
Hybrids as NO Donors



Abstract
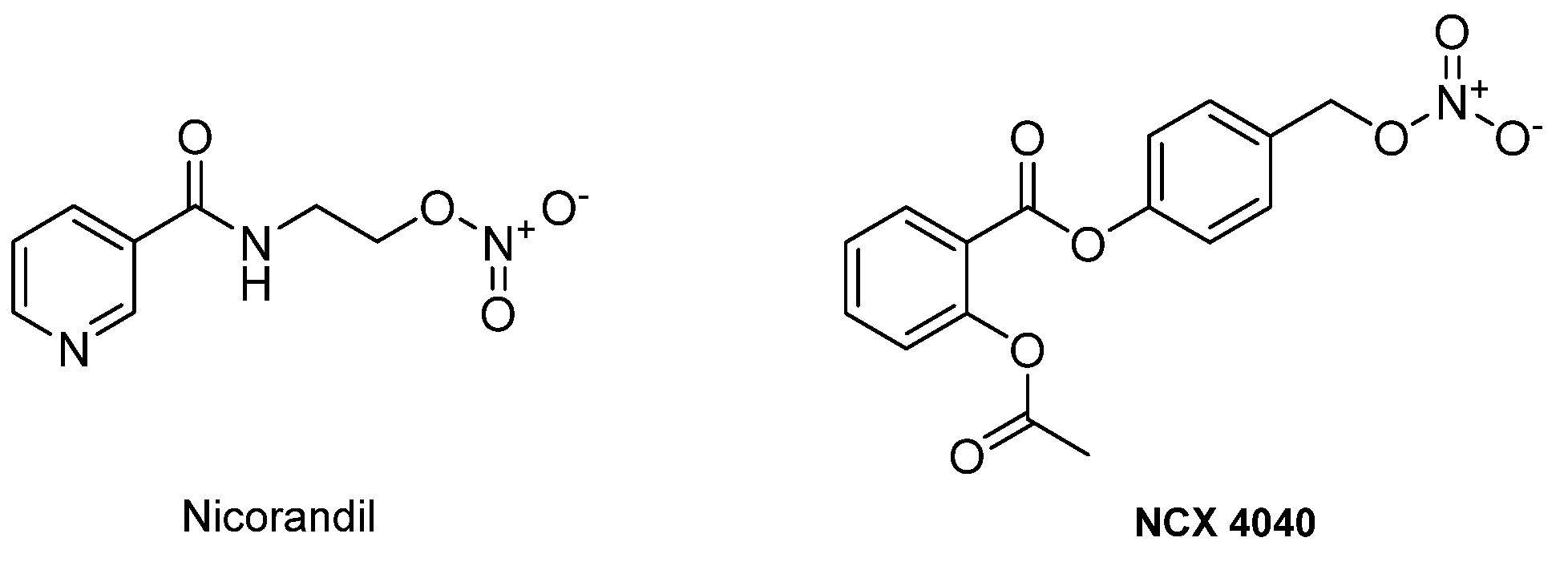
:1. Introduction
- Nitro-oxy esters of cinnamic acids
- (a)
- Nitro-oxy ester hybrids as anti-inflammatory and antioxidant agents;
- (b)
- Nitro-oxy ester hybrids as anti-atherosclerotic agents;
- (c)
- Nitro-oxy ester hybrids as anticancer agents;
- (d)
- Nitro-oxy ester hybrids as multifunctional acetyl- and butyrylcholinesterase inhibitors;
- (e)
- Nitro-oxy ester hybrids as antiplatelet and antithrombotic agents.
- Furoxan-cinnamic acid hybrids
- (a)
- Furoxan hybrids as anti-atherosclerotic agents;
- (b)
- Furoxan hybrids as anticancer agents;
- (c)
- Furoxan hybrids as anti-diabetic agents.
2. Nitro-Oxy Esters of Cinnamic Acids
2.1. Nitro-Oxy Ester Hybrids as Anti-Inflammatory and Antioxidant Agents
2.2. Nitro-Oxy Ester Hybrids as Antiatherosclerotic Agents
2.3. Nitro-Oxy Ester Hybrids as Anticancer Agents
2.4. Nitro-Oxy Ester Hybrids as Multifunctional Acetyl- and Butyrylcholinesterase Inhibitors
2.5. Nitro-Oxy Ester Hybrids as Antiplatelet and Antithrombotic Agents
3. Furoxan–Cinnamic Acid Hybrids
3.1. Furoxan Hybrids as Antiatherosclerotic Agents
3.2. Furoxan Hybrids as Anticancer Agents
3.3. Furoxan Hybrids as Antidiabetic Agents
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| NO | Nitric Oxide |
| eNOS | Endothelial Nitric Oxide Synthase |
| nNOS | Neuronal Nitric Oxide Synthase |
| iNOS | Inducible Nitric Oxide Synthase |
| COX | Cyclooxygenase |
| LOX | Lipoxygenase |
| ATP | Adenosine Triphosphate |
| NSAIDs | Non-Steroidal Anti-Inflammatory Drugs |
| NDGA | Nordihydroguaiaretic Acid |
| ROS | Reactive Oxygen Species |
| LDL | Low-Density Lipoprotein |
| DPPH | 2,2-diphenyl-1-picrylhydrazyl |
| SNAP | S-nitroso-N-acetylpenicillamine |
| PGF2a | Prostaglandin F2alpha |
| ISDN | Isosorbide Ninitrate |
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| BuChE | Butyrylcholinesterase |
| NBP | 3-n-butylphtalide |
| SFDA | State Food and Drug Administration |
| PRP | Platelet-Rich Plasma |
| A-V | Arteriovenous |
| CVD | Cardiovascular Disease |
| T2DM | Type 2 Diabetes Mellitus |
| AGEs | Advanced Glycation End-Products |
| BSA | Bovine Serum Albumin |
| MGO | Methylglyoxal |
| ADP | Adenosine Diphosphate |
References
- Viegas-junior, C.; Danuello, A.; Bolzani, S.; Barreiro, E.J.; Alberto, C.; Fraga, M. Molecular Hybridization: A Useful Tool in the Design of New Drug Prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Ignarro, L.J.; Murad, F.; Prize, N. Nobel Laureate series Nitric oxide discovery. Eur. Heart J. 2019, 40, 1747–1755. [Google Scholar]
- Strijdom, H.; Chamane, N.; Lochner, A. Nitric oxide in the cardiovascular system: A simple molecule with complex actions. Cardiovasc. J. Afr. 2009, 20, 303–310. [Google Scholar] [PubMed]
- Esplugues, J.V. NO as a signalling molecule in the nervous system. Br. J. Pharmacol. 2002, 135, 1079–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef]
- Follmann, M.; Griebenow, N.; Hahn, M.G.; Hartung, I.; Mais, F.J.; Mittendorf, J.; Schäfer, M.; Schirok, H.; Stasch, J.P.; Stoll, F.; et al. The chemistry and biology of soluble guanylate cyclase stimulators and activators. Angew. Chem. Int. Ed. 2013, 52, 9442–9462. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Lee, S.G.; Song, S.H.; Heo, D.S.; Park, B.J.; Lee, D.W.; Kim, K.H.; Sung, M.W. The effect of nitric oxide on cyclooxygenase-2 (COX-2) overexpression in head and neck cancer cell lines. Int. J. Cancer 2003, 107, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Needleman, P.; Manning, P.T. Interactions between the inducible cyclooxygenase (COX-2) and nitric oxide synthase (iNOS) pathways: Implications for therapeutic intervention in osteoarthritis. Osteoarthr. Cartil. 1999, 7, 367–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachmilewitz, D.; Karmeli, F.; Okon, E.; Bursztyn, M. Experimental colitis is ameliorated by inhibition of nitric oxide synthase activity. Gut 1995, 37, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Clancy, R.; Varenika, B.; Huang, W.; Ballou, L.; Attur, M.; Amin, A.R.; Abramson, S.B. Nitric Oxide Synthase/COX Cross-Talk: Nitric Oxide Activates COX-1 But Inhibits COX-2-Derived Prostaglandin Production. J. Immunol. 2000, 165, 1582–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.; Megson, I. Recent developments in nitric oxide donor drugs. Br. J. Pharmacol. 2007, 151, 305–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frampton, J.E.; Buckley, M.M.; Fitton, A. Nicorandil: A Review of its Pharmacology and Therapeutic Efficacy in Angina Pectoris. Drugs 1992, 44, 625–655. [Google Scholar] [CrossRef] [PubMed]
- Serafim, R.A.M.; Pernichelle, F.G.; Ferreira, E.I.; Serafim, R.A.M.; Pernichelle, F.G.; The, E.I.F. The latest advances in the discovery of nitric oxide hybrid drug compounds. Expert Opin. Drug Discov. 2017, 12, 941–953. [Google Scholar] [CrossRef]
- Hoskins, J.A. The Occurrence, Metabolism and Toxicity of Cinnamic Acid and Related Compounds. J. Appl. Toxicol. 1984, 4, 283–292. [Google Scholar] [CrossRef]
- Guzman, J.D. Natural Cinnamic Acids, Synthetic Derivatives and Hybrids with Antimicrobial Activity. Molecules 2014, 19, 19292–19349. [Google Scholar] [CrossRef] [PubMed]
- Clifford, M.N. Chlorogenic acids and other cinnamates—Nature, occurrence, dietary burden, absorption and metabolism. J. Sci. Food Agric. 2000, 80, 1033–1043. [Google Scholar] [CrossRef]
- Sova, M. Antioxidant and Antimicrobial Activities of Cinnamic Acid Derivatives. Mini-Rev. Med. Chem. 2012, 12, 749–767. [Google Scholar] [CrossRef] [PubMed]
- Baltas, M. Cinnamic Acid Derivatives as Anticancer Agents—A Review. Curr. Med. Chem. 2011, 18, 1672–1703. [Google Scholar]
- Cássia, R.D.; Andrade, L.N.; Barreto, R.; Sousa, D.P.D. A Review on Anti-Inflammatory Activity of Phenylpropanoids Found in Essential Oils. Molecules 2014, 19, 1459–1480. [Google Scholar]
- Hafizur, R.M.; Hameed, A.; Shukrana, M.; Raza, S.A.; Chishti, S.; Kabir, N.; Siddiqui, R.A.; Hameed, A. Cinnamic acid exerts anti-diabetic activity by improving glucose tolerance in vivo and by stimulating insulin secretion in vitro. Phytomedicine 2015, 22, 297–300. [Google Scholar] [CrossRef]
- Bobadilla, R.A.; Muriel, P. Structure—Hepatoprotective Activity Relationship of 3,4-Dihydroxycinnamic Acid (Caffeic Acid) Derivatives. J. Appl. Toxicol. 2001, 21, 527–531. [Google Scholar]
- Gayam, V.; Ravi, S. Cinnamoylated chloroquine analogues: A new structural class of antimalarial agents. Eur. J. Med. Chem. 2017, 135, 382–391. [Google Scholar] [CrossRef] [PubMed]
- De, P.; De, K.; Veau, D.; Bedos-belval, F.; Chassaing, S.; Baltas, M. Recent advances in the development of cinnamic-like derivatives as antituberculosis agents. Expert Opin. 2012, 22, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, F.A.; Mascoti, L.A.; Sanchez, C.E.; Garibotto, F.R.; Giannini, F.E.; Sanz, M.A.K.; Enriz, R.I. Structure—Antifungal Activity Relationship of Cinnamic Acid Derivatives. J. Agric. Food Chem. 2007, 55, 10635–10640. [Google Scholar] [CrossRef] [PubMed]
- Pontiki, E.; Peperidou, A.; Fotopoulos, I.; Hadjipavlou Litina, D. Cinnamate Hybrids: A Unique Family of Compounds with Multiple Biological Activities Review. Curr. Pharm. Biotechnol. 2018, 19, 1019–1048. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef]
- Dubois, R.N.; Abramson, S.B.; Crofford, L. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar] [CrossRef] [Green Version]
- Rådmark, O.; Samuelsson, B. 5-Lipoxygenase: Mechanisms of regulation. J. Lipid Res. 2009, 50, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Pontiki, E.; Hadjipavlou-Litina, D. Synthesis and pharmacochemical evaluation of novel aryl-acetic acid inhibitors of lipoxygenase, antioxidants, and anti-inflammatory agents. Bioorg. Med. Chem. 2007, 15, 5819–5827. [Google Scholar] [CrossRef]
- Peperidou, A.; Pontiki, E.; Hadjipavlou-litina, D. Multifunctional Cinnamic Acid Derivatives. Molecules 2017, 22, 1247. [Google Scholar] [CrossRef]
- Bandarage, U.K.; Janero, D.R. Nitric Oxide-Releasing Nonsteroidal Anti-inflammatory Drugs: Novel Gastrointestinal-Sparing Drugs. Mini Rev. Med. Chem. 2001, 1, 57–70. [Google Scholar] [CrossRef]
- Arulselvan, P.; Fard, M.T.; Tan, W.S.; Gothai, S.; Fakurazi, S.; Norhaizan, M.E.; Kumar, S.S. Role of Antioxidants and Natural Products in Inflammation. Oxid. Med. Cell. Longev. 2016, 2016, 5276130. [Google Scholar] [CrossRef] [Green Version]
- Fotopoulos, I.; Pontiki, E.; Hadjipavlou Litina, D. Targeting Inflammation with Conjugated Cinnamic Amides, Ethers and Esters. Lett. Drug Des. Discov. 2020, 17, 3–11. [Google Scholar] [CrossRef]
- Theodosis-Nobelos, P.; Papagiouvanis, G.; Pantelidou, M.; Kourounakis, P.N.; Athanasekou, C.; Rekka, E.A. Design, synthesis and study of nitrogen monoxide donors as potent hypolipidaemic and anti-inflammatory agents. Molecules 2020, 25, 19. [Google Scholar] [CrossRef] [Green Version]
- Frostegård, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef] [Green Version]
- Li, N.G.; Wang, R.; Shi, Z.H.; Tang, Y.P.; Li, B.Q.; Wang, Z.J.; Song, S.L.; Qian, L.H.; Wei, L.; Yang, J.P.; et al. Design and synthesis of novel NO-donor-ferulic acid hybrids as potential antiatherosclerotic drug candidates. Drug Dev. Res. 2011, 72, 405–415. [Google Scholar]
- Li, N.G.; Wang, R.; Shi, Z.H.; Tang, Y.P.; Li, B.Q.; Wang, Z.J.; Song, S.L.; Qian, L.H.; Wei, L.; Yang, J.P.; et al. Design, Synthesis and Biological Study of Novel NO-Donor-Caffeic Acid Hybrids as Potential Anti-Atherosclerotic Drug Candidates. Lett. Drug Des. Discov. 2011, 72, 550–557. [Google Scholar] [CrossRef]
- Ling, Y.; Gao, W.J.; Ling, C.; Liu, J.; Meng, C.; Qian, J.; Liu, S.; Gan, H.; Wu, H.; Tao, J.; et al. β-Carboline and N-hydroxycinnamamide hybrids as anticancer agents for drug-resistant hepatocellular carcinoma. Eur. J. Med. Chem. 2019, 168, 515–526. [Google Scholar] [CrossRef]
- Wu, X.; Cheng, J.; Wang, X. Dietary Antioxidants: Potential Anticancer Agents. Nutr. Cancer 2017, 69, 521–533. [Google Scholar] [CrossRef]
- Hu, Y.; Xiang, J.; Su, L.; Tang, X. The regulation of nitric oxide in tumor progression and therapy. J. Int. Med. Res. 2020, 48, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, N.; Tang, Y.; Li, B.; Liu, L.; Zhang, X.; Fu, H.; Duan, J.A. Biological activity evaluation and structure-activity relationships analysis of ferulic acid and caffeic acid derivatives for anticancer. Bioorg. Med. Chem. Lett. 2012, 22, 6085–6088. [Google Scholar] [CrossRef]
- Holzgrabe, U.; Kapková, P.; Alptüzün, V.; Scheiber, J.; Kugelmann, E. Targeting acetylcholinesterase to treat neurodegeneration. Expert Opin. Ther. Targets 2007, 11, 161–179. [Google Scholar] [CrossRef]
- Arendt, T.; Brückner, M.K.; Lange, M.; Bigl, V. Changes in acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease resemble embryonic development-A study of molecular forms. Neurochem. Int. 1992, 21, 381–396. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, J.; Fang, L.; Liu, M.; Peng, S.; Liao, H.; Lehmann, J.; Zhang, Y. Tacrine-ferulic acid-nitric oxide (NO) donor trihybrids as potent, multifunctional acetyl- and butyrylcholinesterase inhibitors. J. Med. Chem. 2012, 55, 4309–4321. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Zhao, Q.; Min, Z.; Zhang, C.; Lai, Y.; Ji, H.; Peng, S.; Zhang, Y. Design, synthesis and evaluation of nitric oxide releasing derivatives of 3-n-butylphthalide as antiplatelet and antithrombotic agents. Org. Biomol. Chem. 2011, 9, 5670–5681. [Google Scholar] [CrossRef]
- Lu, M.D.; Zhou, X.; Yu, Y.J.; Li, P.H.; Sun, W.J.; Zhao, C.G.; Zheng, Z.Q.; You, T.; Wang, F.H. Synthesis and in vitro biological evaluation of nitric oxide-releasing derivatives of hydroxylcinnamic acids as anti-tumor agents. Chin. Chem. Lett. 2013, 24, 415–418. [Google Scholar] [CrossRef]
- Reddy, P.; Lent-schochet, D.; Ramakrishnan, N.; Mclaughlin, M. Metabolic syndrome is an inflammatory disorder: A conspiracy between adipose tissue and phagocytes. Clin. Chim. Acta 2019, 496, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, J.C.; Cardona, F.; Tinahones, F.J. Inflammation, Oxidative Stress and Metabolic Syndrome: Dietary Modulation. Curr. Vasc. Pharmacol. 2013, 11, 906–919. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Yang, Y.; Li, S.; Xu, Y.; Lu, W.; Chen, Z.; Yang, G.; Li, Y.; Cao, Y.; Bian, X. Phenylsulfonylfuroxan NO-donor phenols: Synthesis and multifunctional activities evaluation. Bioorg. Med. Chem. 2017, 25, 4407–4413. [Google Scholar] [CrossRef]
- Xie, Y.D.; Shao, L.H.; Wang, Q.T.; Bai, Y.; Li, N.; Yang, G.; Li, Y.P.; Bian, X.L. Design, synthesis and evaluation of phenylfuroxan nitric oxide-donor phenols as potential anti-diabetic agents. Bioorg. Chem. 2019, 89, 103000. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybrid | A549 | H157 | H460 | 1792 | H266 | Hop62 | 1299 | 292G | Calu1 |
|---|---|---|---|---|---|---|---|---|---|
| 9a | 13.2 | 20.5 | 29.0 | 28.5 | 44.0 | 19.4 | 28.3 | 39.4 | >50 |
| 9b | 8.82 | 12.9 | 19.5 | 20.8 | 24.8 | 13.1 | 4.51 | 28.4 | 30.1 |
| 9c | 6.39 | 13.4 | 12.3 | 13.0 | 21.2 | 12.5 | 20.8 | 24.9 | 26.3 |
| 9d | 37.4 | 32.1 | >50 | 48.7 | >50 | >50 | >50 | >50 | >50 |
| 9e | 5.95 | 4.29 | 4.98 | 4.86 | 9.79 | 5.13 | 2.34 | 21.9 | 2.48 |
| 10a | 10.8 | 4.34 | 3.31 | 18.1 | >50 | 10.0 | 8.03 | 13.5 | 10.7 |
| 10b | 22.6 | 5.19 | 3.52 | 15.4 | 23.3 | 10.0 | 3.14 | 16.7 | 21.7 |
| 10e | 0.40 | 1.36 | 2.90 | 0.41 | 10.3 | 4.65 | 0.41 | 7.95 | 0.42 |
| 11a | 15.8 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | 1.42 |
| 11b | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 11c | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 11d | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | 27.8 |
| 12a | 0.69 | 5.71 | 9.58 | 6.61 | >50 | 10.0 | >50 | 4.98 | 12.5 |
| 12b | 0.41 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 12c | 0.45 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 12d | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| Hybrid | Melanona | Cervical | Neck and Head | Breast | |
|---|---|---|---|---|---|
| LOX-IMVI | M14 | Hela | M4E | SKBR | |
| 9a | 29.5 | 42.6 | 6.70 | 18.4 | 17.2 |
| 9b | 20.4 | 32.5 | 6.35 | 20.3 | 8.92 |
| 9c | >50 | 24.5 | 13.4 | 21.8 | 8.06 |
| 9d | >50 | >50 | 16.3 | >50 | 15.2 |
| 9e | 2.36 | 3.89 | 1.00 | 9.38 | 6.53 |
| 10a | 18.9 | 24.8 | 3.11 | 20.0 | 5.77 |
| 10b | >50 | 19.9 | 2.25 | 17.5 | 4.55 |
| 10e | 0.43 | 1.49 | 0.40 | 4.31 | 0.41 |
| 11a | >50 | >50 | 3.62 | >50 | >50 |
| 11b | >50 | >50 | 0.74 | >50 | >50 |
| 11c | >50 | >50 | >50 | >50 | >50 |
| 11d | >50 | >50 | >50 | >50 | >50 |
| 12a | 6.94 | 8.96 | >50 | 14.1 | 4.81 |
| 12b | >50 | >50 | 3.93 | >50 | 3.23 |
| 12c | 7.23 | >50 | 5.73 | >50 | 2.71 |
| 12d | >50 | >50 | 1.35 | >50 | 39.7 |
| Hybrid | A549 | H157 | H460 | 1792 | H266 | Hop62 | 1299 | 292G | Calu1 |
|---|---|---|---|---|---|---|---|---|---|
| 17a | 2.06 | 4.53 | 7.03 | 11.1 | 13.4 | 8.66 | 12.9 | 16.5 | 22.9 |
| 17b | 0.72 | 24.7 | 11.6 | >50 | >50 | >50 | >50 | >50 | >50 |
| 17c | 3.39 | 43.1 | 15.8 | 8.15 | >50 | 30.4 | 28.5 | 31.0 | 28.8 |
| 17d | >50 | >50 | 29.4 | >50 | >50 | >50 | >50 | >50 | >50 |
| 17e | 17.2 | >50 | 44.2 | >50 | >50 | >50 | >50 | >50 | >50 |
| 18a | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 18b | 0.42 | 0.41 | 0.42 | 0.40 | 0.43 | 0.42 | 0.41 | 1.77 | 0.45 |
| 18c | 0.40 | 0.42 | 0.45 | 0.42 | 0.40 | 0.41 | 0.42 | 1.93 | 0.41 |
| 18d | 0.41 | 0.44 | 0.43 | 1.07 | 0.42 | 0.48 | 0.49 | 2.88 | 0.45 |
| 19a | 2.71 | >50 | 2.54 | 3.78 | 14.2 | 6.12 | >50 | 9.17 | 7.68 |
| Hybrid | Melanona | Cervical | Neck and Head | Breast | |
|---|---|---|---|---|---|
| LOX-IMVI | M14 | Hela | M4E | SKBR | |
| 17a | 13.8 | 14.0 | 7.82 | 3.37 | 4.98 |
| 17b | 49.4 | >50 | >50 | 44.6 | 14.2 |
| 17c | >50 | >50 | 12.1 | >50 | 11.0 |
| 17d | >50 | >50 | 5.64 | >50 | 4.18 |
| 17e | >50 | >50 | >50 | >50 | 11.7 |
| 18a | >50 | >50 | >50 | >50 | >50 |
| 18b | 0.41 | 0.41 | 0.40 | 1.18 | 0.40 |
| 18c | 0.44 | 0.40 | 0.46 | 0.84 | 0.42 |
| 18d | 0.42 | 0.41 | 0.43 | 1.12 | 0.41 |
| 19a | 13.3 | 12.5 | 1.23 | 12.9 | 0.70 |
| Compound | In Vitro Cytotoxicity | ||
|---|---|---|---|
| SMMC-7721 | HepG2 | MCF-7 | |
| 21 | 6.1 | 7.3 | 3.8 |
| 22 | 7.9 | 8.2 | 6.5 |
| 23 | 8.9 | 7.8 | 5.0 |
| 24 | 10.8 | 7.7 | 6.3 |
| 25 | 20.3 | 18.8 | 11.2 |
| Adriamycin | 0.9 | 2.1 | 1.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsopka, I.-C.; Hadjipavlou-Litina, D. Hybrids as NO Donors. Int. J. Mol. Sci. 2021, 22, 9788. https://doi.org/10.3390/ijms22189788
Tsopka I-C, Hadjipavlou-Litina D. Hybrids as NO Donors. International Journal of Molecular Sciences. 2021; 22(18):9788. https://doi.org/10.3390/ijms22189788
Chicago/Turabian StyleTsopka, Ioanna-Chrysoula, and Dimitra Hadjipavlou-Litina. 2021. "Hybrids as NO Donors" International Journal of Molecular Sciences 22, no. 18: 9788. https://doi.org/10.3390/ijms22189788
APA StyleTsopka, I.-C., & Hadjipavlou-Litina, D. (2021). Hybrids as NO Donors. International Journal of Molecular Sciences, 22(18), 9788. https://doi.org/10.3390/ijms22189788