
AMPK–mTOR Signaling and Cellular Adaptations in Hypoxia


Abstract
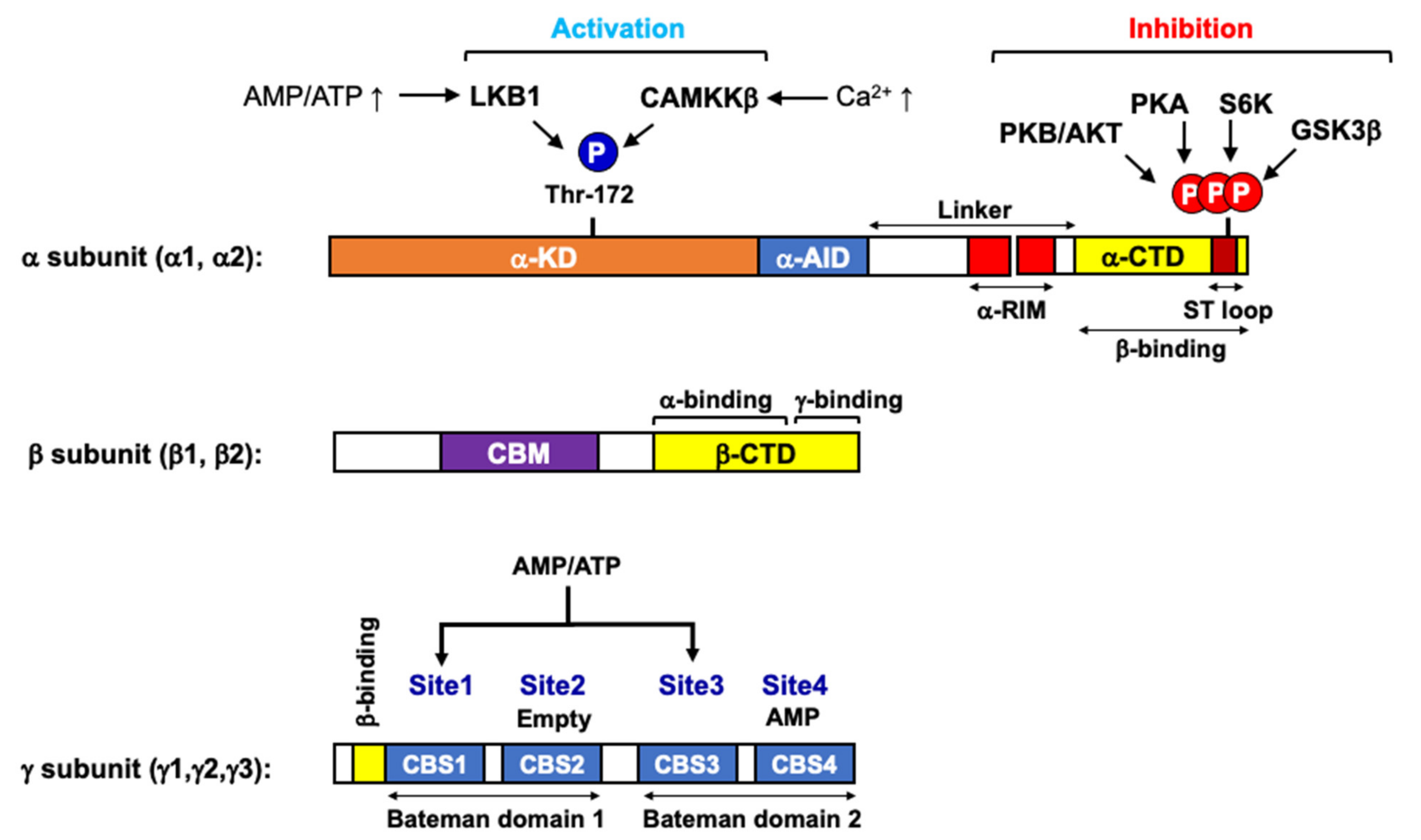
:1. AMPK as a Cellular Energy Gauge
2. mTOR Complex and Its Amino Acid Sensing Modules
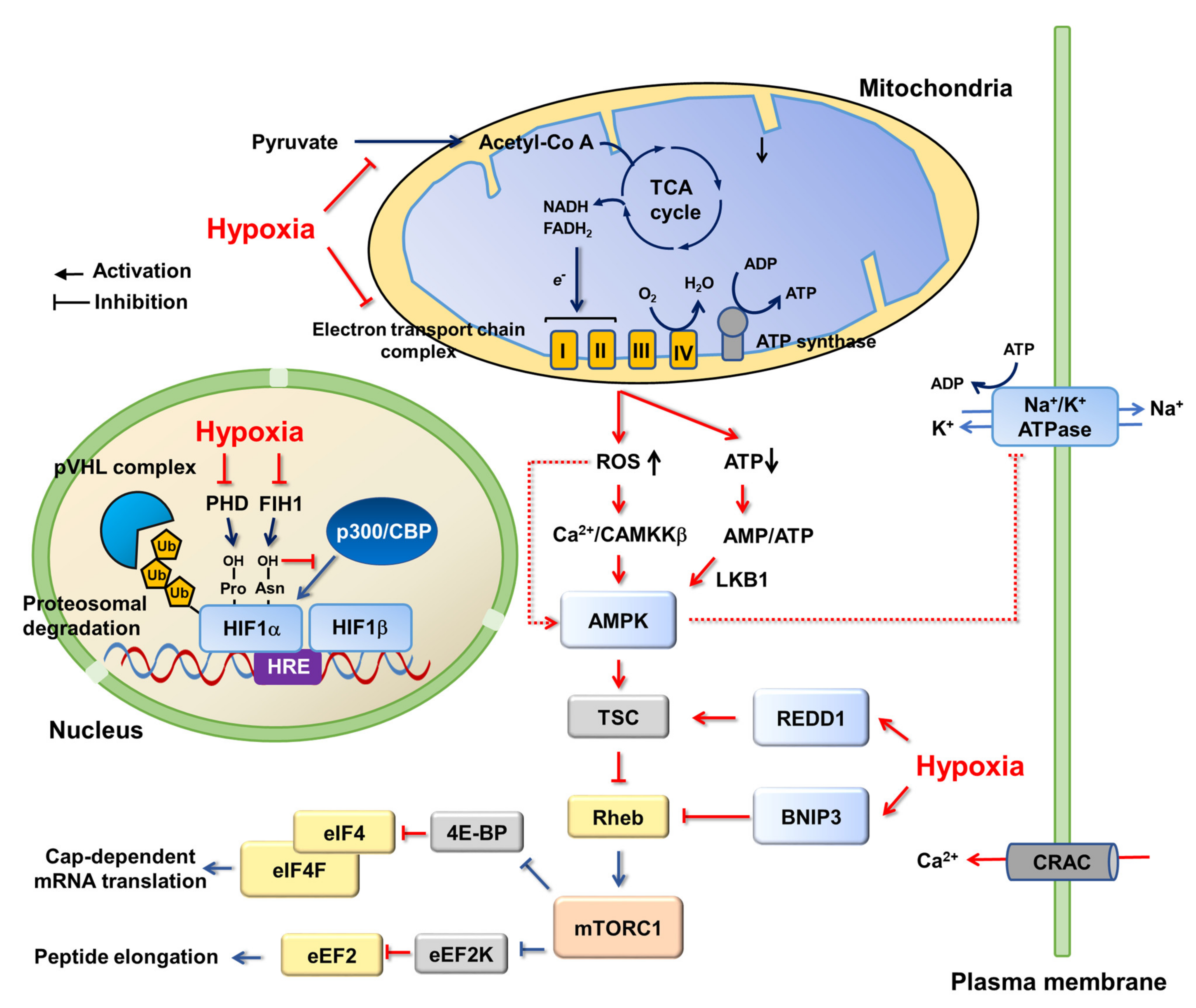
3. Cellular Adaptions to Hypoxia
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramirez, J.-M.; Folkow, L.P.; Blix, A.S. Hypoxia Tolerance in Mammals and Birds: From the Wilderness to the Clinic. Annu. Rev. Physiol. 2007, 69, 113–143. [Google Scholar] [CrossRef]
- Gu, C.; Jun, J.C. Does Hypoxia Decrease the Metabolic Rate? Front. Endocrinol. 2018, 9, 668. [Google Scholar] [CrossRef] [Green Version]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Yang, G.; Ha, J. Targeting of AMP-activated protein kinase: Prospects for computer-aided drug design. Expert Opin. Drug Discov. 2017, 12, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Hawley, S.A. AMP-activated protein kinase: The energy charge hypothesis revisited. BioEssays News Rev. Mol. Cell. Dev. Biol. 2001, 23, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, B.; Heath, R.; Saiu, P.; Leiper, F.C.; Leone, P.; Jing, C.; Walker, P.A.; Haire, L.; Eccleston, J.F.; Davis, C.T.; et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 2007, 449, 496–500. [Google Scholar] [CrossRef]
- Scott, J.W.; Hawley, S.A.; Green, K.A.; Anis, M.; Stewart, G.; Scullion, G.A.; Norman, D.G.; Hardie, D.G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. 2004, 113, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wang, J.; Zhang, Y.Y.; Yan, S.F.; Neumann, D.; Schlattner, U.; Wang, Z.X.; Wu, J.W. AMP-activated protein kinase undergoes nucleotide-dependent conformational changes. Nat. Struct. Mol. Biol. 2012, 19, 716–718. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xin, F.J.; Wang, J.; Hu, J.; Zhang, Y.Y.; Wan, S.; Cao, L.S.; Lu, C.; Li, P.; Yan, S.F.; et al. Conserved regulatory elements in AMPK. Nature 2013, 498, E8–E10. [Google Scholar] [CrossRef]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [Green Version]
- Oakhill, J.S.; Steel, R.; Chen, Z.P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011, 332, 1433–1435. [Google Scholar] [CrossRef]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Mäkelä, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Fagotto, F.; Zhang, T.; Hsu, W.; Vasicek, T.J.; Perry, W.L., 3rd; Lee, J.J.; Tilghman, S.M.; Gumbiner, B.M.; Costantini, F. The mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell 1997, 90, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.L.; Guo, H.; Zhang, C.S.; Lin, S.Y.; Yin, Z.; Peng, Y.; Luo, H.; Shi, Y.; Lian, G.; Zhang, C.; et al. AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab. 2013, 18, 546–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.L.; Wu, Y.Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Zhang, D.; Dyck, J.R.B.; Li, Y.; Zhang, H.; Morishima, M.; Mann, D.L.; Taffet, G.E.; Baldini, A.; Khoury, D.S.; et al. A pivotal role for endogenous TGF-β-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 17378. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMPK--sensing energy while talking to other signaling pathways. Cell Metab. 2014, 20, 939–952. [Google Scholar] [CrossRef] [Green Version]
- Hurley, R.L.; Barré, L.K.; Wood, S.D.; Anderson, K.A.; Kemp, B.E.; Means, A.R.; Witters, L.A. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J. Biol. Chem. 2006, 281, 36662–36672. [Google Scholar] [CrossRef] [Green Version]
- Horman, S.; Morel, N.; Vertommen, D.; Hussain, N.; Neumann, D.; Beauloye, C.; El Najjar, N.; Forcet, C.; Viollet, B.; Walsh, M.P.; et al. AMP-activated protein kinase phosphorylates and desensitizes smooth muscle myosin light chain kinase. J. Biol. Chem. 2008, 283, 18505–18512. [Google Scholar] [CrossRef] [Green Version]
- Dagon, Y.; Hur, E.; Zheng, B.; Wellenstein, K.; Cantley, L.C.; Kahn, B.B. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab. 2012, 16, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Bridges, D.; Nakada, D.; Skiniotis, G.; Morrison, S.J.; Lin, J.D.; Saltiel, A.R.; Inoki, K. Inhibition of AMPK catabolic action by GSK3. Mol. Cell 2013, 50, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coughlan, K.A.; Valentine, R.J.; Sudit, B.S.; Allen, K.; Dagon, Y.; Kahn, B.B.; Ruderman, N.B.; Saha, A.K. PKD1 Inhibits AMPKα2 through Phosphorylation of Serine 491 and Impairs Insulin Signaling in Skeletal Muscle Cells. J. Biol. Chem. 2016, 291, 5664–5675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heathcote, H.R.; Mancini, S.J.; Strembitska, A.; Jamal, K.; Reihill, J.A.; Palmer, T.M.; Gould, G.W.; Salt, I.P. Protein kinase C phosphorylates AMP-activated protein kinase α1 Ser487. Biochem. J. 2016, 473, 4681–4697. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMPK: A target for drugs and natural products with effects on both diabetes and cancer. Diabetes 2013, 62, 2164–2172. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.; Abbott, M.J.; Tang, T.; Hudak, C.S.; Kim, Y.; Bruss, M.; Hellerstein, M.K.; Lee, H.Y.; Samuel, V.T.; Shulman, G.I.; et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011, 13, 739–748. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Tang, T.; Abbott, M.; Viscarra, J.A.; Wang, Y.; Sul, H.S. AMPK Phosphorylates Desnutrin/ATGL and Hormone-Sensitive Lipase To Regulate Lipolysis and Fatty Acid Oxidation within Adipose Tissue. Mol. Cell. Biol. 2016, 36, 1961–1976. [Google Scholar] [CrossRef] [Green Version]
- Koo, S.H.; Flechner, L.; Qi, L.; Zhang, X.; Screaton, R.A.; Jeffries, S.; Hedrick, S.; Xu, W.; Boussouar, F.; Brindle, P.; et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 2005, 437, 1109–1111. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Vasquez, D.S.; Ravnskjaer, K.; Denechaud, P.D.; Yu, R.T.; Alvarez, J.G.; Downes, M.; Evans, R.M.; Montminy, M.; Shaw, R.J. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 2011, 145, 607–621. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.H.; Varanasi, U.S.; Yang, W.; Leff, T. AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J. Biol. Chem. 2003, 278, 27495–27501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, T.; Osatomi, K.; Yamashita, H.; Kabashima, T.; Uyeda, K. Mechanism for fatty acid "sparing" effect on glucose-induced transcription: Regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J. Biol. Chem. 2002, 277, 3829–3835. [Google Scholar] [CrossRef] [Green Version]
- Jäger, S.; Handschin, C.; St.-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010, 11, 213–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamura, K.; Ogura, T.; Kishimoto, A.; Kaminishi, M.; Esumi, H. Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole- 4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem. Biophys. Res. Commun. 2001, 287, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Igata, M.; Motoshima, H.; Tsuruzoe, K.; Kojima, K.; Matsumura, T.; Kondo, T.; Taguchi, T.; Nakamaru, K.; Yano, M.; Kukidome, D.; et al. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ. Res. 2005, 97, 837–844. [Google Scholar] [CrossRef] [Green Version]
- Banko, M.R.; Allen, J.J.; Schaffer, B.E.; Wilker, E.W.; Tsou, P.; White, J.L.; Villén, J.; Wang, B.; Kim, S.R.; Sakamoto, K.; et al. Chemical genetic screen for AMPKα2 substrates uncovers a network of proteins involved in mitosis. Mol. Cell 2011, 44, 878–892. [Google Scholar] [CrossRef] [Green Version]
- Karuman, P.; Gozani, O.; Odze, R.D.; Zhou, X.C.; Zhu, H.; Shaw, R.; Brien, T.P.; Bozzuto, C.D.; Ooi, D.; Cantley, L.C.; et al. The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol. Cell 2001, 7, 1307–1319. [Google Scholar] [CrossRef]
- Tiainen, M.; Vaahtomeri, K.; Ylikorkala, A.; Mäkelä, T.P. Growth arrest by the LKB1 tumor suppressor: Induction of p21(WAF1/CIP1). Hum. Mol. Genet. 2002, 11, 1497–1504. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, M.; Tian, Y.; Li, J.; Qi, Y.; Zhao, D.; Wu, Z.; Huang, M.; Wong, C.C.L.; Wang, H.W.; et al. Cryo-EM structure of human mTOR complex 2. Cell Res. 2018, 28, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuttfeld, E.; Aylett, C.H.; Imseng, S.; Boehringer, D.; Scaiola, A.; Sauer, E.; Hall, M.N.; Maier, T.; Ban, N. Architecture of the human mTORC2 core complex. Elife 2018, 7, e33101. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [Green Version]
- Toschi, A.; Lee, E.; Xu, L.; Garcia, A.; Gadir, N.; Foster, D.A. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: Competition with rapamycin. Mol. Cell. Biol. 2009, 29, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Foster, D.A.; Toschi, A. Targeting mTOR with rapamycin: One dose does not fit all. Cell Cycle 2009, 8, 1026–1029. [Google Scholar] [CrossRef]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001, 294, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Park, I.H.; Wu, A.L.; Du, G.; Huang, P.; Frohman, M.A.; Walker, S.J.; Brown, H.A.; Chen, J. PLD1 regulates mTOR signaling and mediates Cdc42 activation of S6K1. Curr. Biol. 2003, 13, 2037–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Fang, Y.; Yoon, M.S.; Zhang, C.; Roccio, M.; Zwartkruis, F.J.; Armstrong, M.; Brown, H.A.; Chen, J. Phospholipase D1 is an effector of Rheb in the mTOR pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 8286–8291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zheng, Y.; Foster, D.A. Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene 2003, 22, 3937–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björklund, M.; Taipale, M.; Varjosalo, M.; Saharinen, J.; Lahdenperä, J.; Taipale, J. Identification of pathways regulating cell size and cell-cycle progression by RNAi. Nature 2006, 439, 1009–1013. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. CB 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Saqcena, M.; Chatterjee, A.; Garcia, A.; Frias, M.A.; Foster, D.A. Reciprocal regulation of AMP-activated protein kinase and phospholipase D. J. Biol. Chem. 2015, 290, 6986–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, D.A.; Xu, L. Phospholipase D in cell proliferation and cancer. Mol. Cancer Res. MCR 2003, 1, 789–800. [Google Scholar]
- Mukhopadhyay, S.; Chatterjee, A.; Kogan, D.; Patel, D.; Foster, D.A. 5-Aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) enhances the efficacy of rapamycin in human cancer cells. Cell Cycle 2015, 14, 3331–3339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rae, C.; Mairs, R.J. AMPK activation by AICAR sensitizes prostate cancer cells to radiotherapy. Oncotarget 2019, 10, 749–759. [Google Scholar] [CrossRef]
- Guo, D.; Hildebrandt, I.J.; Prins, R.M.; Soto, H.; Mazzotta, M.M.; Dang, J.; Czernin, J.; Shyy, J.Y.J.; Watson, A.D.; Phelps, M.; et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 12932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, R.-Y.; Chao, Y.; Chen, T.-Y.; Huang, D.-Y.; Lin, W.-W. 5-Aminoimidazole-4-carboxamide riboside sensitizes TRAIL- and TNFα-induced cytotoxicity in colon cancer cells through AMP-activated protein kinase signaling. Mol. Cancer Ther. 2007, 6, 1562. [Google Scholar] [CrossRef] [Green Version]
- Saxena, M.; Balaji, S.A.; Deshpande, N.; Ranganathan, S.; Pillai, D.M.; Hindupur, S.K.; Rangarajan, A. AMP-activated protein kinase promotes epithelial-mesenchymal transition in cancer cells through Twist1 upregulation. J. Cell Sci. 2018, 131, 14. [Google Scholar] [CrossRef] [Green Version]
- Zadra, G.; Photopoulos, C.; Tyekucheva, S.; Heidari, P.; Weng, Q.P.; Fedele, G.; Liu, H.; Scaglia, N.; Priolo, C.; Sicinska, E.; et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 2014, 6, 519–538. [Google Scholar] [CrossRef]
- Plews, R.L.; Mohd Yusof, A.; Wang, C.; Saji, M.; Zhang, X.; Chen, C.S.; Ringel, M.D.; Phay, J.E. A novel dual AMPK activator/mTOR inhibitor inhibits thyroid cancer cell growth. J. Clin. Endocrinol. Metab. 2015, 100, E748–E756. [Google Scholar] [CrossRef]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The beneficial effects of metformin on cancer prevention and therapy: A comprehensive review of recent advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morran, D.C.; Wu, J.; Jamieson, N.B.; Mrowinska, A.; Kalna, G.; Karim, S.A.; Au, A.Y.M.; Scarlett, C.J.; Chang, D.K.; Pajak, M.Z.; et al. Targeting mTOR dependency in pancreatic cancer. Gut 2014, 63, 1481. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-L.; Nokin, M.-J.; Egorov, M.; Tomé, M.; Bodineau, C.; Di Primo, C.; Minder, L.; Wdzieczak-Bakala, J.; Garcia-Alvarez, M.C.; Bignon, J.; et al. mTOR Inhibition via Displacement of Phosphatidic Acid Induces Enhanced Cytotoxicity Specifically in Cancer Cells. Cancer Res. 2018, 78, 5384–5397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendell, J.C.; Varghese, A.M.; Hyman, D.M.; Bauer, T.M.; Pant, S.; Callies, S.; Lin, J.; Martinez, R.; Wickremsinhe, E.; Fink, A.; et al. A First-in-Human Phase 1 Study of LY3023414, an Oral PI3K/mTOR Dual Inhibitor, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 3253–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caumanns, J.J.; Berns, K.; Wisman, G.B.A.; Fehrmann, R.S.N.; Tomar, T.; Klip, H.; Meersma, G.J.; Hijmans, E.M.; Gennissen, A.M.C.; Duiker, E.W.; et al. Integrative Kinome Profiling Identifies mTORC1/2 Inhibition as Treatment Strategy in Ovarian Clear Cell Carcinoma. Clin. Cancer Res. 2018, 24, 3928–3940. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [Green Version]
- Efeyan, A.; Zoncu, R.; Chang, S.; Gumper, I.; Snitkin, H.; Wolfson, R.L.; Kirak, O.; Sabatini, D.D.; Sabatini, D.M. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 2013, 493, 679–683. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef] [Green Version]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Yin, N.; Li, M.O. SZT2 dictates GATOR control of mTORC1 signalling. Nature 2017, 543, 433–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfson, R.L.; Chantranupong, L.; Wyant, G.A.; Gu, X.; Orozco, J.M.; Shen, K.; Condon, K.J.; Petri, S.; Kedir, J.; Scaria, S.M.; et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 2017, 543, 438–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Knockenhauer, K.E.; Wolfson, R.L.; Chantranupong, L.; Pacold, M.E.; Wang, T.; Schwartz, T.U.; Sabatini, D.M. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 2016, 351, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Chantranupong, L.; Knockenhauer, K.E.; Schwartz, T.U.; Sabatini, D.M. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature 2016, 536, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Rebsamen, M.; Pochini, L.; Stasyk, T.; de Araújo, M.E.; Galluccio, M.; Kandasamy, R.K.; Snijder, B.; Fauster, A.; Rudashevskaya, E.L.; Bruckner, M.; et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015, 519, 477–481. [Google Scholar] [CrossRef]
- Wang, S.; Tsun, Z.Y.; Wolfson, R.L.; Shen, K.; Wyant, G.A.; Plovanich, M.E.; Yuan, E.D.; Jones, T.D.; Chantranupong, L.; Comb, W.; et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015, 347, 188–194. [Google Scholar] [CrossRef] [Green Version]
- Wyant, G.A.; Abu-Remaileh, M.; Wolfson, R.L.; Chen, W.W.; Freinkman, E.; Danai, L.V.; Vander Heiden, M.G.; Sabatini, D.M. mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 2017, 171, 642–654.e12. [Google Scholar] [CrossRef]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Petit, C.S.; Roczniak-Ferguson, A.; Ferguson, S.M. Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J. Cell Biol. 2013, 202, 1107–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsun, Z.Y.; Bar-Peled, L.; Chantranupong, L.; Zoncu, R.; Wang, T.; Kim, C.; Spooner, E.; Sabatini, D.M. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell 2013, 52, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Stracka, D.; Jozefczuk, S.; Rudroff, F.; Sauer, U.; Hall, M.N. Nitrogen source activates TOR (target of rapamycin) complex 1 via glutamine and independently of Gtr/Rag proteins. J. Biol. Chem. 2014, 289, 25010–25020. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.-X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.-L. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194. [Google Scholar] [CrossRef] [Green Version]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Durán, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Han, J.M.; Jeong, S.J.; Park, M.C.; Kim, G.; Kwon, N.H.; Kim, H.K.; Ha, S.H.; Ryu, S.H.; Kim, S. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 2012, 149, 410–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.D.; Gong, W.; Zhang, J.N.; Nie, J.; Yao, C.F.; Guo, F.S.; Lin, Y.; Wu, X.H.; Li, F.; Li, J.; et al. Sensing and Transmitting Intracellular Amino Acid Signals through Reversible Lysine Aminoacylations. Cell Metab. 2018, 27, 151–166.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polak, P.; Cybulski, N.; Feige, J.N.; Auwerx, J.; Rüegg, M.A.; Hall, M.N. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 2008, 8, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Carnevalli, L.S.; Masuda, K.; Frigerio, F.; Le Bacquer, O.; Um, S.H.; Gandin, V.; Topisirovic, I.; Sonenberg, N.; Thomas, G.; Kozma, S.C. S6K1 plays a critical role in early adipocyte differentiation. Dev. Cell 2010, 18, 763–774. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Chen, J. Regulation of Peroxisome Proliferator–Activated Receptor-γ Activity by Mammalian Target of Rapamycin and Amino Acids in Adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [Green Version]
- Robitaille, A.M.; Christen, S.; Shimobayashi, M.; Cornu, M.; Fava, L.L.; Moes, S.; Prescianotto-Baschong, C.; Sauer, U.; Jenoe, P.; Hall, M.N. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013, 339, 1320–1323. [Google Scholar] [CrossRef]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Gomes, A.P.; Wang, X.; Yoon, S.O.; Lee, G.; Nagiec, M.J.; Cho, S.; Chavez, A.; Islam, T.; Yu, Y.; et al. mTORC1 Promotes Metabolic Reprogramming by the Suppression of GSK3-Dependent Foxk1 Phosphorylation. Mol. Cell 2018, 70, 949–960.e4. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Peterson, T.R.; Laplante, M.; Oh, S.; Sabatini, D.M. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 2010, 468, 1100–1104. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Zid, B.M.; Rogers, A.N.; Katewa, S.D.; Vargas, M.A.; Kolipinski, M.C.; Lu, T.A.; Benzer, S.; Kapahi, P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell 2009, 139, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dengler, F. Activation of AMPK under Hypoxia: Many Roads Leading to Rome. Int. J. Mol. Sci. 2020, 21, 2428. [Google Scholar] [CrossRef] [Green Version]
- Moral-Sanz, J.; Lewis, S.A.; MacMillan, S.; Ross, F.A.; Thomson, A.; Viollet, B.; Foretz, M.; Moran, C.; Hardie, D.G.; Evans, A.M. The LKB1-AMPK-α1 signaling pathway triggers hypoxic pulmonary vasoconstriction downstream of mitochondria. Sci. Signal. 2018, 11, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.D.; Smolenski, R.T.; Harhun, M.I.; Patel, H.K.; Ahmed, S.G.; Wanisch, K.; Yáñez-Muñoz, R.J.; Baines, D.L. AMP-activated protein kinase (AMPK)-dependent and -independent pathways regulate hypoxic inhibition of transepithelial Na+ transport across human airway epithelial cells. Br. J. Pharm. 2012, 167, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Sallé-Lefort, S.; Miard, S.; Nolin, M.-A.; Boivin, L.; Paré, M.-È.; Debigaré, R.; Picard, F. Hypoxia upregulates Malat1 expression through a CaMKK/AMPK/HIF-1α axis. Int. J. Oncol. 2016, 49, 1731–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsin, A.S.; Bertrand, L.; Rider, M.H.; Deprez, J.; Beauloye, C.; Vincent, M.F.; Van den Berghe, G.; Carling, D.; Hue, L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr. Biol. 2000, 10, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.A.; Waypa, G.B.; Schumacker, P.T. Redox signaling during hypoxia in mammalian cells. Redox Biol. 2017, 13, 228–234. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [Green Version]
- Emerling, B.M.; Weinberg, F.; Snyder, C.; Burgess, Z.; Mutlu, G.M.; Viollet, B.; Budinger, G.R.; Chandel, N.S. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic. Biol. Med. 2009, 46, 1386–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusarova, G.A.; Trejo, H.E.; Dada, L.A.; Briva, A.; Welch, L.C.; Hamanaka, R.B.; Mutlu, G.M.; Chandel, N.S.; Prakriya, M.; Sznajder, J.I. Hypoxia leads to Na,K-ATPase downregulation via Ca(2+) release-activated Ca(2+) channels and AMPK activation. Mol. Cell. Biol. 2011, 31, 3546–3556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mungai, P.T.; Waypa, G.B.; Jairaman, A.; Prakriya, M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol. Cell. Biol. 2011, 31, 3531–3545. [Google Scholar] [CrossRef] [Green Version]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Oka, S.; Liu, T.; Zhai, P.; Ago, T.; Sciarretta, S.; Li, H.; Sadoshima, J. A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 2014, 19, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine Hydroxylation of the HIF Transactivation Domain: A Hypoxic Switch. Science 2002, 295, 858. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Brière, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Wang, B.; Liu, H.; Shah, H.; Carmona-Fontaine, C.; Rustenburg, A.S.; Salah, S.; Gunner, M.R.; Chodera, J.D.; Cross, J.R.; et al. L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat. Chem. Biol. 2017, 13, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhang, D.X.; Shi, X.; Zhang, Q.; Huang, Y.S. Activation of the prolyl-hydroxylase oxygen-sensing signal cascade leads to AMPK activation in cardiomyocytes. J. Cell Mol. Med. 2012, 16, 2049–2059. [Google Scholar] [CrossRef] [Green Version]
- Dengler, F.; Gäbel, G. The Fast Lane of Hypoxic Adaptation: Glucose Transport Is Modulated via A HIF-Hydroxylase-AMPK-Axis in Jejunum Epithelium. Int. J. Mol. Sci. 2019, 20, 4993. [Google Scholar] [CrossRef] [Green Version]
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F.; Godson, C.; Nielsen, J.E.; Moynagh, P.; Pouyssegur, J.; et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl. Acad. Sci. USA 2006, 103, 18154–18159. [Google Scholar] [CrossRef] [Green Version]
- Moser, S.C.; Bensaddek, D.; Ortmann, B.; Maure, J.F.; Mudie, S.; Blow, J.J.; Lamond, A.I.; Swedlow, J.R.; Rocha, S. PHD1 links cell-cycle progression to oxygen sensing through hydroxylation of the centrosomal protein Cep192. Dev. Cell 2013, 26, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Köditz, J.; Nesper, J.; Wottawa, M.; Stiehl, D.P.; Camenisch, G.; Franke, C.; Myllyharju, J.; Wenger, R.H.; Katschinski, D.M. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood 2007, 110, 3610–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cockman, M.E.; Lippl, K.; Tian, Y.M.; Pegg, H.B.; Figg, W.D.J.; Abboud, M.I.; Heilig, R.; Fischer, R.; Myllyharju, J.; Schofield, C.J.; et al. Lack of activity of recombinant HIF prolyl hydroxylases (PHDs) on reported non-HIF substrates. Elife 2019, 8, e46490. [Google Scholar] [CrossRef]
- Cockman, M.E.; Webb, J.D.; Kramer, H.B.; Kessler, B.M.; Ratcliffe, P.J. Proteomics-based identification of novel factor inhibiting hypoxia-inducible factor (FIH) substrates indicates widespread asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Mol. Cell Proteom. 2009, 8, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Linke, S.; Dias, J.M.; Zheng, X.; Gradin, K.; Wallis, T.P.; Hamilton, B.R.; Gustafsson, M.; Ruas, J.L.; Wilkins, S.; et al. Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cockman, M.E.; Lancaster, D.E.; Stolze, I.P.; Hewitson, K.S.; McDonough, M.A.; Coleman, M.L.; Coles, C.H.; Yu, X.; Hay, R.T.; Ley, S.C.; et al. Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc. Natl. Acad. Sci. USA 2006, 103, 14767–14772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Guan, M.; Townsend, K.L.; Huang, T.L.; An, D.; Yan, X.; Xue, R.; Schulz, T.J.; Winnay, J.; Mori, M.; et al. MicroRNA-455 regulates brown adipogenesis via a novel HIF1an-AMPK-PGC1α signaling network. EMBO Rep. 2015, 16, 1378–1393. [Google Scholar] [CrossRef] [Green Version]
- Scholz, C.C.; Rodriguez, J.; Pickel, C.; Burr, S.; Fabrizio, J.A.; Nolan, K.A.; Spielmann, P.; Cavadas, M.A.; Crifo, B.; Halligan, D.N.; et al. FIH Regulates Cellular Metabolism through Hydroxylation of the Deubiquitinase OTUB1. PLoS Biol. 2016, 14, e1002347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel Malik, R.; Zippel, N.; Frömel, T.; Heidler, J.; Zukunft, S.; Walzog, B.; Ansari, N.; Pampaloni, F.; Wingert, S.; Rieger, M.A.; et al. AMP-Activated Protein Kinase α2 in Neutrophils Regulates Vascular Repair via Hypoxia-Inducible Factor-1α and a Network of Proteins Affecting Metabolism and Apoptosis. Circ. Res. 2017, 120, 99–109. [Google Scholar] [CrossRef]
- Jung, S.N.; Yang, W.K.; Kim, J.; Kim, H.S.; Kim, E.J.; Yun, H.; Park, H.; Kim, S.S.; Choe, W.; Kang, I.; et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 2008, 29, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013, 17, 113–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zadra, G.; Batista, J.L.; Loda, M. Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Mol. Cancer Res. MCR 2015, 13, 1059–1072. [Google Scholar] [CrossRef] [Green Version]
- Seo, K.; Seo, S.; Ki, S.H.; Shin, S.M. Sestrin2 inhibits hypoxia-inducible factor-1α accumulation via AMPK-mediated prolyl hydroxylase regulation. Free Radic. Biol. Med. 2016, 101, 511–523. [Google Scholar] [CrossRef]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofer, A.; Lei, K.; Johannessen, C.M.; Ellisen, L.W. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell. Biol. 2005, 25, 5834–5845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seong, M.; Lee, J.; Kang, H. Hypoxia-induced regulation of mTOR signaling by miR-7 targeting REDD1. J. Cell. Biochem. 2019, 120, 4523–4532. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Kim, E.; Beemiller, P.; Wang, C.Y.; Swanson, J.; You, M.; Guan, K.L. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J. Biol. Chem. 2007, 282, 35803–35813. [Google Scholar] [CrossRef] [Green Version]
- Cam, H.; Easton, J.B.; High, A.; Houghton, P.J. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1α. Mol. Cell 2010, 40, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Feldman, D.E.; Chauhan, V.; Koong, A.C. The unfolded protein response: A novel component of the hypoxic stress response in tumors. Mol. Cancer Res. MCR 2005, 3, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol. Cell. Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef] [Green Version]
- Blais, J.D.; Addison, C.L.; Edge, R.; Falls, T.; Zhao, H.; Wary, K.; Koumenis, C.; Harding, H.P.; Ron, D.; Holcik, M.; et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell. Biol. 2006, 26, 9517–9532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, E.; Braunstein, S.; Formenti, S.; Schneider, R.J. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol. Cell. Biol. 2006, 26, 3955–3965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenney, J.W.; Moore, C.E.; Wang, X.; Proud, C.G. Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv. Biol. Regul. 2014, 55, 15–27. [Google Scholar] [CrossRef]
- Kozak, M. A second look at cellular mRNA sequences said to function as internal ribosome entry sites. Nucleic Acids Res. 2005, 33, 6593–6602. [Google Scholar] [CrossRef] [PubMed]
- King, H.A.; Cobbold, L.C.; Willis, A.E. The role of IRES trans-acting factors in regulating translation initiation. Biochem. Soc. Trans. 2010, 38, 1581–1586. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Cooper, C.E. The steady-state kinetics of cytochrome c oxidation by cytochrome oxidase. Biochim. Et Biophys. Acta 1990, 1017, 187–203. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordóñez, Á.; Corral-Escariz, M.; Soro, I.; López-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameri, K.; Jahangiri, A.; Rajah, A.M.; Tormos, K.V.; Nagarajan, R.; Pekmezci, M.; Nguyen, V.; Wheeler, M.L.; Murphy, M.P.; Sanders, T.A.; et al. HIGD1A Regulates Oxygen Consumption, ROS Production, and AMPK Activity during Glucose Deprivation to Modulate Cell Survival and Tumor Growth. Cell Rep. 2015, 10, 891–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheaton, W.W.; Chandel, N.S. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am. J. Physiol. Cell Physiol. 2011, 300, C385–C393. [Google Scholar] [CrossRef] [Green Version]
- Chance, B.; Williams, G.R. The respiratory chain and oxidative phosphorylation. Adv. Enzymol. Relat. Subj. Biochem. 1956, 17, 65–134. [Google Scholar]
- Milligan, L.P.; McBride, B.W. Energy costs of ion pumping by animal tissues. J. Nutr. 1985, 115, 1374–1382. [Google Scholar] [CrossRef]
- Gusarova, G.A.; Dada, L.A.; Kelly, A.M.; Brodie, C.; Witters, L.A.; Chandel, N.S.; Sznajder, J.I. Alpha1-AMP-activated protein kinase regulates hypoxia-induced Na,K-ATPase endocytosis via direct phosphorylation of protein kinase C zeta. Mol. Cell. Biol. 2009, 29, 3455–3464. [Google Scholar] [CrossRef] [Green Version]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M. The molecular physiology of CRAC channels. Immunol. Rev. 2009, 231, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 2018, 20, 775–781. [Google Scholar] [CrossRef]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, M.S.; Keith, B.; Simon, M.C. Oxygen availability and metabolic adaptations. Nat. Rev. Cancer 2016, 16, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, C.; Hui, S.; Zeng, X.; Cowan, A.J.; Wang, L.; Chen, L.; Morscher, R.J.; Reyes, J.; Frezza, C.; Hwang, H.Y.; et al. Metabolite Exchange between Mammalian Organs Quantified in Pigs. Cell Metab. 2019, 30, 594–606.e3. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Semenza, G.L. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim. Et Biophys. Acta. Rev. Cancer 2018, 1870, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morotti, M.; Bridges, E.; Valli, A.; Choudhry, H.; Sheldon, H.; Wigfield, S.; Gray, N.; Zois, C.E.; Grimm, F.; Jones, D.; et al. Hypoxia-induced switch in SNAT2/SLC38A2 regulation generates endocrine resistance in breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 12452–12461. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Semenza, G.L. Serine Synthesis Helps Hypoxic Cancer Stem Cells Regulate Redox. Cancer Res. 2016, 76, 6458. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Klionsky, D.J. Autophagy and disease: Unanswered questions. Cell Death Differ. 2020, 27, 858–871. [Google Scholar] [CrossRef]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.L. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.M.; Jung, C.H.; Seo, M.; Kim, E.K.; Park, J.M.; Bae, S.S.; Kim, D.H. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol. Cell 2015, 57, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Zheng, Y.; Zhang, X.; Chen, Y.; Wu, X.; Wu, J.; Shen, Z.; Jiang, L.; Wang, L.; Yang, W.; et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 2017, 13, 1754–1766. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Lee, H.Y.; Hanna, R.A.; Gustafsson, Å.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AMPK Inhibitors | Status | Study Model | Reference | |
| AICAR | ZMP, AMP mimetic | Preclinical study | Cancer cell lines (prostate, glioblastoma, colon) | [65,66,67] |
| A769662 | a direct activator | Preclinical study | Cancer cell lines (breast, melanoma, lung) | [68] |
| MT63-78 | a direct activator | Preclinical study | Cancer cell lines (prostate), animal model | [69] |
| OSU-53 | allosteric activator | Preclinical study | Cancer cell lines (thyroid), animal model | [70] |
| Metformin | inhibits mitochondrial electron transport chain complex I | Clinical trials (phase II) | Breast, prostate, pancreatic cancers | [71] |
| mTOR Inhibitors | Status | Study Model | Reference | |
| Rapamycin | Preclinical | Animal model (Pancreatic cancer) | [72] | |
| Everolimus Temsirolimus | a derivative of rapamycin | US FDA approved | Renal cell carcinoma (RCC) | |
| ICSN3250 | targets PA binding to FRB domain on mTOR | Preclinical study | Cancer cell lines (colon) | [73] |
| LY3023414 | a competitive ATP-binding inhibitor | Clinical trials (phase I) | Solid tumor or lymphoma | [74] |
| AZD8055 | a competitive ATP-binding inhibitor | Preclinical study | Animal model (ovarian clear cell carcinoma) | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chun, Y.; Kim, J. AMPK–mTOR Signaling and Cellular Adaptations in Hypoxia. Int. J. Mol. Sci. 2021, 22, 9765. https://doi.org/10.3390/ijms22189765
Chun Y, Kim J. AMPK–mTOR Signaling and Cellular Adaptations in Hypoxia. International Journal of Molecular Sciences. 2021; 22(18):9765. https://doi.org/10.3390/ijms22189765
Chicago/Turabian StyleChun, Yoomi, and Joungmok Kim. 2021. "AMPK–mTOR Signaling and Cellular Adaptations in Hypoxia" International Journal of Molecular Sciences 22, no. 18: 9765. https://doi.org/10.3390/ijms22189765
APA StyleChun, Y., & Kim, J. (2021). AMPK–mTOR Signaling and Cellular Adaptations in Hypoxia. International Journal of Molecular Sciences, 22(18), 9765. https://doi.org/10.3390/ijms22189765