
A Computational Workflow for the Identification of Novel Fragments Acting as Inhibitors of the Activity of Protein Kinase CK1δ


 , , ,
, , ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Protein Kinase CK1δ
1.2. Fragment-Based Drug Discovery (FBDD) Principles
1.3. Fragment-Based Drug Discovery and Kinase Inhibitors
1.4. Computational Methods in FBDD
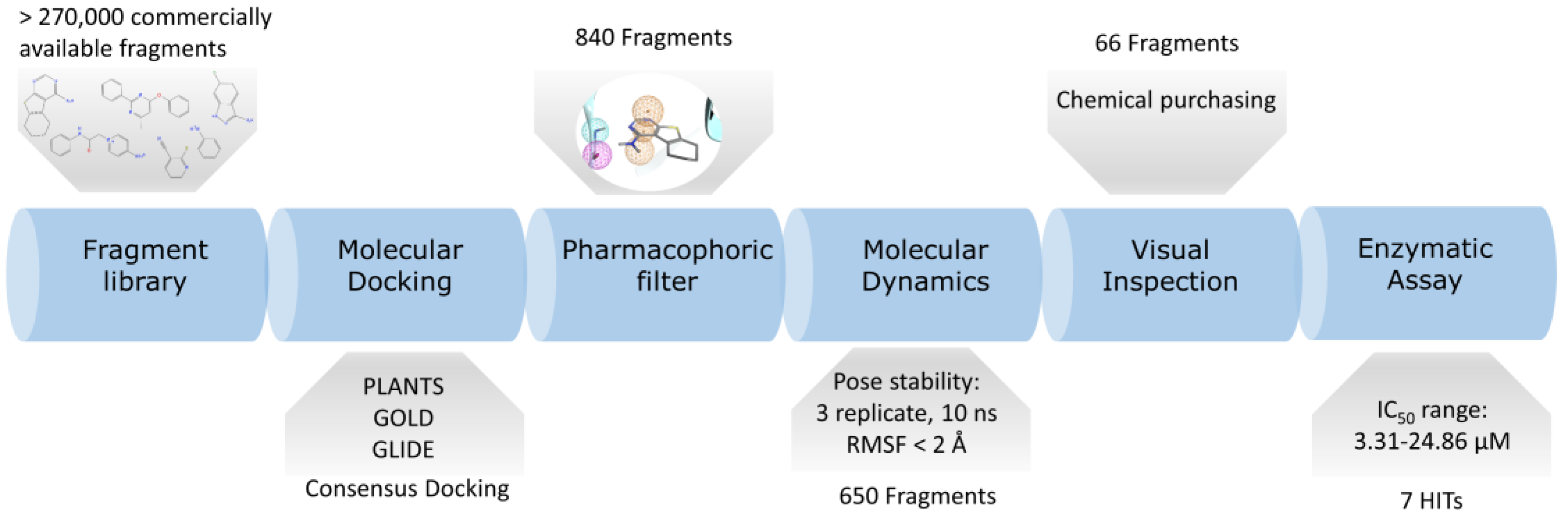
2. Results
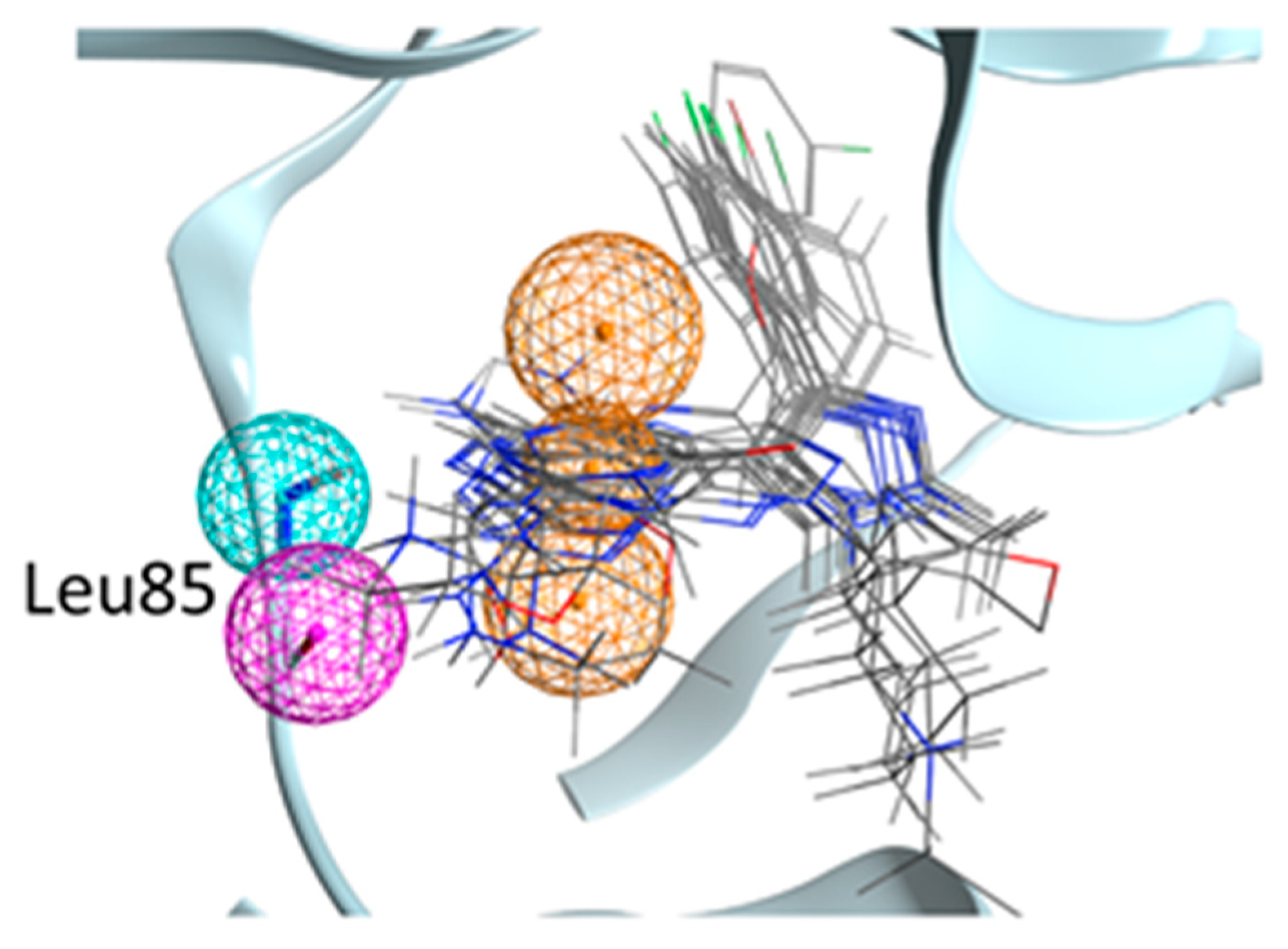
2.1. Computational Results
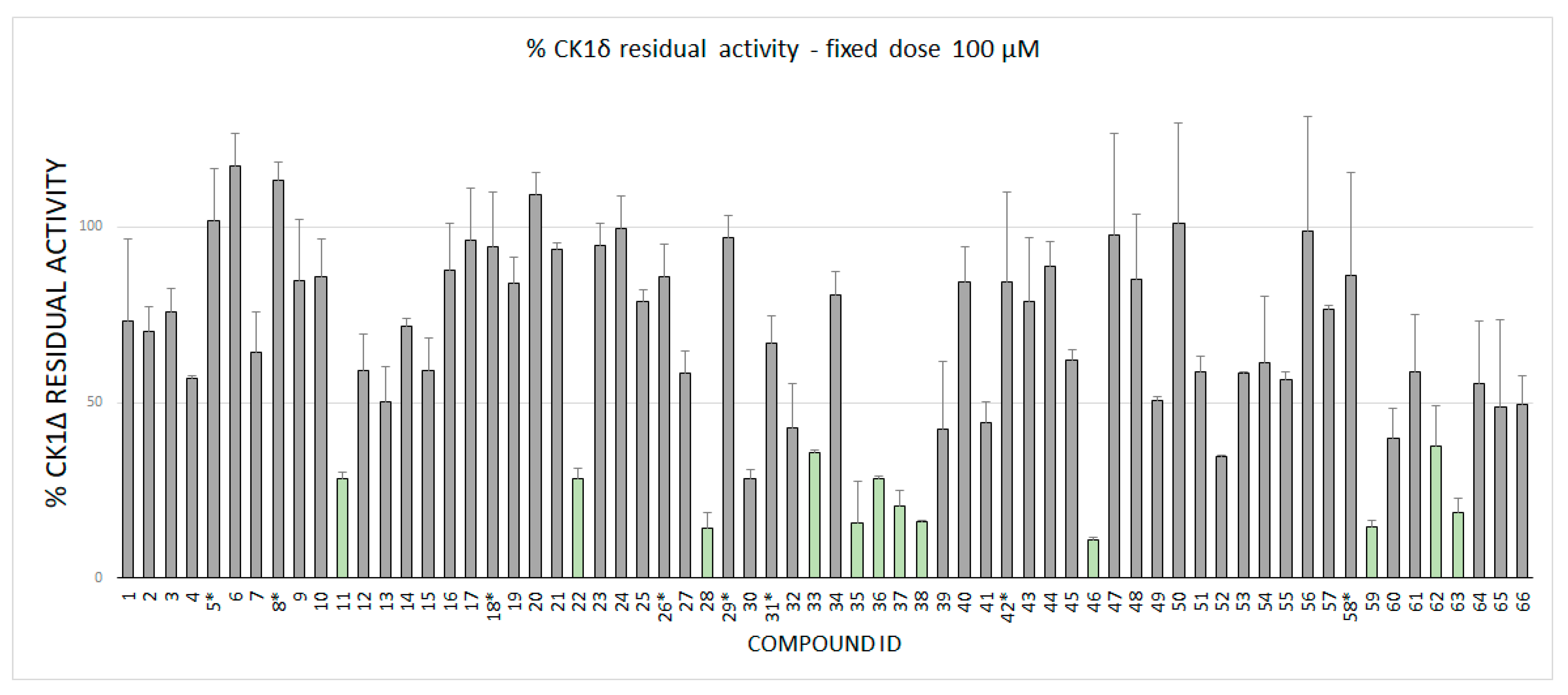
2.2. Enzymatic Assay Results
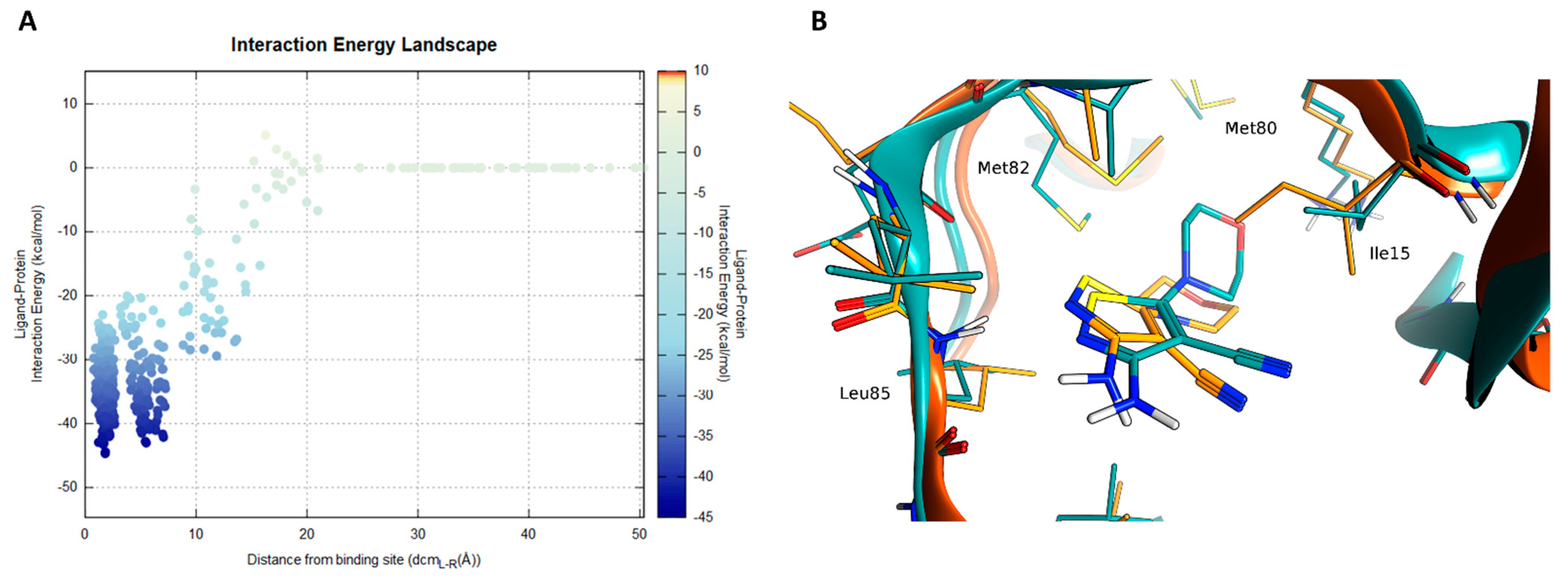
2.3. Molecular Recognition Studies of the Most Promising Fragment
3. Discussion
4. Materials and Methods
4.1. Molecular Modelling and Docking
4.2. Pharmacophore Modeling
4.3. Molecular Dynamics
4.4. Enzymatic Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Knippschild, U.; Krüger, M.; Richter, J.; Xu, P.; García-Reyes, B.; Peifer, C.; Halekotte, J.; Bakulev, V.; Bischof, J. The CK1 family: Contribution to cellular stress response and its role in carcinogenesis. Front. Oncol. 2014, 4, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Meggio, F.; Perich, J.W.; Reynolds, E.C.; Pinna, L.A. A synthetic β-casein phosphopeptide and analogues as model substrates for casein kinase-1, a ubiquitous, phosphate directed protein kinase. FEBS Lett. 1991, 283, 303–306. [Google Scholar] [CrossRef] [Green Version]
- Pulgar, V.; Marin, O.; Meggio, F.; Allende, C.C.; Allende, J.E.; Pinna, L.A. Optimal sequences for non-phosphate-directed phoshorylation by protein kinase CK1 (casein kinase-1)—A re-evaluation. Eur. J. Biochem. 1999, 260, 520–526. [Google Scholar] [CrossRef]
- Marin, O.; Meggio, F.; Sarno, S.; Andretta, M.; Pinna, L.A. Phosphorylation of synthetic fragments of inhibitor-2 of protein phosphatase-1 by casein kinase-1 and -2: Evidence that phosphorylated residues are not strictly required for efficient targeting by casein kinase-1. Eur. J. Biochem. 1994, 223, 647–653. [Google Scholar] [CrossRef]
- Bischof, J.; Randoll, S.J.; Süßner, N.; Henne-Bruns, D.; Pinna, L.A.; Knippschild, U. CK1δ Kinase Activity Is Modulated by Chk1-Mediated Phosphorylation. PLoS ONE 2013, 8, e68803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graves, P.R.; Roach, P.J. Role of COOH-terminal phosphorylation in the regulation of casein kinase Iδ. J. Biol. Chem. 1995, 270, 21689–21694. [Google Scholar] [CrossRef] [Green Version]
- Milne, D.M.; Looby, P.; Meek, D.W. Catalytic activity of protein kinase CK1δ (casein kinase 1 δ) is essential for its normal subcellular localization. Exp. Cell Res. 2001, 263, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Ianes, C.; Gärtner, F.; Liu, C.; Burster, T.; Bakulev, V.; Rachidi, N.; Knippschild, U.; Bischof, J. Structure, Regulation, and (Patho-)Physiological Functions of the Stress-Induced Protein Kinase CK1 Delta (CSNK1D); Elsevier: Amsterdam, The Netherlands, 2019; Volume 715, ISBN 1393428169. [Google Scholar]
- Longenecker, K.L.; Roach, P.J.; Hurley, T.D. Crystallographic studies of casein kinasc I δ: Toward a structural understanding of auto-inhibition. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 473–475. [Google Scholar] [CrossRef]
- Hirner, H.; Günes, C.; Bischof, J.; Wolff, S.; Grothey, A.; Kühl, M.; Oswald, F.; Wegwitz, F.; Bösl, M.R.; Trauzold, A.; et al. Impaired CK1 delta activity attenuates SV40-induced cellular transformation in vitro and mouse mammary carcinogenesis in Vivo. PLoS ONE 2012, 7, e29709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, D.I.; Gil, C.; Martinez, A. Protein kinases CK1 and CK2 as new targets for neurodegenerative diseases. Med. Res. Rev. 2011, 31, 924–954. [Google Scholar] [CrossRef] [Green Version]
- Ghoshal, N.; Smiley, J.F.; DeMaggio, A.J.; Hoekstra, M.F.; Cochran, E.J.; Binder, L.I.; Kuret, J. A new molecular link between the fibrillar and granulovacuolar lesions of Alzheimer’s disease. Am. J. Pathol. 1999, 155, 1163–1172. [Google Scholar] [CrossRef]
- Yasojima, K.; Kuret, J.; Demaggio, A.J.; McGeer, E.; McGeer, P.L. Casein kinase 1 delta mRNA is upregulated in Alzheimer disease brain. Brain Res. 2000, 865, 116–120. [Google Scholar] [CrossRef]
- Kuret, J.; Johnson, G.S.; Cha, D.; Christenson, E.R.; DeMaggio, A.J.; Hoekstra, M.F. Casein kinase 1 is tightly associated with paired-helical filaments isolated from Alzheimer’s disease brain. J. Neurochem. 1997, 69, 2506–2515. [Google Scholar] [CrossRef] [Green Version]
- Schwab, C.; Demaggio, A.J.; Ghoshal, N.; Binder, L.I.; Kuret, J.; McGeer, P.L. Casein kinase 1 delta is associated with pathological accumulation of tau in several neurodegenerative diseases. Neurobiol. Aging 2000, 21, 503–510. [Google Scholar] [CrossRef]
- Li, G.; Yin, H.; Kuret, J. Casein Kinase 1 Delta Phosphorylates Tau and Disrupts Its Binding to Microtubules. J. Biol. Chem. 2004, 279, 15938–15945. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, A.; Chauhan, V.P.S.; Murakami, N.; Brockerhoff, H.; Wisniewski, H.M. Amyloid β-protein stimulates casein kinase I and casein kinase II activities. Brain Res. 1993, 629, 47–52. [Google Scholar] [CrossRef]
- Flajolet, M.; He, G.; Heiman, M.; Lin, A.; Nairn, A.C.; Greengard, P. Regulation of Alzheimer’s disease amyloid-β formation by casein kinase I. Proc. Natl. Acad. Sci. USA 2007, 104, 4159–4164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höttecke, N.; Liebeck, M.; Baumann, K.; Schubenel, R.; Winkler, E.; Steiner, H.; Schmidt, B. Inhibition of γ-secretase by the CK1 inhibitor IC261 does not depend on CK1δ. Bioorganic Med. Chem. Lett. 2010, 20, 2958–2963. [Google Scholar] [CrossRef] [PubMed]
- Kosten, J.; Binolfi, A.; Stuiver, M.; Verzini, S.; Theillet, F.X.; Bekei, B.; Van Rossum, M.; Selenko, P. Efficient modification of alpha-synuclein serine 129 by protein kinase CK1 requires phosphorylation of tyrosine 125 as a priming event. ACS Chem. Neurosci. 2014, 5, 1203–1208. [Google Scholar] [CrossRef]
- Nonaka, T.; Suzuki, G.; Tanaka, Y.; Kametani, F.; Hirai, S.; Okado, H.; Miyashita, T.; Saitoe, M.; Akiyama, H.; Masai, H.; et al. Phosphorylation of TAR DNA-binding protein of 43 kDa (TDP-43) by truncated casein kinase 1δ triggers mislocalization and accumulation of TDP-43. J. Biol. Chem. 2016, 291, 5473–5483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salado, I.G.; Redondo, M.; Bello, M.L.; Perez, C.; Liachko, N.F.; Kraemer, B.C.; Miguel, L.; Lecourtois, M.; Gil, C.; Martinez, A.; et al. Protein kinase CK-1 inhibitors as new potential drugs for amyotrophic lateral sclerosis. J. Med. Chem. 2014, 57, 2755–2772. [Google Scholar] [CrossRef]
- Hall, R.J.; Mortenson, P.N.; Murray, C.W. Efficient exploration of chemical space by fragment-based screening. Prog. Biophys. Mol. Biol. 2014, 116, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Yasothan, U.; Kirkpatrick, P. Vemurafenib. Nat. Rev. Drug Discov. 2011, 10, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Romero, D. Initial results with asciminib in CML. Nat. Rev. Clin. Oncol. 2020, 17, 135. [Google Scholar] [CrossRef] [Green Version]
- Schoepfer, J.; Jahnke, W.; Berellini, G.; Buonamici, S.; Cotesta, S.; Cowan-Jacob, S.W.; Dodd, S.; Drueckes, P.; Fabbro, D.; Gabriel, T.; et al. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J. Med. Chem. 2018, 18, 8120–8135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef]
- Miranker, A.; Karplus, M. Functionality maps of binding sites: A multiple copy simultaneous search method. Proteins Struct. Funct. Bioinform. 1991, 11, 29–34. [Google Scholar] [CrossRef]
- Clark, M.; Meshkat, S.; Talbot, G.T.; Carnevali, P.; Wiseman, J.S. Fragment-based computation of binding free energies by systematic sampling. J. Chem. Inf. Model. 2009, 49, 1901–1913. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H.J. The computer program LUDI: A new method for the de novo design of enzyme inhibitors. J. Comput. Aided. Mol. Des. 1992, 6, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Eisen, M.B.; Wiley, D.C.; Karplus, M.; Hubbard, R.E. HOOK: A program for finding novel molecular architectures that satisfy the chemical and steric requirements of a macromolecule binding site. Proteins Struct. Funct. Bioinform. 1994, 19, 199–211. [Google Scholar] [CrossRef]
- Lauri, G.; Bartlett, P.A. CAVEAT: A program to facilitate the design of organic molecules. J. Comput. Aided Mol. Des. 1994, 8, 51–66. [Google Scholar] [CrossRef]
- Maass, P.; Schulz-Gasch, T.; Stahl, M.; Rarey, M. Recore: A fast and versatile method for scaffold hopping based on small molecule crystal structure conformations. J. Chem. Inf. Model. 2007, 47, 390–399. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-4: Maestro; Schrödinger LLC: New York, NY, USA, 2020.
- Chemical Computing Group ULC, Molecular Operating Environment (MOE), 2019.01. 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7. 2019. Available online: https://www.chemcomp.com/release_notes/moe201901/rnotes.htm (accessed on 29 June 2021).
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006, 26, 531–568. [Google Scholar] [CrossRef]
- Gill, S.C.; Lim, N.M.; Grinaway, P.B.; Rustenburg, A.S.; Fass, J.; Ross, G.A.; Chodera, J.D.; Mobley, D.L. Binding Modes of Ligands Using Enhanced Sampling (BLUES): Rapid Decorrelation of Ligand Binding Modes via Nonequilibrium Candidate Monte Carlo. J. Phys. Chem. B 2018, 122, 5579–5598. [Google Scholar] [CrossRef]
- Lim, N.M.; Osato, M.; Warren, G.L.; Mobley, D.L. Fragment Pose Prediction Using Non-equilibrium Candidate Monte Carlo and Molecular Dynamics Simulations. J. Chem. Theory Comput. 2020, 16, 2778–2794. [Google Scholar] [CrossRef] [PubMed]
- Linker, S.M.; Magarkar, A.; Köfinger, J.; Hummer, G.; Seeliger, D. Fragment Binding Pose Predictions Using Unbiased Simulations and Markov-State Models. J. Chem. Theory Comput. 2019, 15, 4974–4981. [Google Scholar] [CrossRef] [PubMed]
- Sabbadin, D.; Moro, S. Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale. J. Chem. Inf. Model. 2014, 54, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, F.; Bissaro, M.; Fabbian, S.; De Almeida Roger, J.; Mammi, S.; Moro, S.; Bellanda, M.; Sturlese, M. HT-SuMD: Making molecular dynamics simulations suitable for fragment-based screening. A comparative study with NMR. J. Enzyme Inhib. Med. Chem. 2021, 36, 1–14. [Google Scholar] [CrossRef]
- Chaput, L.; Mouawad, L. Efficient conformational sampling and weak scoring in docking programs? Strategy of the wisdom of crowds. J. Cheminform. 2017, 9, 1–18. [Google Scholar] [CrossRef]
- de Souza Neto, L.R.; Moreira-Filho, J.T.; Neves, B.J.; Maidana, R.L.B.R.; Guimarães, A.C.R.; Furnham, N.; Andrade, C.H.; Silva, F.P. In silico Strategies to Support Fragment-to-Lead Optimization in Drug Discovery. Front. Chem. 2020, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Houston, D.R.; Walkinshaw, M.D. Consensus docking: Improving the reliability of docking in a virtual screening context. J. Chem. Inf. Model. 2013, 53, 384–390. [Google Scholar] [CrossRef]
- Fischer, A.; Smieško, M.; Sellner, M.; Lill, M.A. Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results. J. Med. Chem. 2021, 64, 2489–2500. [Google Scholar] [CrossRef]
- Metz, J.T.; Johnson, E.F.; Soni, N.B.; Merta, P.J.; Kifle, L.; Hajduk, P.J. Navigating the kinome. Nat. Chem. Biol. 2011, 7, 200–202. [Google Scholar] [CrossRef]
- Sabbatini, P.; Korenchuk, S.; Roward, J.L.; Groy, A.; Liu, Q.; Leperi, D.; Atkins, C.; Dumble, M.; Yang, J.; Anderson, K.; et al. GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Mol. Cancer Ther. 2009, 8, 2811–2820. [Google Scholar] [CrossRef] [Green Version]
- QUACPAC 2.1.1.0; OpenEye Scientific Software: Santa Fe, NM, USA, 2020.
- Sadowski, J.; Gasteiger, J.; Klebe, G. Comparison of Automatic Three-Dimensional Model Builders Using 639 X-ray Structures. J. Chem. Inf. Comput. Sci. 1994, 34, 1000–1008. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. PLANTS: Application of Ant Colony Optimization to Structure-Based Drug Design. In Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer: Berlin, Gemany, 2006. [Google Scholar]
- Korb, O.; Stützle, T.; Exner, T.E. An ant colony optimization approach to flexible protein–ligand docking. Swarm Intell. 2007, 1, 115–134. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Sándor, M.; Kiss, R.; Keseru, G.M. Virtual fragment docking by glide: A validation study on 190 protein-fragment complexes. J. Chem. Inf. Model. 2010, 50, 1165–1172. [Google Scholar] [CrossRef]
- Harvey, M.J.; Giupponi, G.; De Fabritiis, G. ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Cuzzolin, A.; Sturlese, M.; Deganutti, G.; Salmaso, V.; Sabbadin, D.; Ciancetta, A.; Moro, S. Deciphering the Complexity of Ligand-Protein Recognition Pathways Using Supervised Molecular Dynamics (SuMD) Simulations. J. Chem. Inf. Model. 2016, 56, 687–705. [Google Scholar] [CrossRef] [PubMed]
- Badura, L.; Swanson, T.; Adamowicz, W.; Adams, J.; Cianfrogna, J.; Fisher, K.; Holland, J.; Kleiman, R.; Nelson, F.; Reynolds, L.; et al. An inhibitor of casein kinase Iε induces phase delays in circadian rhythms under free-running and entrained conditions. J. Pharmacol. Exp. Ther. 2007, 322, 730–738. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolcato, G.; Cescon, E.; Pavan, M.; Bissaro, M.; Bassani, D.; Federico, S.; Spalluto, G.; Sturlese, M.; Moro, S. A Computational Workflow for the Identification of Novel Fragments Acting as Inhibitors of the Activity of Protein Kinase CK1δ. Int. J. Mol. Sci. 2021, 22, 9741. https://doi.org/10.3390/ijms22189741
Bolcato G, Cescon E, Pavan M, Bissaro M, Bassani D, Federico S, Spalluto G, Sturlese M, Moro S. A Computational Workflow for the Identification of Novel Fragments Acting as Inhibitors of the Activity of Protein Kinase CK1δ. International Journal of Molecular Sciences. 2021; 22(18):9741. https://doi.org/10.3390/ijms22189741
Chicago/Turabian StyleBolcato, Giovanni, Eleonora Cescon, Matteo Pavan, Maicol Bissaro, Davide Bassani, Stephanie Federico, Giampiero Spalluto, Mattia Sturlese, and Stefano Moro. 2021. "A Computational Workflow for the Identification of Novel Fragments Acting as Inhibitors of the Activity of Protein Kinase CK1δ" International Journal of Molecular Sciences 22, no. 18: 9741. https://doi.org/10.3390/ijms22189741
APA StyleBolcato, G., Cescon, E., Pavan, M., Bissaro, M., Bassani, D., Federico, S., Spalluto, G., Sturlese, M., & Moro, S. (2021). A Computational Workflow for the Identification of Novel Fragments Acting as Inhibitors of the Activity of Protein Kinase CK1δ. International Journal of Molecular Sciences, 22(18), 9741. https://doi.org/10.3390/ijms22189741