzafibrate (0.02% in diet) | MPTP | Mouse | Fenofibrate safeguarded DArgic nerve cells in the SN and TH-immunoreactive endings within the striatal region, however benzafibrate displayed no such safeguarding action. | [177] |
| Fenofibrate (oral, 100 mg/kg) | Rat | Reduced MPTP provoked hypo locomotion, and depressive behavioral patterns after neurotoxin administration. Safeguarded from elevation in the ROS generation and decrease in levels of DA following surgical procedure. | [178] | |
| 4. PPAR-α/γ dual agonist | ||||
| MHY908 | MPTP | Mouse | Reduced MPTP-prompted DArgic nerve cell deprivation, and motor impairment. Alleviation of MPTP-instigated activation of glial cells in the nigrostriatal region. Suppression of MPP+ prompted activation of astroglia by inhibition of NF-signaling in primary cultured astrocytes. Suppression of MPP+ provoked cellular damage and ROS generation in SH-SY5Y neuroblastoma cells. | [179] |
| 5. NSAIDs | ||||
| Paracetamol (1 mM), and Ibuprofen (0.1 mM) | 6-OHDA, MPP+, and glutamate | Mesencephalic cultured cells | Effectively mitigated 6-OHDA, MPP+, and glutamate prompted DArgic nerve cell death. Ibuprofen individually elevated the quantity of DArgic nerve cells by nearly 47%. | [180,181] |
| 1-DNJ + Ibuprofen | MPTP | Mice | Impedes mesencephalic DArgic nerve cell death. Minimizes the levels of IL-6, TNF-α, total microglia markers namely CD68+/Iba-1+ cells, and interaction between microglia cells and nerve cells. | [182] |
| GST + Ibuprofen | Mouse | Exhibited a synergistic action in ameliorating DArgic nerve cell death and reducing the activation of macrophages. Reduced NO levels in LPS-activated macrophages. GST alone reduced DArgic nerve cell death, levels of iNOS, IL-6, IL-1β, and COX-2, and relieved PD-concerned behavioral impairment. | [183] | |
| Indomethacin | Mice | Extended safeguard towards MPTP-prompted nerve cell destruction. Diminished infiltration of lymphocytes, and microglia activation. | [184] | |
| Celecoxib (< 20 uM) | Paraquat, and 6-OHDA | SH-SY5Y cells | Reinstated SH-SY5Y cells from damage caused by exposure to paraquat and 6-OHDA. Resulted in prolonged overexpression of APOD, MITF, and TFEB, and safeguarded DArgic nerve cell from damage. | [185] |
| 6. Leukotriene receptor antagonist | ||||
| Montelukast | 6-OHDA | Mouse | Safeguarded DA nerve cells against microglia cells activation, and reduced the generation of IL-1β and TNF-α. | [186] |
| Montelukast | Rotenone | Rat | Reduced microglia cells activation and upgraded motor activities. Decreased p53 protein, oxidative damage, thereby strongly influences life span of nerve cells. | [187,188] |
| 7. PGC-1α | ||||
| PGC-1α | MPTP | PGC-1α genetically inactivated mice | Elevated proneness to MPTP prompted degeneration of DArgic nerve cells in SN-PC. Up-regulation of PGC-1α provoked mitochondrial biogenesis, and safeguarded nerve cells from oxidative damage. | [189] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behl, T.; Madaan, P.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Chigurupati, S.; Alrashdi, I.; Bungau, S.G. Elucidating the Neuroprotective Role of PPARs in Parkinson’s Disease: A Neoteric and Prospective Target. Int. J. Mol. Sci. 2021, 22, 10161. https://doi.org/10.3390/ijms221810161
Behl T, Madaan P, Sehgal A, Singh S, Sharma N, Bhatia S, Al-Harrasi A, Chigurupati S, Alrashdi I, Bungau SG. Elucidating the Neuroprotective Role of PPARs in Parkinson’s Disease: A Neoteric and Prospective Target. International Journal of Molecular Sciences. 2021; 22(18):10161. https://doi.org/10.3390/ijms221810161
Chicago/Turabian StyleBehl, Tapan, Piyush Madaan, Aayush Sehgal, Sukhbir Singh, Neelam Sharma, Saurabh Bhatia, Ahmed Al-Harrasi, Sridevi Chigurupati, Ibrahim Alrashdi, and Simona Gabriela Bungau. 2021. "Elucidating the Neuroprotective Role of PPARs in Parkinson’s Disease: A Neoteric and Prospective Target" International Journal of Molecular Sciences 22, no. 18: 10161. https://doi.org/10.3390/ijms221810161
APA StyleBehl, T., Madaan, P., Sehgal, A., Singh, S., Sharma, N., Bhatia, S., Al-Harrasi, A., Chigurupati, S., Alrashdi, I., & Bungau, S. G. (2021). Elucidating the Neuroprotective Role of PPARs in Parkinson’s Disease: A Neoteric and Prospective Target. International Journal of Molecular Sciences, 22(18), 10161. https://doi.org/10.3390/ijms221810161