
Drugs Interfering with Insulin Resistance and Their Influence on the Associated Hypermetabolic State in Severe Burns: A Narrative Review




 ,
, 
Abstract
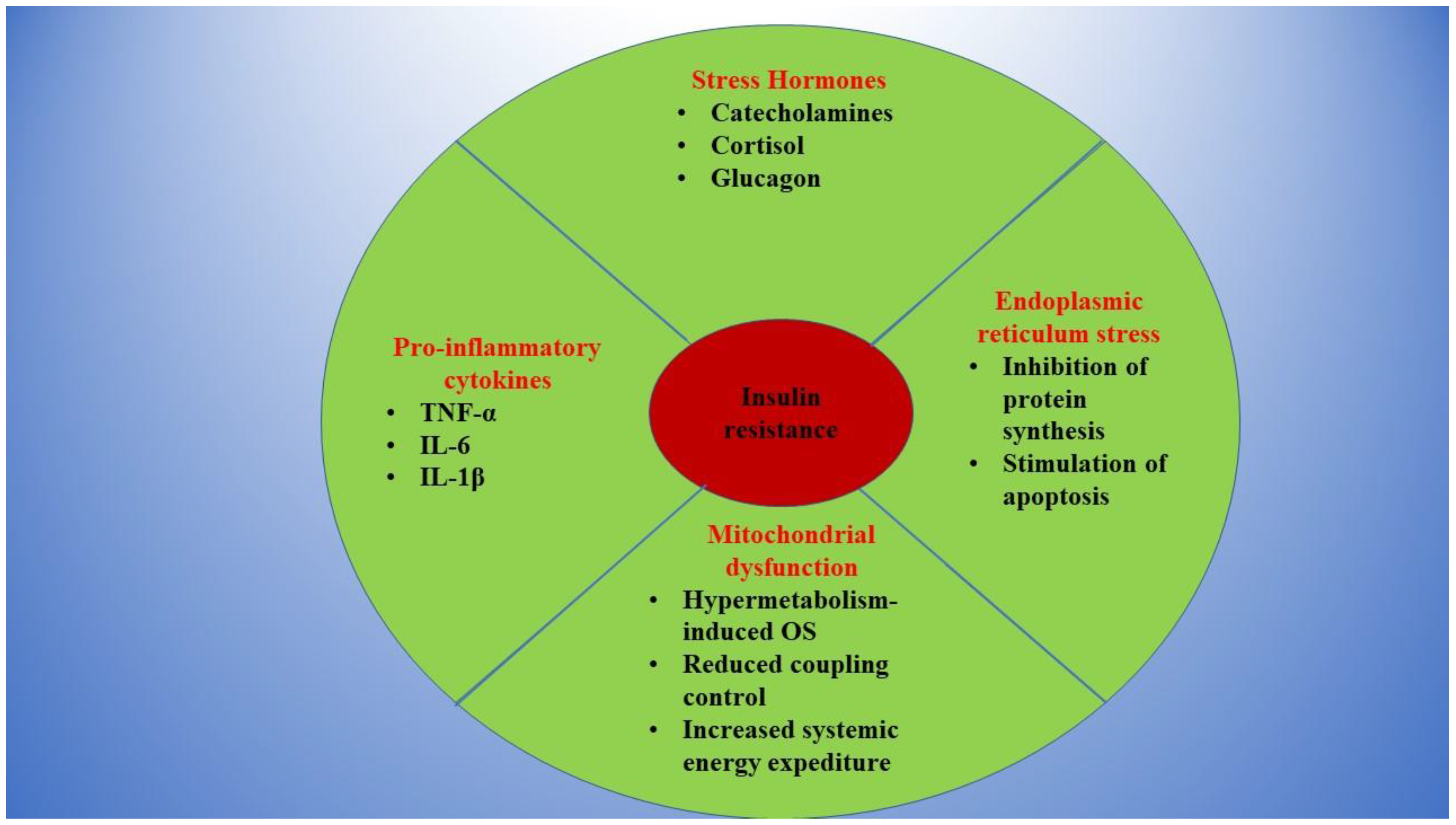
:1. Burns and Insulin Resistance: Background of a Toxic Relationship
2. Drugs That Might Interfere with Insulin Resistance in Burns
- -
- β blockers, angiotensin-converting enzyme inhibitors, and angiotensin II receptor blockers for the treatment of cardio-vascular conditions;
- -
- Antipsychotic drugs for the treatment of psychiatric conditions;
- -
- Vitamin B3 and statins for the treatment of dyslipidemias;
- -
- Thiazolidinediones and metformin for the treatment of type II diabetes mellitus.
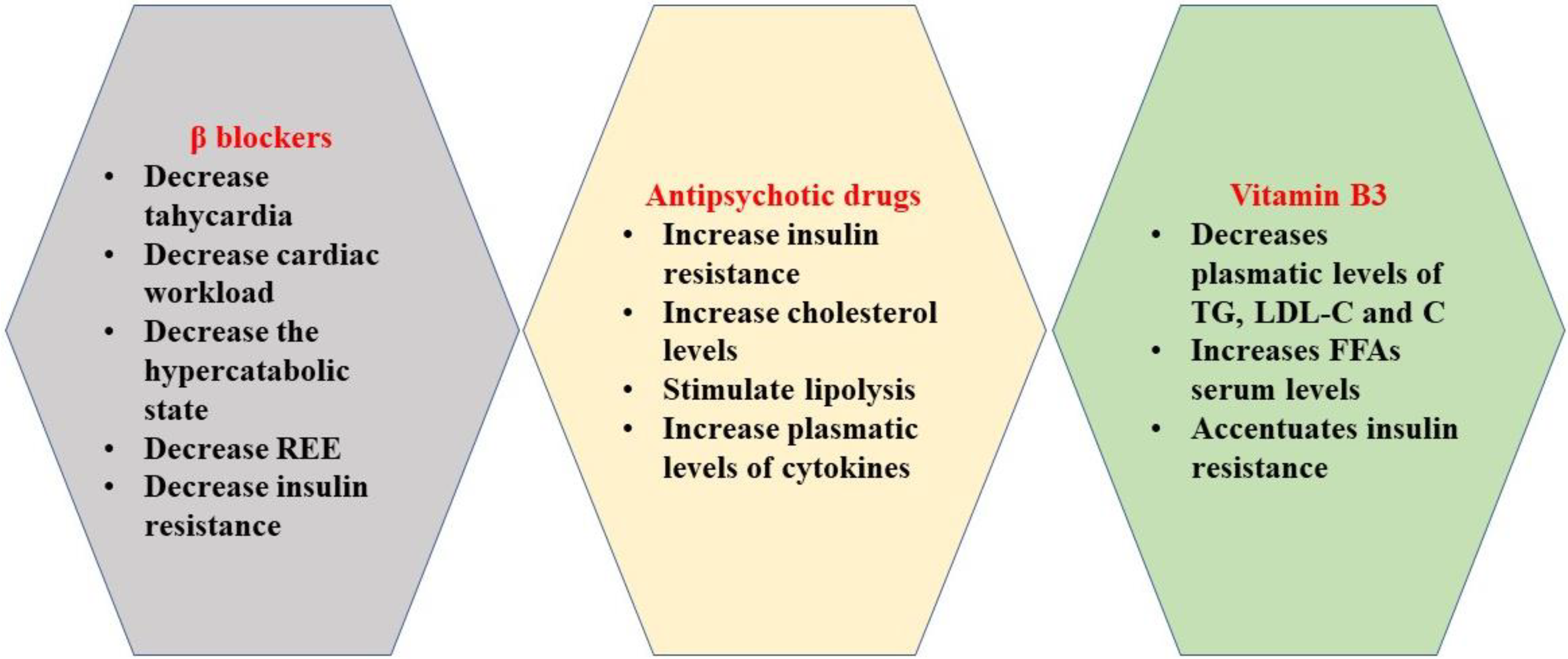
2.1. β Blockers
2.2. Antipsychotic Drugs
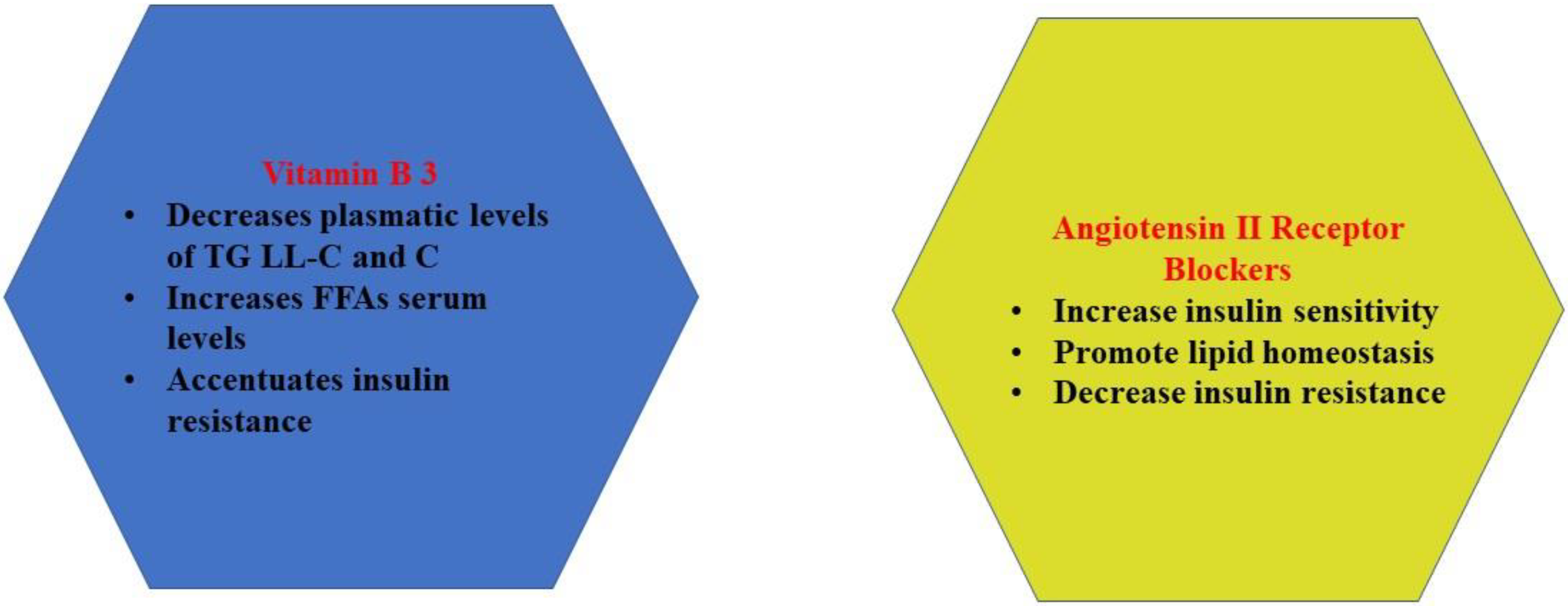
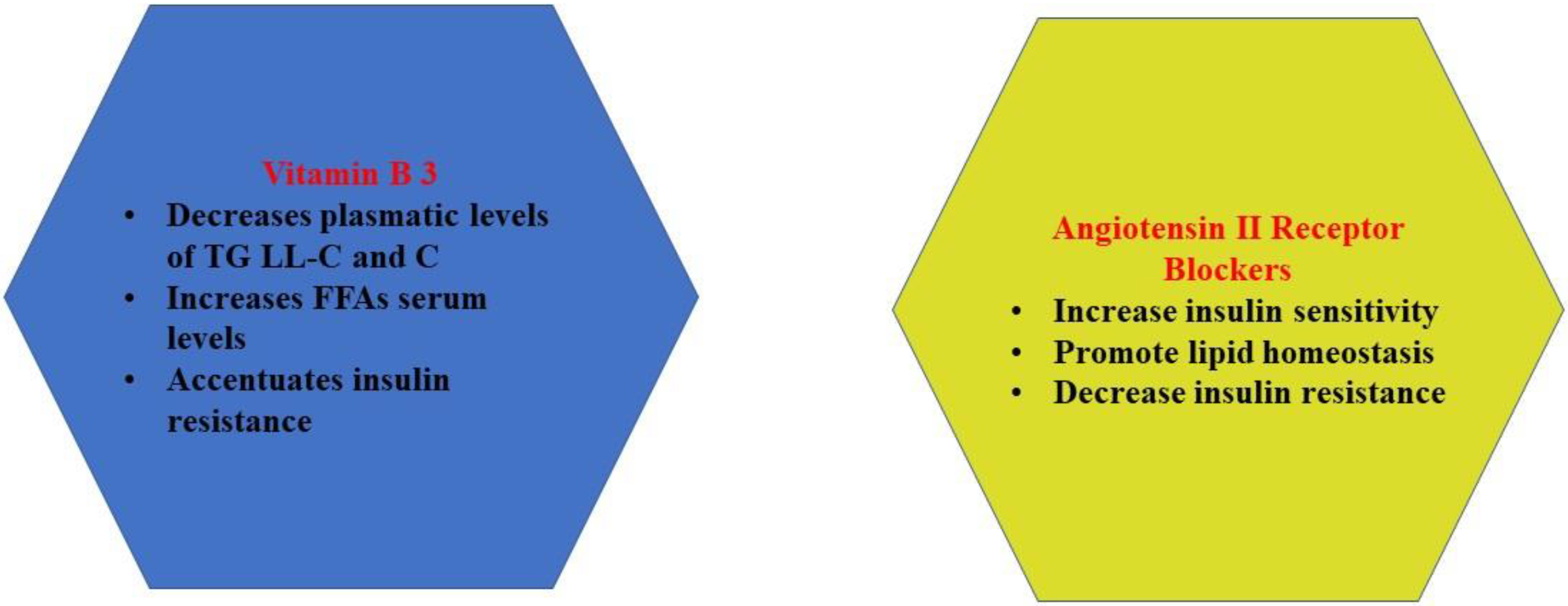
2.3. Vitamin B3 (Niacin)
2.4. Angiotensin-Converting Enzyme Inhibitors (ACEI)
2.5. Angiotensin II Receptor Blockers
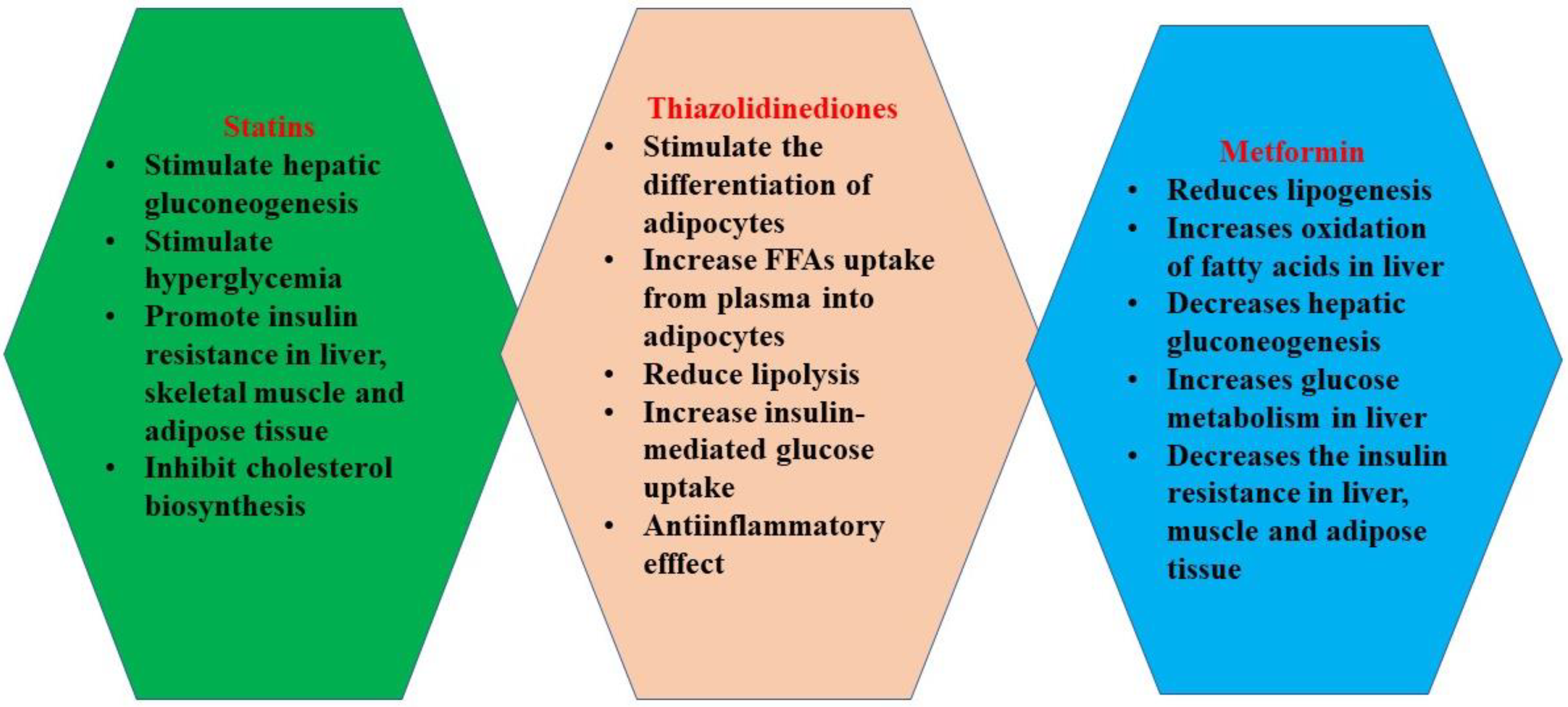
2.6. Statins and Insulin Resistance in Burns
- -
- Increase in hepatocyte gluconeogenesis through upregulation of the expression of genes codifying the synthesis of nodal enzymes [95];
- -
- -
- Decrease in insulin production due to β-insular cell damage [98];
- -
- Accumulation of FFAs in hepatocytes [93];
- -
- Decrease in the production of adiponectin and leptin by adipose tissue [99];
- -
- Alteration of the expression profile of microRNAs involved in the regulation of glucose metabolism and lipid metabolism [100];
- -
- Induction of insulin resistance [101].
2.6.1. Statins and Hepatocyte Insulin Sensitivity
2.6.2. Statins and Skeletal Muscle Insulin Sensitivity
- It appears that simvastatin impairs the phosphorylation of mTOR (mammalian target of rapamycin), which prevents the phosphorylation of AKT/PKB (protein kinase B) at Ser473 [119]; this results in a lack of activation of GSK3β, thus determining an impairment of GLUT4 translocation to the skeletal muscles’ cell membranes [117,119], finally decreasing glucose uptake by the cells.
- Simvastatin inhibits the translocation of GLUT4 to the plasma membrane through inhibition of prenylation of RabGTPases [120].
- c.
- Statins inhibit the biosynthesis of cholesterol [91], acting as hypolipemiant drugs [91]. By decreasing intracellular cholesterol, Atorvastatin decreases the translocation of GLUT4 [124], a partially cholesterol-dependent process [125]. The result is a decrease in glucose uptake in the skeletal muscle.
- d.
- It has been demonstrated that the accumulation of FFAs in the skeletal muscle inhibits glucose uptake through inhibition of glucose transport and glucose phosphorylation [126]. FFAs in excess compete with glucose oxidation by decreasing glucose-6-phosphate formation [126], which is the form that permits glucose to be metabolized in the cell.
2.6.3. Statins and Adipocyte Insulin Sensitivity
- Statins inhibit the insulin-induced translocation of GLUT4 [127]. By inhibiting isoprenoid synthesis [96], statins impede on the active membrane fraction of Rab4 GTPase and RhoA (small GTPases involved in transmembrane transport) [127], because their active forms are dependent on isoprenilation [128]. Therefore, the insulin-stimulated translocation of GLUT4 is decreased [127], which results in reduced glucose uptake in the adipocytes.
- Statins impede caveolae formation [129].
- c.
- Statins indirectly reduce the secretion of high-molecular-weight oligomers of adiponectin, resulting in insulin resistance [133].
2.7. Thiazolidinediones
- -
- Liver toxicity: This was noticed in troglitazone, which has been withdrawn from the market [143];
- -
- -
- Increase in body adipose tissue: Thiazolidinediones increase the uptake of FFAs from blood into the adipocytes, diminish the lipid stores in extra-adipose tissues (liver, skeletal muscle), and augment lipid storage in adipose tissue (especially subcutaneous adipose tissue) [146];
- -
- -
2.8. Metformin
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Leclerc, T.; Thepenier, C.; Jault, P.; Bey, E.; Peltzer, J.; Trouillas, M.; Duhamel, P.; Bargues, L.; Prat, M.; Bonderriter, M. Cell therapy of burns. Cell Prolif. 2011, 44, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Norman, G.; Christie, J.; Liu, Z.; Westby, M.J.; Jefferies, J.M.; Hudson, T.; Edwards, J.; Mohapatra, D.P.; Hassan, I.A.; Dumville, J.C. Antiseptics for burns. Cochrane Database Syst. Rev. 2017, 2017, 011821. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Mehrotra, M.; Kumar, P.; Gogia, A.R.; Prasad, A.; Fisher, J.A. Smoke inhalation injury: Etiopathogenesis, diagnosis, and management. Indian J. Crit. Care Med. Peer-Rev. Off. Publ. Indian Soc. Crit. Care Med. 2018, 22, 180. [Google Scholar] [CrossRef]
- Hartford, C.E. Care of outpatient burns. In Total Burn Care, 4th ed.; Saunders: Philadelphia, PA, USA, 2012; pp. 81e82–92e82. [Google Scholar]
- Jeschke, M.G.; Patsouris, D.; Stanojcic, M.; Abdullahi, A.; Rehou, S.; Pinto, R.; Chen, P.; Burnett, M.; Amini-Nik, S. Pathophysiologic response to burns in the elderly. EBioMedicine 2015, 2, 1536–1548. [Google Scholar] [CrossRef] [Green Version]
- Gauglitz, G.G.; Halder, S.; Boehning, D.F.; Kulp, G.A.; Herndon, D.N.; Barral, J.M.; Jeschke, M.G. Post-burn hepatic insulin resistance is associated with ER stress. Shock 2010, 33. [Google Scholar] [CrossRef] [Green Version]
- Pruskowski, K.A.; Shields, B.A.; Ainsworth, C.R.; Cancio, L.C. Evaluation of the use of sitagliptin for insulin resistance in burn patients. Int. J. Burn Trauma 2020, 10, 237. [Google Scholar]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19. [Google Scholar] [PubMed]
- Kido, Y.; Nakae, J.; Accili, D. The insulin receptor and its cellular targets. J. Clin. Endocrinol. Metab. 2001, 86, 972–979. [Google Scholar] [CrossRef]
- Li, L.; Messina, J.L. Acute insulin resistance following injury. Trends Endocrinol. Metab. 2009, 20, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posner, B.I. Insulin signalling: The inside story. Can. J. Diabetes 2017, 41, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, M.A.; Alessi, D.R. PKB/Akt: A key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 2001, 114, 2903–2910. [Google Scholar] [CrossRef]
- Meijles, D.N.; Fuller, S.J.; Cull, J.J.; Alharbi, H.O.; Cooper, S.T.; Sugden, P.H.; Clerk, A. The insulin receptor family and protein kinase B (Akt) are activated in the heart by alkaline pH and α1-adrenergic receptors. Biochem. J. 2021, 478, 2059–2079. [Google Scholar] [CrossRef]
- Ballian, N.; Rabiee, A.; Andersen, D.K.; Elahi, D.; Gibson, B.R. Glucose metabolism in burn patients: The role of insulin and other endocrine hormones. Burns 2010, 36, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Bouche, C.; Serdy, S.; Kahn, C.R.; Goldfine, A.B. The cellular fate of glucose and its relevance in type 2 diabetes. Endocr. Rev. 2004, 25, 807–830. [Google Scholar] [CrossRef]
- Hanke, S.; Mann, M. The phosphotyrosine interactome of the insulin receptor family and its substrates IRS-1 and IRS-2. Mol. Cell. Proteom. 2009, 8, 519–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copps, K.; White, M. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [Green Version]
- Lennicke, C.; Cochemé, H.M. Redox regulation of the insulin signalling pathway. Redox Biol. 2021, 42, 101964. [Google Scholar] [CrossRef]
- Deng, H.-P.; Chai, J.-K. The effects and mechanisms of insulin on systemic inflammatory response and immune cells in severe trauma, burn injury, and sepsis. Int. Immunopharmacol. 2009, 9, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Auger, C.; Samadi, O.; Jeschke, M.G. The biochemical alterations underlying post-burn hypermetabolism. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2017, 1863, 2633–2644. [Google Scholar] [CrossRef]
- Van den Berghe, G.; Wouters, P.; Weekers, F.; Verwaest, C.; Bruyninckx, F.; Schetz, M.; Vlasselaers, D.; Ferdinande, P.; Lauwers, P.; Bouillon, R. Intensive insulin therapy in critically ill patients. N. Engl. J. Med. 2001, 345, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Berlanga-Acosta, J.; Mendoza-Marí, Y.; Rodríguez-Rodríguez, N.; del Barco Herrera, D.G.; García-Ojalvo, A.; Fernández-Mayola, M.; Guillén-Nieto, G.; Valdés-Sosa, P.A. Burn injury insulin resistance and central nervous system complications: A review. Burn Open 2020, 4, 41–52. [Google Scholar] [CrossRef]
- Jeschke, M.G.; Finnerty, C.C.; Herndon, D.N.; Song, J.; Boehning, D.; Tompkins, R.G.; Baker, H.V.; Gauglitz, G.G. Severe injury is associated with insulin resistance, endoplasmic reticulum stress response, and unfolded protein response. Ann. Surg. 2012, 255, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herndon, D.N.; Rodriguez, N.A.; Diaz, E.C.; Hegde, S.; Jennings, K.; Mlcak, R.P.; Suri, J.S.; Lee, J.O.; Williams, F.N.; Meyer, W. Long-term propranolol use in severely burned pediatric patients: A randomized controlled study. Ann. Surg. 2012, 256, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauglitz, G.G.; Williams, F.N.; Herndon, D.N.; Jeschke, M.G. Burns: Where are we standing with propranolol, oxandrolone, rhGH, and the new incretin analogues? Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 176. [Google Scholar] [CrossRef] [Green Version]
- Manzano-Nunez, R.; García-Perdomo, H.A.; Ferrada, P.; Delgado, C.A.O.; Gomez, D.A.; Foianini, J.E. Safety and effectiveness of propranolol in severely burned patients: Systematic review and meta-analysis. World J. Emerg. Surg. 2017, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- Hassoun-Kheir, N.; Henig, O.; Avni, T.; Leibovici, L.; Paul, M. The Effect of β-Blockers for Burn Patients on Clinical Outcomes: Systematic Review and Meta-Analysis. J. Intensive Care Med. 2020, 36, 945–953. [Google Scholar] [CrossRef]
- Jeschke, M.G.; Chinkes, D.L.; Finnerty, C.C.; Kulp, G.; Suman, O.E.; Norbury, W.B.; Branski, L.K.; Gauglitz, G.G.; Mlcak, R.P.; Herndon, D.N. The pathophysiologic response to severe burn injury. Ann. Surg. 2008, 248, 387. [Google Scholar] [CrossRef] [Green Version]
- Guillory, A.N.; Clayton, R.P.; Herndon, D.N.; Finnerty, C.C. Cardiovascular dysfunction following burn injury: What we have learned from rat and mouse models. Int. J. Mol. Sci. 2016, 17, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnerty, C.C.; Jeschke, M.G.; Herndon, D.N.; Gamelli, R.; Gibran, N.; Klein, M.; Silver, G.; Arnoldo, B.; Remick, D.; Tompkins, R.G. Temporal cytokine profiles in severely burned patients: A comparison of adults and children. Mol. Med. 2008, 14, 553–560. [Google Scholar] [CrossRef]
- Hartmann, C.; Radermacher, P.; Wepler, M.; Nußbaum, B. Non-hemodynamic effects of catecholamines. Shock Inj. Inflamm. Sepsis Lab. Clin. Approaches 2017, 48, 390–400. [Google Scholar] [CrossRef]
- Brooks, N.C.; Song, J.; Boehning, D.; Kraft, R.; Finnerty, C.C.; Herndon, D.N.; Jeschke, M.G. Propranolol improves impaired hepatic phosphatidylinositol 3-kinase/akt signaling after burn injury. Mol. Med. 2012, 18, 707–711. [Google Scholar] [CrossRef]
- Flores, O.; Stockton, K.; Roberts, J.A.; Muller, M.J.; Paratz, J.D. The efficacy and safety of adrenergic blockade after burn injury: A systematic review and meta-analysis. J. Trauma Acute Care Surg. 2016, 80, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Jacob, S. Beta-blocking agents in patients with insulin resistance: Effects of vasodilating beta-blockers. Blood Press. 1999, 8, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Carella, A.M.; Antonucci, G.; Conte, M.; Di Pumpo, M.; Giancola, A.; Antonucci, E. Antihypertensive treatment with beta-blockers in the metabolic syndrome: A review. Curr. Diabetes Rev. 2010, 6, 215–221. [Google Scholar] [CrossRef]
- Di Bari, M.; Marchionni, N.; Pahor, M. β-Blockers after Acute Myocardial Infarction in Elderly Patients with Diabetes Mellitus. Drugs Aging 2003, 20, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Fares, H.; Lavie, C.J.; Ventura, H.O. Vasodilating versus first-generation β-blockers for cardiovascular protection. Postgrad. Med. 2012, 124, 7–15. [Google Scholar] [CrossRef]
- Toda, N. Vasodilating β-adrenoceptor blockers as cardiovascular therapeutics. Pharmacol. Ther. 2003, 100, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Fergus, I.V.; Connell, K.L.; Ferdinand, K.C. A comparison of vasodilating and non-vasodilating beta-blockers and their effects on cardiometabolic risk. Curr. Cardiol. Rep. 2015, 17, 38. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.; Fagan, S.; Nejad, S.; Bilodeau, M.; Goverman, L.; Ibrahim, A.; Beresneva, O.; Sarhane, K.; Goverman, J. The role of a dedicated staff psychiatrist in modern burn centers. Ann. Burn Fire Disasters 2013, 26, 213. [Google Scholar]
- Ren, Z.; Zhang, P.; Wang, H.; Wang, H. Qualitative research investigating the mental health care service gap in Chinese burn injury patients. BMC Health Serv. Res. 2018, 18, 902. [Google Scholar] [CrossRef] [Green Version]
- Wiechman, S.; Ptacek, J.; Patterson, D.; Gibran, N.; Engrav, L.; Heimbach, D. Rates, trends, and severity of depression after burn injuries. J. Burn Care Rehabil. 2001, 22, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Pervaiz, M. Depression in adults post burn injury: A descriptive study conducted in the burn centre of a tertiary care hospital in Karachi. Ann. Burn Fire Disasters 2019, 32, 33. [Google Scholar]
- Rummel-Kluge, C.; Komossa, K.; Schwarz, S.; Hunger, H.; Schmid, F.; Lobos, C.A.; Kissling, W.; Davis, J.M.; Leucht, S. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: A systematic review and meta-analysis. Schizophr. Res. 2010, 123, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.M.; Stahl, S.M. The metabolic syndrome and schizophrenia. Acta Psychiatr. Scand. 2009, 119, 4–14. [Google Scholar] [CrossRef] [PubMed]
- De Hert, M.; Detraux, J.; Van Winkel, R.; Yu, W.; Correll, C.U. Metabolic and cardiovascular adverse effects associated with antipsychotic drugs. Nat. Rev. Endocrinol. 2012, 8, 114–126. [Google Scholar] [CrossRef]
- Manu, P.; Correll, C.U.; Wampers, M.; van Winkel, R.; Yu, W.; Shiffeldrim, D.; Kane, J.M.; De Hert, M. Insulin secretion in patients receiving clozapine, olanzapine, quetiapine and risperidone. Schizophr. Res. 2013, 143, 358–362. [Google Scholar] [CrossRef]
- Yuen, J.W.; Kim, D.D.; Procyshyn, R.M.; Panenka, W.J.; Honer, W.G.; Barr, A.M. A focused review of the metabolic side-effects of clozapine. Front. Endocrinol. 2021, 12, 67. [Google Scholar] [CrossRef]
- Lau, S.L.; Muir, C.; Assur, Y.; Beach, R.; Tran, B.; Bartrop, R.; McLean, M.; Caetano, D. Predicting weight gain in patients treated with clozapine: The role of sex, body mass index, and smoking. J. Clin. Psychopharmacol. 2016, 36, 120–124. [Google Scholar] [CrossRef]
- Henderson, D.C.; Daley, T.B.; Kunkel, L.; Rodrigues-Scott, M.; Koul, P.; Hayden, D. Clozapine and hypertension: A chart review of 82 patients. J. Clin. Psychiatry 2004, 65, 686–689. [Google Scholar] [CrossRef]
- Kim, D.D.; Barr, A.M.; Fredrikson, D.H.; Honer, W.G.; Procyshyn, R.M. Association between serum lipids and antipsychotic response in schizophrenia. Curr. Neuropharmacol. 2019, 17, 852–860. [Google Scholar] [CrossRef]
- Yazici, K.; Erbas, T.; Yazici, A. The effect of clozapine on glucose metabolism. Exp. Clin. Endocrinol. Diabetes 1998, 106, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Lindenmayer, J.-P.; Nathan, A.-M.; Smith, R.C. Hyperglycemia associated with the use of atypical antipsychotics. J. Clin. Psychiatry 2001, 62, 30–38. [Google Scholar]
- Boyda, H.N.; Ho, A.A.; Tse, L.; Procyshyn, R.M.; Yuen, J.W.; Kim, D.D.; Honer, W.G.; Barr, A.M. Differential effects of acute treatment with antipsychotic drugs on peripheral catecholamines. Front. Psychiatry 2020, 11, 617428. [Google Scholar] [CrossRef] [PubMed]
- Yuen, J.; Wu, C.; Wang, C.; Kim, D.; Procyshyn, R.; Honer, W.; Barr, A. A comparison of the effects of clozapine and its metabolite norclozapine on metabolic dysregulation in rodent models. Neuropharmacology 2019, 175, 107717. [Google Scholar] [CrossRef] [PubMed]
- Chintoh, A.F.; Mann, S.W.; Lam, L.; Giacca, A.; Fletcher, P.; Nobrega, J.; Remington, G. Insulin resistance and secretion in vivo: Effects of different antipsychotics in an animal model. Schizophr. Res. 2009, 108, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Houseknecht, K.L.; Robertson, A.S.; Zavadoski, W.; Gibbs, E.M.; Johnson, D.E.; Rollema, H. Acute effects of atypical antipsychotics on whole-body insulin resistance in rats: Implications for adverse metabolic effects. Neuropsychopharmacology 2007, 32, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondelli, V.; Anacker, C.; Vernon, A.; Cattaneo, A.; Natesan, S.; Modo, M.; Dazzan, P.; Kapur, S.; Pariante, C. Haloperidol and olanzapine mediate metabolic abnormalities through different molecular pathways. Transl. Psychiatry 2013, 3, e208. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, E.L.; Kim, H.; Kim, M.; Jacobson, M. Niacin: Vitamin and antidyslipidemic drug. In Water Soluble Vitamins; Springer: Dordrecht, The Netherlands, 2012; pp. 37–47. [Google Scholar]
- Kirkland, J.B.; Meyer-Ficca, M.L. Niacin. In Advances in Food and Nutrition Research; Elsevier: Amsterdam, The Netherlands, 2018; Volume 83, pp. 83–149. [Google Scholar]
- Makarov, M.V.; Trammell, S.A.; Migaud, M.E. The chemistry of the vitamin B3 metabolome. Biochem. Soc. Trans. 2019, 47, 131–147. [Google Scholar] [CrossRef]
- Garg, A.; Sharma, A.; Krishnamoorthy, P.; Garg, J.; Virmani, D.; Sharma, T.; Stefanini, G.; Kostis, J.B.; Mukherjee, D.; Sikorskaya, E. Role of niacin in current clinical practice: A systematic review. Am. J. Med. 2017, 130, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Dias, S.; Paredes, S.; Ribeiro, L. Drugs involved in dyslipidemia and obesity treatment: Focus on adipose tissue. Int. J. Endocrinol. 2018, 2018, 2673418. [Google Scholar] [CrossRef]
- Heemskerk, M.M.; van den Berg, S.A.; Pronk, A.C.; van Klinken, J.-B.; Boon, M.R.; Havekes, L.M.; Rensen, P.C.; van Dijk, K.W.; van Harmelen, V. Long-term niacin treatment induces insulin resistance and adrenergic responsiveness in adipocytes by adaptive downregulation of phosphodiesterase 3B. Am. J. Physiol.-Endocrinol. Metab. 2014, 306, E808–E813. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.T.; Oh, K.-S.; Choi, Y.M.; Jokiaho, A.; Donovan, C.; Choi, S.; Kang, I.; Youn, J.H. Continuous 24-h nicotinic acid infusion in rats causes FFA rebound and insulin resistance by altering gene expression and basal lipolysis in adipose tissue. Am. J. Physiol.-Endocrinol. Metab. 2011, 300, E1012–E1021. [Google Scholar] [CrossRef] [Green Version]
- Page, M.R. The JNC 8 hypertension guidelines: An in-depth guide. Am. J. Manag. Care 2014, 20, E8. [Google Scholar]
- Investigators, H.O.P.E.S. Effects of an angiotensin-converting–enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N. Engl. J. Med. 2000, 342, 145–153. [Google Scholar]
- Williams, B. Drug discovery in renin–angiotensin system intervention: Past and future. Ther. Adv. Cardiovasc. Dis. 2016, 10, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Teng, T.-H.K.; Tromp, J.; Tay, W.T.; Anand, I.; Ouwerkerk, W.; Chopra, V.; Wander, G.S.; Yap, J.J.; MacDonald, M.R.; Xu, C.F. Prescribing patterns of evidence-based heart failure pharmacotherapy and outcomes in the ASIAN-HF registry: A cohort study. Lancet Glob. Health 2018, 6, e1008–e1018. [Google Scholar] [CrossRef] [Green Version]
- Silvariño, R.; Rios, P.; Baldovinos, G.; Chichet, M.A.; Perg, N.; Sola, L.; Saona, G.; De Souza, N.; Lamadrid, V.; Gadola, L. Is chronic kidney disease progression influenced by the type of renin-angiotensin-system blocker used? Nephron 2019, 143, 100–107. [Google Scholar] [CrossRef]
- Peng, H.; Carretero, O.A.; Alfie, M.E.; Masura, J.A.; Rhaleb, N.-E. Effects of angiotensin-converting enzyme inhibitor and angiotensin type 1 receptor antagonist in deoxycorticosterone acetate–salt hypertensive mice lacking Ren-2 gene. Hypertension 2001, 37, 974–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhaleb, N.-E.; Peng, H.; Yang, X.-P.; Liu, Y.-H.; Mehta, D.; Ezan, E.; Carretero, O.A. Long-term effect of N-acetyl-seryl-aspartyl-lysyl-proline on left ventricular collagen deposition in rats with 2-kidney, 1-clip hypertension. Circulation 2001, 103, 3136–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uehara, M.; Kishikawa, H.; Isami, S.; Kisanuki, K.; Ohkubo, Y.; Miyamura, N.; Miyata, T.; Yano, T.; Shichiri, M. Effect on insulin sensitivity of angiotensin converting enzyme inhibitors with or without a sulphydryl group: Bradykinin may improve insulin resistance in dogs and humans. Diabetologia 1994, 37, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Rizos, C.V.; Elisaf, M.S. Antihypertensive drugs and glucose metabolism. World J. Cardiol. 2014, 6, 517. [Google Scholar] [CrossRef] [PubMed]
- Abuissa, H.; Jones, P.G.; Marso, S.P.; O’Keefe, J.H. Angiotensin-converting enzyme inhibitors or angiotensin receptor blockers for prevention of type 2 diabetes: A meta-analysis of randomized clinical trials. J. Am. Coll. Cardiol. 2005, 46, 821–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiuchi, T.; Cui, T.-X.; Wu, L.; Nakagami, H.; Takeda-Matsubara, Y.; Iwai, M.; Horiuchi, M. ACE inhibitor improves insulin resistance in diabetic mouse via bradykinin and NO. Hypertension 2002, 40, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Farsang, C. Indications for and utilization of angiotensin receptor II blockers in patients at high cardiovascular risk. Vasc. Health Risk Manag. 2011, 7, 605. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Hernández, R.; Sosa-Canache, B.; Velasco, M.; Armas-Hernandez, M.; Armas-Padilla, M.; Cammarata, R. Angiotensin II receptor antagonists role in arterial hypertension. J. Hum. Hypertens. 2002, 16, S93–S99. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.Y.; Park, H.K.; Lee, B.-S.; Jeong, S.; Hyun, S.; Choi, J.-W.; Kim, S.W.; Lee, S.; Lim, S.; Hwang, K.-C. Novel Therapeutic Effects of Pterosin B on Ang II-Induced Cardiomyocyte Hypertrophy. Molecules 2020, 25, 5279. [Google Scholar] [CrossRef]
- Schupp, M.; Janke, J.r.; Clasen, R.; Unger, T.; Kintscher, U. Angiotensin type 1 receptor blockers induce peroxisome proliferator–activated receptor-γ activity. Circulation 2004, 109, 2054–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, S.C.; Pershadsingh, H.A.; Ho, C.I.; Chittiboyina, A.; Desai, P.; Pravenec, M.; Qi, N.; Wang, J.; Avery, M.A.; Kurtz, T.W. Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARγ–modulating activity. Hypertension 2004, 43, 993–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Qiao, S.; Han, D.-W.; Rong, X.-R.; Wang, Y.-X.; Xue, J.-J.; Yang, J. Telmisartan improves insulin resistance: A meta-analysis. Am. J. Ther. 2018, 25, e642–e651. [Google Scholar] [CrossRef]
- Sugden, M.C.; Caton, P.W.; Holness, M.J. Peroxisome Proliferator-Activated Receptors. In Encyclopedia of Biological Chemistry, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 229–235. [Google Scholar] [CrossRef]
- Smith, U. Pioglitazone: Mechanism of action. Int. J. Clin. Pr. Suppl. 2001, 121, 13–18. [Google Scholar]
- Henriksen, E.J.; Jacob, S.; Kinnick, T.R.; Teachey, M.K.; Krekler, M. Selective angiotensin II receptor antagonism reduces insulin resistance in obese Zucker rats. Hypertension 2001, 38, 884–890. [Google Scholar] [CrossRef] [Green Version]
- Tamori, Y.; Masugi, J.; Nishino, N.; Kasuga, M. Role of peroxisome proliferator-activated receptor-γ in maintenance of the characteristics of mature 3T3-L1 adipocytes. Diabetes 2002, 51, 2045–2055. [Google Scholar] [CrossRef] [Green Version]
- Takada, I.; Makishima, M. Peroxisome proliferator-activated receptor agonists and antagonists: A patent review (2014-present). Expert Opin. Ther. Pat. 2020, 30, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, P.S. American Association of Clinical Endocrinologists/American College of Endocrinology Management of Dyslipidemia and Prevention of Cardiovascular Disease Clinical Practice Guidelines. Diabetes Spectr 2018, 31, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colhoun, H.M.; Betteridge, D.J.; Durrington, P.N.; Hitman, G.A.; Neil, H.A.; Livingstone, S.J.; Thomason, M.J.; Mackness, M.I.; Charlton-Menys, V.; Fuller, J.H.; et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): Multicentre randomised placebo-controlled trial. Lancet 2004, 364, 685–696. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Jebari, S.; Larrea-Sebal, A.; Uribe, K.B.; Siddiqi, H.; Ostolaza, H.; Benito-Vicente, A.; Martín, C. Statin treatment-induced development of type 2 diabetes: From clinical evidence to mechanistic insights. Int. J. Mol. Sci. 2020, 21, 4725. [Google Scholar] [CrossRef]
- Endo, A. A gift from nature: The birth of the statins. Nat. Med. 2008, 14, 1050–1052. [Google Scholar] [CrossRef]
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B 2010, 86, 484–493. [Google Scholar] [CrossRef] [Green Version]
- Betteridge, D.J.; Carmena, R. The diabetogenic action of statins—Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2016, 12, 99–110. [Google Scholar] [CrossRef]
- Navarese, E.P.; Buffon, A.; Andreotti, F.; Kozinski, M.; Welton, N.; Fabiszak, T.; Caputo, S.; Grzesk, G.; Kubica, A.; Swiatkiewicz, I. Meta-analysis of impact of different types and doses of statins on new-onset diabetes mellitus. Am. J. Cardiol. 2013, 111, 1123–1130. [Google Scholar] [CrossRef]
- Wang, H.J.; Park, J.Y.; Kwon, O.; Choe, E.Y.; Kim, C.H.; Hur, K.Y.; Lee, M.-S.; Yun, M.; Cha, B.S.; Kim, Y.-B. Chronic HMGCR/HMG-CoA reductase inhibitor treatment contributes to dysglycemia by upregulating hepatic gluconeogenesis through autophagy induction. Autophagy 2015, 11, 2089–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakata, M.; Nagasaka, S.; Kusaka, I.; Matsuoka, H.; Ishibashi, S.; Yada, T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): Implications in glycaemic control. Diabetologia 2006, 49, 1881–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Zhao, S.-P. Different effects of statins on induction of diabetes mellitus: An experimental study. Drug Des. Dev. Ther. 2015, 9, 6211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brault, M.; Ray, J.; Gomez, Y.-H.; Mantzoros, C.S.; Daskalopoulou, S.S. Statin treatment and new-onset diabetes: A review of proposed mechanisms. Metabolism 2014, 63, 735–745. [Google Scholar] [CrossRef]
- Mancini, G.J.; Baker, S.; Bergeron, J.; Fitchett, D.; Frohlich, J.; Genest, J.; Gupta, M.; Hegele, R.A.; Ng, D.; Pearson, G.J. Diagnosis, prevention, and management of statin adverse effects and intolerance: Canadian Consensus Working Group Update (2016). Can. J. Cardiol. 2016, 32, S35–S65. [Google Scholar] [CrossRef]
- Fernández-Hernando, C.; Ramírez, C.M.; Goedeke, L.; Suárez, Y. MicroRNAs in metabolic disease. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Simmons, R.A. Cell glucose transport and glucose handling during fetal and neonatal development. In Fetal and Neonatal Physiology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 428–435. [Google Scholar]
- Ling, Z.; Shu, N.; Xu, P.; Wang, F.; Zhong, Z.; Sun, B.; Li, F.; Zhang, M.; Zhao, K.; Tang, X. Involvement of pregnane X receptor in the impaired glucose utilization induced by atorvastatin in hepatocytes. Biochem. Pharmacol. 2016, 100, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Hatting, M.; Tavares, C.D.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2018, 1411, 21. [Google Scholar] [CrossRef]
- Gotoh, S.; Negishi, M. Statin-activated nuclear receptor PXR promotes SGK2 dephosphorylation by scaffolding PP2C to induce hepatic gluconeogenesis. Sci. Rep. 2015, 5, 14076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuemmerle, N.B.; Kinlaw, W.B. THRSP (thyroid hormone responsive). Atlas Genet. Cytogenet. Oncol. Haematol. 2011, 15, 480. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wang, C.; Li, S.; Li, S.; Wang, W.; Li, J.; Chi, Y.; Yang, H.; Kong, X.; Zhou, Y. Thyroid hormone-responsive SPOT 14 homolog promotes hepatic lipogenesis, and its expression is regulated by Liver X receptor α through a sterol regulatory element-binding protein 1c–dependent mechanism in mice. Hepatology 2013, 58, 617–628. [Google Scholar] [CrossRef]
- Cogburn, L.A.; Trakooljul, N.; Wang, X.; Ellestad, L.E.; Porter, T.E. Transcriptome analyses of liver in newly-hatched chicks during the metabolic perturbation of fasting and re-feeding reveals THRSPA as the key lipogenic transcription factor. BMC Genom. 2020, 21, 109. [Google Scholar] [CrossRef] [PubMed]
- LaFave, L.T.; Augustin, L.B.; Mariash, C.N. S14: Insights from knockout mice. Endocrinology 2006, 147, 4044–4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Anderson, G.W.; Mucha, G.T.; Parks, E.J.; Metkowski, J.K.; Mariash, C.N. The Spot 14 protein is required for de novo lipid synthesis in the lactating mammary gland. Endocrinology 2005, 146, 3343–3350. [Google Scholar] [CrossRef] [Green Version]
- Colbert, C.L.; Kim, C.-W.; Moon, Y.-A.; Henry, L.; Palnitkar, M.; McKean, W.B.; Fitzgerald, K.; Deisenhofer, J.; Horton, J.D.; Kwon, H.J. Crystal structure of Spot 14, a modulator of fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18820–18825. [Google Scholar] [CrossRef] [Green Version]
- Tolman, K.G.; Fonseca, V.; Dalpiaz, A.; Tan, M.H. Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease. Diabetes Care 2007, 30, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Wakil, S.J.; Abu-Elheiga, L.A. Fatty acid metabolism: Target for metabolic syndrome. J. Lipid Res. 2009, 50, S138–S143. [Google Scholar] [CrossRef] [Green Version]
- Carnagarin, R.; Dharmarajan, A.M.; Dass, C.R. Molecular aspects of glucose homeostasis in skeletal muscle–A focus on the molecular mechanisms of insulin resistance. Mol. Cell. Endocrinol. 2015, 417, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Sears, B.; Perry, M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015, 14, 121. [Google Scholar] [CrossRef] [Green Version]
- Cree, M.G.; Aarsland, A.; Herndon, D.N.; Wolfe, R.R. Role of fat metabolism in burn trauma-induced skeletal muscle insulin resistance. Crit. Care Med. 2007, 35, S476–S483. [Google Scholar] [CrossRef]
- Yaluri, N.; Modi, S.; Kokkola, T. Simvastatin induces insulin resistance in L6 skeletal muscle myotubes by suppressing insulin signaling, GLUT4 expression and GSK-3β phosphorylation. Biochem. Biophys. Res. Commun. 2016, 480, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kain, V.; Kapadia, B.; Misra, P.; Saxena, U. Simvastatin may induce insulin resistance through a novel fatty acid mediated cholesterol independent mechanism. Sci. Rep. 2015, 5, 13823. [Google Scholar] [CrossRef] [Green Version]
- Sanvee, G.M.; Panajatovic, M.V.; Bouitbir, J.; Krähenbühl, S. Mechanisms of insulin resistance by simvastatin in C2C12 myotubes and in mouse skeletal muscle. Biochem. Pharmacol. 2019, 164, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liang, X.; Zeng, Z.; Yu, K.; Zhan, S.; Su, Q.; Yan, Y.; Mansai, H.; Qiao, W.; Yang, Q. Simvastatin inhibits glucose uptake activity and GLUT4 translocation through suppression of the IR/IRS-1/Akt signaling in C2C12 myotubes. Biomed. Pharmacother. 2016, 83, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacal, J.C. GTPase. In Encyclopedic Reference of Cancer; Schwab, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2001; pp. 375–378. [Google Scholar]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.R. Rab GTPase regulation of membrane identity. Curr. Opin. Cell Biol. 2013, 25, 414–419. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Zhong, Z.; Wang, F.; Xu, J.; Xu, F.; Kong, W.; Ling, Z.; Shu, N.; Li, Y.; Wu, T. Atorvastatin impaired glucose metabolism in C2C12 cells partly via inhibiting cholesterol-dependent glucose transporter 4 translocation. Biochem. Pharmacol. 2018, 150, 108–119. [Google Scholar] [CrossRef]
- Stöckli, J.; Fazakerley, D.J.; James, D.E. GLUT4 exocytosis. J. Cell Sci. 2011, 124, 4147–4159. [Google Scholar] [CrossRef] [Green Version]
- Roden, M. How free fatty acids inhibit glucose utilization in human skeletal muscle. Physiology 2004, 19, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Takaguri, A.; Satoh, K.; Itagaki, M.; Tokumitsu, Y.; Ichihara, K. Effects of atorvastatin and pravastatin on signal transduction related to glucose uptake in 3T3L1 adipocytes. J. Pharmacol. Sci. 2008, 107, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira-Leal, J.B.; Hume, A.N.; Seabra, M.C. Prenylation of Rab GTPases: Molecular mechanisms and involvement in genetic disease. FEBS Lett. 2001, 498, 197–200. [Google Scholar] [CrossRef]
- Breen, M.R.; Camps, M.; Carvalho-Simoes, F.; Zorzano, A.; Pilch, P.F. Cholesterol depletion in adipocytes causes caveolae collapse concomitant with proteosomal degradation of cavin-2 in a switch-like fashion. PLoS ONE 2012, 7, e34516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustavsson, J.; Parpal, S.; Strålfors, P. Insulin-stimulated glucose uptake involves the transition of glucose transporters to a caveolae-rich fraction within the plasma membrane: Implications for type II diabetes. Mol. Med. 1996, 2, 367–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, M.; Peränen, J.; Schreiner, R.; Wieland, F.; Kurzchalia, T.V.; Simons, K. VIP21/caveolin is a cholesterol-binding protein. Proc. Natl. Acad. Sci. USA 1995, 92, 10339–10343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, A.W.; Razani, B.; Wang, X.B.; Combs, T.P.; Williams, T.M.; Scherer, P.E.; Lisanti, M.P. Caveolin-1-deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am. J. Physiol.-Cell Physiol. 2003, 285, C222–C235. [Google Scholar] [CrossRef] [Green Version]
- Krautbauer, S.; Neumeier, M.; Eisinger, K.; Hader, Y.; Dada, A.; Schmitz, G.; Aslanidis, C.; Buechler, C. LDL but not HDL increases adiponectin release of primary human adipocytes. Exp. Mol. Pathol. 2013, 95, 325–329. [Google Scholar] [CrossRef]
- Yadav, A.; Kataria, M.A.; Saini, V.; Yadav, A. Role of leptin and adiponectin in insulin resistance. Clin. Chim. Acta 2013, 417, 80–84. [Google Scholar] [CrossRef]
- Li, X.; Zhang, D.; Vatner, D.F.; Goedeke, L.; Hirabara, S.M.; Zhang, Y.; Perry, R.J.; Shulman, G.I. Mechanisms by which adiponectin reverses high fat diet-induced insulin resistance in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 32584–32593. [Google Scholar] [CrossRef]
- Nanjan, M.; Mohammed, M.; Kumar, B.P.; Chandrasekar, M. Thiazolidinediones as antidiabetic agents: A critical review. Bioorganic Chem. 2018, 77, 548–567. [Google Scholar] [CrossRef]
- Moini, J. Epidemiology of Diabetes; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- DeFronzo, R.A.; Tripathy, D.; Schwenke, D.C.; Banerji, M.; Bray, G.A.; Buchanan, T.A.; Clement, S.C.; Henry, R.R.; Hodis, H.N.; Kitabchi, A.E.; et al. Pioglitazone for Diabetes Prevention in Impaired Glucose Tolerance. N. Engl. J. Med. 2011, 364, 1104–1115. [Google Scholar] [CrossRef]
- Taira, B.R.; Singer, A.J.; McClain, S.A.; Lin, F.; Rooney, J.; Zimmerman, T.; Clark, R.A. Rosiglitazone, a PPAR-γ ligand, reduces burn progression in rats. J. Burn Care Res. 2009, 30, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Şener, G.; Şehirli, A.Ö.; Gedik, N.; Dülger, G.A. Rosiglitazone, a PPAR-γ ligand, protects against burn-induced oxidative injury of remote organs. Burns 2007, 33, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Mukohda, M.; Ozaki, H. Anti-inflammatory mechanisms of the vascular smooth muscle PPARγ. J. Smooth Muscle Res. 2021, 57, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hauner, H. The mode of action of thiazolidinediones. Diabetes/Metab. Res. Rev. 2002, 18, S10–S15. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A. Thiazolidinediones and liver toxicity. Diabetes Metab. 2001, 27, 305–313. [Google Scholar]
- Nesto, R.W.; Bell, D.; Bonow, R.O.; Fonseca, V.; Grundy, S.M.; Horton, E.S.; Le Winter, M.; Porte, D.; Semenkovich, C.F.; Smith, S. Thiazolidinedione use, fluid retention, and congestive heart failure: A consensus statement from the American Heart Association and American Diabetes Association. Circulation 2003, 108, 2941–2948. [Google Scholar] [CrossRef] [Green Version]
- Bell, D.S.; Goncalves, E. Heart failure in the patient with diabetes: Epidemiology, aetiology, prognosis, therapy and the effect of glucose-lowering medications. Diabetes Obes. Metab. 2019, 21, 1277–1290. [Google Scholar] [CrossRef] [Green Version]
- Punthakee, Z.; Alméras, N.; Després, J.P.; Dagenais, G.; Anand, S.; Hunt, D.; Sharma, A.; Jung, H.; Yusuf, S.; Gerstein, H. Impact of rosiglitazone on body composition, hepatic fat, fatty acids, adipokines and glucose in persons with impaired fasting glucose or impaired glucose tolerance: A sub-study of the DREAM trial. Diabet. Med. 2014, 31, 1086–1092. [Google Scholar] [CrossRef]
- Dore, D.D.; Trivedi, A.N.; Mor, V.; Lapane, K.L. Association between extent of thiazolidinedione exposure and risk of acute myocardial infarction. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2009, 29, 775–783. [Google Scholar] [CrossRef] [Green Version]
- Graham, D.J.; Ouellet-Hellstrom, R.; MaCurdy, T.E.; Ali, F.; Sholley, C.; Worrall, C.; Kelman, J.A. Risk of acute myocardial infarction, stroke, heart failure, and death in elderly Medicare patients treated with rosiglitazone or pioglitazone. JAMA 2010, 304, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Rawson, N.S. Review of the quality of observational studies of the association between rosiglitazone and acute myocardial infarction. J. Popul. Ther. Clin. Pharmacol. 2014, 21, 214–232. [Google Scholar]
- Shin, S.; Kim, H. The effect of sitagliptin on cardiovascular risk profile in Korean patients with type 2 diabetes mellitus: A retrospective cohort study. Ther. Clin. Risk Manag. 2016, 12, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, S.E.; Zinman, B.; Lachin, J.M.; Haffner, S.M.; Herman, W.H.; Holman, R.R.; Kravitz, B.G.; Yu, D.; Heise, M.A.; Aftring, R.P.; et al. Rosiglitazone-Associated Fractures in Type 2 Diabetes. Anal. A Diabetes Outcome Progress. Trial (ADOPT) 2008, 31, 845–851. [Google Scholar] [CrossRef] [Green Version]
- Bray, G.; Smith, S.; Banerji, M.; Tripathy, D.; Clement, S.; Buchanan, T.; Henry, R.; Kitabchi, A.; Mudaliar, S.; Musi, N. Effect of pioglitazone on body composition and bone density in subjects with prediabetes in the ACT NOW trial. Diabetes Obes. Metab. 2013, 15, 931–937. [Google Scholar] [CrossRef]
- Schwartz, A.V.; Sellmeyer, D.E.; Vittinghoff, E.; Palermo, L.; Lecka-Czernik, B.; Feingold, K.R.; Strotmeyer, E.S.; Resnick, H.E.; Carbone, L.; Beamer, B.A. Thiazolidinedione use and bone loss in older diabetic adults. J. Clin. Endocrinol. Metab. 2006, 91, 3349–3354. [Google Scholar] [CrossRef] [PubMed]
- Watts, N.B. Effects on the skeleton from medications used to treat nonskeletal disorders. In Marcus and Feldman’s Osteoporosis; Elsevier: Amsterdam, The Netherlands, 2021; pp. 1061–1068. [Google Scholar]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Viollet, B.; Foretz, M. Revisiting the mechanisms of metformin action in the liver. In Annales D’endocrinologie; Elsevier: Masson, France, 2013; pp. 123–129. [Google Scholar]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, J.J.; Rhinehart, A.S.; Shaefer, C.F., Jr.; Neuman, A. Diagnosis and management of diabetes: Synopsis of the 2016 American diabetes association standards of medical care in diabetes. Ann. Intern. Med. 2016, 164, 542. [Google Scholar] [CrossRef]
- Yu, H.M.; Kim, S.J.; Chun, S.W.; Park, K.Y.; Lim, D.M.; Lee, J.M.; Hong, J.H.; Park, K.S. A comparison study on efficacy, insulin sensitivity and safety of glimepiride/metformin fixed dose combination versus glimepiride single therapy on type 2 diabetes mellitus patients with basal insulin therapy. Diabetes Res. Clin. Pract. 2019, 155, 107796. [Google Scholar] [CrossRef]
- Seifarth, C.; Schehler, B.; Schneider, H. Effectiveness of metformin on weight loss in non-diabetic individuals with obesity. Exp. Clin. Endocrinol. Diabetes 2013, 121, 27–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cree-Green, M.; Bergman, B.C.; Cengiz, E.; Fox, L.A.; Hannon, T.S.; Miller, K.; Nathan, B.; Pyle, L.; Kahn, D.; Tansey, M. Metformin improves peripheral insulin sensitivity in youth with type 1 diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 3265–3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikorskaya, K.; Zarzecka, I.; Ejikeme, U.; Russell, J. The use of metformin as an add-on therapy to insulin in the treatment of poorly controlled type 1 diabetes mellitus in adolescents. Metab. Open 2021, 9, 100080. [Google Scholar] [CrossRef]
- Jeschke, M.G.; Abdullahi, A.; Burnett, M.; Rehou, S.; Stanojcic, M. Glucose control in severely burned patients using metformin: An interim safety and efficacy analysis of a phase II randomized controlled trial. Ann. Surg. 2016, 264, 518. [Google Scholar] [CrossRef]
- Gus, E.I.; Shahrokhi, S.; Jeschke, M.G. Anabolic and anticatabolic agents used in burn care: What is known and what is yet to be learned. Burns 2020, 46, 19–32. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Cardiac Effects | Blood Pressure | Hypermetabolism | Insulin Resistance | Lipid Metabolism | Catecholamines |
|---|---|---|---|---|---|---|
| β blockers | cardio protective effects/decrease tachycardia and workload | ↓ hypertension | ↓ hypercatabolic state ↓ hyperglycemia | ↓ | inhibit lipolysis ↓ FFAs, ↓ cholesterol, ↓ TG levels | - |
| Antipsychotics | - | - | ↑ hyperglycemia | ↑ | stimulate lipolysis ↑ FFAs, ↑ cholesterol, ↑ TG levels | ↑ Plasmatic levels of catecholamines |
| Vitamin B3 | - | - | ↑ hyperglycemia | ↑ | stimulate lipolysis ↑ FFAs, ↑ cholesterol, ↑ TG levels | ↑ plasmatic levels of catecholamines |
| ACEI | cardio protective effects | ↓ hypertension | - | ↓ | ameliorate lipid profile | ↓ plasmatic levels of catecholamines |
| ARBs | cardio protective effects | ↓ hypertension | - | ↓ | ameliorate lipid profile | |
| Statins | - | - | ↑ hypercatabolic state ↑ hyperglycemia | ↑ | ↓ cholesterol levels ↑ accumulation of FFAs in muscles and the liver ↓ LDL, VLDL | - |
| Thiazolidinediones | might be linked to myocardial ischemia | might precipitate congestive heart failure in diabetic patients | ↓ hyperglycemia | ↓ | ↑ FFA accumulation in adipose tissue | - |
| Metformin | - | - | ↓ hypercatabolic state ↓ hyperglycemia | ↓ | ↑ oxidation of FFA in the liver reduces lipogenesis | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greabu, M.; Badoiu, S.C.; Stanescu-Spinu, I.-I.; Miricescu, D.; Totan, A.R.; Badoiu, S.E.; Costagliola, M.; Jinga, V. Drugs Interfering with Insulin Resistance and Their Influence on the Associated Hypermetabolic State in Severe Burns: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 9782. https://doi.org/10.3390/ijms22189782
Greabu M, Badoiu SC, Stanescu-Spinu I-I, Miricescu D, Totan AR, Badoiu SE, Costagliola M, Jinga V. Drugs Interfering with Insulin Resistance and Their Influence on the Associated Hypermetabolic State in Severe Burns: A Narrative Review. International Journal of Molecular Sciences. 2021; 22(18):9782. https://doi.org/10.3390/ijms22189782
Chicago/Turabian StyleGreabu, Maria, Silviu Constantin Badoiu, Iulia-Ioana Stanescu-Spinu, Daniela Miricescu, Alexandra Ripszky Totan, Silvia Elena Badoiu, Michel Costagliola, and Viorel Jinga. 2021. "Drugs Interfering with Insulin Resistance and Their Influence on the Associated Hypermetabolic State in Severe Burns: A Narrative Review" International Journal of Molecular Sciences 22, no. 18: 9782. https://doi.org/10.3390/ijms22189782
APA StyleGreabu, M., Badoiu, S. C., Stanescu-Spinu, I.-I., Miricescu, D., Totan, A. R., Badoiu, S. E., Costagliola, M., & Jinga, V. (2021). Drugs Interfering with Insulin Resistance and Their Influence on the Associated Hypermetabolic State in Severe Burns: A Narrative Review. International Journal of Molecular Sciences, 22(18), 9782. https://doi.org/10.3390/ijms22189782