
Role of Glycosylation in Vascular Calcification


Abstract
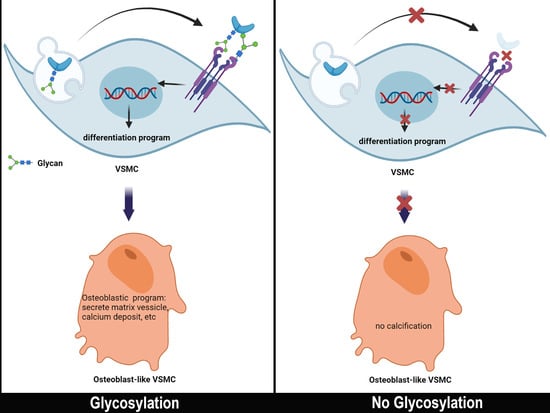
1. Introduction
2. Classification, Clinical Consequences, and Risk Factors of Vascular Calcification
3. Mechanism of Vascular Calcification
4. Regulatory Factors in Vascular Calcification
4.1. Calcification Inhibitors
4.2. Calcification Inducer
5. Glycosylation
6. N-glycosylation and Vascular Calcification
7. O-glycosylation and Vascular Calcification
8. Proteoglycan and Vascular Calcification
9. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Wilson, P.W.F.; Kauppila, L.I.; O’Donnell, C.J.; Kiel, D.P.; Hannan, M.; Polak, J.M.; Cupples, L.A. Abdominal aortic calcific deposits are an important predictor of vascular morbidity and mortality. Circulation 2001, 103, 1529–1534. [Google Scholar] [CrossRef]
- Budoff, M.J.; Shaw, L.J.; Liu, S.T.; Weinstein, S.R.; Mosler, T.P.; Tseng, P.H.; Flores, F.R.; Callister, T.Q.; Raggi, P.; Berman, D.S. Long-Term Prognosis Associated With Coronary Calcification. Observations From a Registry of 25,253 Patients. J. Am. Coll. Cardiol. 2007, 49, 1860–1870. [Google Scholar] [CrossRef]
- Garrison, L.P.; Lewin, J.; Young, C.H.; Généreux, P.; Crittendon, J.; Mann, M.R.; Brindis, R.G. The clinical and cost burden of coronary calcification in a Medicare cohort: An economic model to address under-reporting and misclassification. Cardiovasc. Revascularization Med. 2015, 16, 406–412. [Google Scholar] [CrossRef][Green Version]
- Hofmann Bowman, M.A.; McNally, E.M. Genetic Pathways of Vascular Calcification. Trends Cardiovasc. Med. 2012, 22, 93–98. [Google Scholar] [CrossRef]
- Lee, D. Vascular calcification: Inducers and inhibitors. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol. 2011, 176, 1133–1141. [Google Scholar] [CrossRef]
- Ngai, D.; Lino, M.; Bendeck, M.P. Cell-Matrix Interactions and Matricrine Signaling in the Pathogenesis of Vascular Calcification. Front. Cardiovasc. Med. 2018, 5, 1–16. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, I.K.; Jeon, J.H. Vascular calcification—New insights into its mechanism. Int. J. Mol. Sci. 2020, 21, 2685. [Google Scholar] [CrossRef] [PubMed]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.; Long, D.A.; Shanahan, C. Mechanistic insights into vascular calcification in CKD. J. Am. Soc. Nephrol. 2013, 24, 179–189. [Google Scholar] [CrossRef]
- Yang, P.; Troncone, L.; Augur, Z.M.; Kim, S.S.J.; McNeil, M.E.; Yu, P.B. The role of bone morphogenetic protein signaling in vascular calcification. Bone 2020, 141, 115542. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Dweck, M.R.; Narula, N.; Pisapia, D.; Narula, J.; Strauss, H.W. Coronary Artery Calcification: From Mechanism to Molecular Imaging. JACC Cardiovasc. Imaging 2017, 10, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Maehara, A.; Stone, G.W.; Généreux, P. Coronary Artery Calcification Pathogenesis and Prognostic Implications. J. Am. Coll. Cardiol. 2014, 63, 1703–1714. [Google Scholar] [CrossRef]
- Leopold, J.A. Vascular calcification: Mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc. Med. 2015, 25, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Shanahan, C.M. Medial Arterial Calcification: An Overlooked Player in Peripheral Arterial Disease. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1475–1482. [Google Scholar] [CrossRef]
- Demer, L.L.; Tintut, Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 715–723. [Google Scholar] [CrossRef]
- Giachelli, C.M. Vascular calcification mechanisms. J. Am. Soc. Nephrol. 2004, 15, 2959–2964. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.C.; Leopold, J.A.; Loscalzo, J. Vascular calcification: Pathobiological mechanisms and clinical implications. Circ. Res. 2006, 99, 1044–1059. [Google Scholar] [CrossRef]
- Sage, A.P.; Tintut, Y.; Demer, L.L. Regulatory mechanisms in vascular calcification. Nat. Rev. Cardiol. 2010, 7, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Westenfeld, R.; Schäfer, C.; Krüger, T.; Haarmann, C.; Schurgers, L.J.; Reutelingsperger, C.; Ivanosvski, O.; Drueke, T.; Massy, Z.A.; Ketteler, M.; et al. Fetuin-A protects against atherosclerotic calcification in CKD. J. Am. Soc. Nephrol. 2009, 20, 1264–1274. [Google Scholar] [CrossRef]
- Yamada, S.; Tokumoto, M.; Tsuruya, K.; Tatsumoto, N.; Noguchi, H.; Kitazono, T.; Ooboshi, H. Fetuin-A decrease induced by a low-protein diet enhances vascular calcification in uremic rats with hyperphosphatemia. Am. J. Physiol.-Ren. Physiol. 2015, 309, F744–F754. [Google Scholar] [CrossRef] [PubMed]
- Uedono, H.; Mori, K.; Ochi, A.; Nakatani, S.; Miki, Y.; Tsuda, A.; Morioka, T.; Nagata, Y.; Imanishi, Y.; Shoji, T.; et al. Effects of fetuin-A-containing calciprotein particles on posttranslational modifications of fetuin-A in HepG2 cells. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Jahnen-Dechent, W.; Heiss, A.; Schäfer, C.; Ketteler, M. Fetuin-A regulation of calcified matrix metabolism. Circ. Res. 2011, 108, 1494–1509. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.X.; O’Neill, K.D.; Chen, X.; Duan, D.; Wang, E.; Sturek, M.S.; Edwards, J.M.; Moe, S.M. Fetuin-A uptake in bovine vascular smooth muscle cells is calcium dependent and mediated by annexins. Am. J. Physiol.-Ren. Physiol. 2007, 292, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.T.; Chen, P.S.; Chen, P.W.; Tsai, L.M.; Liu, P.Y. Fetuin A adds prognostic value for cardiovascular outcomes among patients with coronary artery disease with moderate calcification. Int. J. Cardiol. 2015, 185, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.T.; Tsai, W.C.; Wu, C.H.; Lee, Y.W.; Tai, Y.L.; Li, Y.H.; Tsai, L.M.; Chen, J.H.; Liu, P.Y. Fetuin-A as a predicator of sarcopenic left ventricular dysfunction. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef]
- Hruska, K.A.; Mathew, S.; Saab, G. Bone morphogenetic proteins in vascular calcification. Circ. Res. 2005, 97, 105–114. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Wang, X.; Millán, J.L.; Dubyak, G.R.; O’Neill, W.C. Extracellular pyrophosphate metabolism and calcification in vascular smooth muscle. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, 61–68. [Google Scholar] [CrossRef]
- Yin, X.; Zhou, C.; Li, J.; Liu, R.; Shi, B.; Yuan, Q.; Zou, S. Autophagy in bone homeostasis and the onset of osteoporosis. Bone Res. 2019, 7, 1–16. [Google Scholar] [CrossRef]
- Peng, Y.Q.; Xiong, D.; Lin, X.; Cui, R.R.; Xu, F.; Zhong, J.Y.; Zhu, T.; Wu, F.; Mao, M.Z.; Liao, X.B.; et al. Oestrogen Inhibits Arterial Calcification by Promoting Autophagy. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Liang, J.; Huang, J.; He, W.; Shi, G.; Chen, J. β-Hydroxybutyric Inhibits Vascular Calcification via Autophagy Enhancement in Models Induced by High Phosphate. Front. Cardiovasc. Med. 2021, 8, 1–10. [Google Scholar] [CrossRef]
- Poniatowski, L.A.; Wojdasiewicz, P.; Gasik, R.; Szukiewicz, D. Transforming growth factor beta family: Insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediat. Inflamm. 2015, 1–17. [Google Scholar] [CrossRef]
- Watson, K.E.; Bostrom, K.; Ravindranath, R.; Lam, T.; Norton, B.; Demer, L.L. Osteoblast-like Vascular Cells to Calcify. J. Clin. Investig. 1994, 93, 2106–2113. [Google Scholar] [CrossRef]
- Jian, B.; Narula, N.; Li, Q.Y.; Mohler, E.R.; Levy, R.J. Progression of aortic valve stenosis: TGF-β1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann. Thorac. Surg. 2003, 75, 457–465. [Google Scholar] [CrossRef]
- Shimokado, A.; Sun, Y.; Nakanishi, M.; Sato, F.; Oikawa, K.; Akasaka, T.; Muragaki, Y. Smad3 plays an inhibitory role in phosphate-induced vascular smooth muscle cell calcification. Exp. Mol. Pathol. 2014, 97, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chen, G.; Li, Y.P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Warburton, D. Matrix Gla protein gene expression is induced by transforming growth factor-β in embryonic lung culture. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1997, 273, L282–L287. [Google Scholar] [CrossRef]
- Guerrero, F.; Herencia, C.; Almadén, Y.; Martínez-Moreno, J.M.; Montes De Oca, A.; Rodriguez-Ortiz, M.E.; Diaz-Tocados, J.M.; Canalejo, A.; Florio, M.; Lopez, I.; et al. TGF-β prevents phosphate-induced osteogenesis through inhibition of BMP and Wnt/β-catenin pathways. PLoS ONE 2014, 9, e89179. [Google Scholar] [CrossRef]
- Halling Linder, C.; Narisawa, S.; Millán, J.L.; Magnusson, P. Glycosylation differences contribute to distinct catalytic properties among bone alkaline phosphatase isoforms. Bone 2009, 45, 987–993. [Google Scholar] [CrossRef]
- Jimbo, R.; Kawakami-Mori, F.; Mu, S.; Hirohama, D.; Majtan, B.; Shimizu, Y.; Yatomi, Y.; Fukumoto, S.; Fujita, T.; Shimosawa, T. Fibroblast growth factor 23 accelerates phosphate-induced vascular calcification in the absence of Klotho deficiency. Kidney Int. 2014, 85, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Donate-Correa, J.; Martín-Núñez, E.; Hernández-Carballo, C.; Ferri, C.; Tagua, V.G.; Delgado-Molinos, A.; Lopez-Castillo, A.; Rodriguez-Ramoz, S.; Cerro-Lopez, P.; Lopez-Tarruella, V.C.; et al. Fibroblast growth factor 23 expression in human calcified vascular tissues. Aging 2019, 11, 7899–7913. [Google Scholar] [CrossRef]
- Alves, R.D.A.M.; Eijken, M.; van de Peppel, J.; van Leeuwen, J.P.T.M. Calcifying vascular smooth muscle cells and osteoblasts: Independent cell types exhibiting extracellular matrix and biomineralization-related mimicries. BMC Genomics 2014, 15, 1–14. [Google Scholar] [CrossRef]
- Chellan, B.; Rojas, E.; Zhang, C.; Hofmann Bowman, M.A. Enzyme-modified non-oxidized LDL (ELDL) induces human coronary artery smooth muscle cell transformation to a migratory and osteoblast-like phenotype. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Collett, G.D.M.; Canfield, A.E. Angiogenesis and pericytes in the initiation of ectopic calcification. Circ. Res. 2005, 96, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, D.; Hsu, C.Y.; Tian, Y.; Zhang, L.; Wang, Y.; Tower, R.J.; Chang, L.; Meyers, C.A.; Gao, Y.; et al. Comparison of skeletal and soft tissue pericytes identifies CXCR4+ bone forming mural cells in human tissues. Bone Res. 2020, 8, 1–14. [Google Scholar] [CrossRef]
- Doehring, L.C.; Heeger, C.; Aherrahrou, Z.; Kaczmarek, P.M.; Erdmann, J.; Schunkert, H.; Ehlers, E.M. Myeloid CD34+CD13+ precursor cells transdifferentiate into chondrocyte-like cells in atherosclerotic intimal calcification. Am. J. Pathol. 2010, 177, 473–480. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- van Tol, W.; Wessels, H.; Lefeber, D.J. O-glycosylation disorders pave the road for understanding the complex human O-glycosylation machinery. Curr. Opin. Struct. Biol. 2019, 56, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 729–749. [Google Scholar] [CrossRef]
- Bektas, M.; Rubenstein, D.S. The role of intracellular protein O-glycosylation in cell adhesion and disease. J. Biomed. Res. 2011, 25, 227–236. [Google Scholar] [CrossRef]
- Gamblin, D.P.; Scanlan, E.M.; Davis, B.G. Glycoprotein synthesis: An update. Chem. Rev. 2009, 109, 131–163. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef]
- Moremen, K.W.; Haltiwanger, R.S. Emerging structural insights into glycosyltransferase-mediated synthesis of glycans. Nat. Chem. Biol. 2019, 15, 853–864. [Google Scholar] [CrossRef]
- Clerc, F.; Reiding, K.R.; Jansen, B.C.; Kammeijer, G.S.M.; Bondt, A.; Wuhrer, M. Human plasma protein N-glycosylation. Glycoconj. J. 2016, 33, 309–343. [Google Scholar] [CrossRef] [PubMed]
- Spiro, R.G. Protein glycosylation: Nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 2002, 12, 43R–56R. [Google Scholar] [CrossRef]
- Wu, Y.M.; Liu, C.H.; Hu, R.H.; Huang, M.J.; Lee, J.; Chen, C.H.; Huang, J.; Lai, H.S.; Lee, P.H.; Hsu, W.M.; et al. Mucin glycosylating enzyme GALNT2 regulates the malignant character of hepatocellular carcinoma by modifying the EGF receptor. Cancer Res. 2011, 71, 7270–7279. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.G.; Pucci, M.; Venturi, G.; Malagolini, N.; Chiricolo, M.; Dall’Olio, F. Glycosylation as a main regulator of growth and death factor receptors signaling. Int. J. Mol. Sci. 2018, 19, 580. [Google Scholar] [CrossRef]
- Bagdonaite, I.; Pallesen, E.M.; Ye, Z.; Vakhrushev, S.Y.; Marinova, I.N.; Nielsen, M.I.; Kramer, S.H.; Pedersen, S.F.; Joshi, H.J.; Bennet, E.P.; et al. O-glycan initiation directs distinct biological pathways and controls epithelial differentiation. EMBO Rep. 2020, 21, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Klaver, E.; Zhao, P.; May, M.; Flanagan-Steet, H.; Freeze, H.H.; Gilmore, R.; Wells, L.; Contessa, J.; Steet, R. Selective inhibition of N-linked glycosylation impairs receptor tyrosine kinase processing. DMM Dis. Models Mech. 2019, 12, dmm039602. [Google Scholar] [CrossRef]
- Steentoft, C.; Vakhrushev, S.Y.; Joshi, H.J.; Kong, Y.; Vester-Christensen, M.B.; Schjoldager, K.T.B.G.; Lavrsen, K.; Dabelsteen, S.; Pedersen, N.B.; Marcos-Silva, L.; et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013, 32, 1478–1488. [Google Scholar] [CrossRef]
- Tajadura-Ortega, V.; Gambardella, G.; Skinner, A.; Halim, A.; Van Coillie, J.; Schjoldager, K.T.B.G.; Beatson, R.; Graham, R.; Achkova, D.; Taylor-Papadimitriou, J.; et al. O-linked mucin-type glycosylation regulates the transcriptional programme downstream of EGFR. Glycobiology 2021, 31, 200–210. [Google Scholar] [CrossRef]
- Tian, E.; Hoffman, M.P.; Ten Hagen, K.G. O-glycosylation modulates integrin and FGF signalling by influencing the secretion of basement membrane components. Nat. Commun. 2012, 3, 1–10. [Google Scholar] [CrossRef]
- Prydz, K.; Dalen, K.T. Synthesis and sorting of proteoglycans. J. Cell Sci. 2000, 113, 193–205. [Google Scholar] [CrossRef]
- Mikami, T.; Kitagawa, H. Biosynthesis and function of chondroitin sulfate. Biochim. Biophys. Acta-Gen. Subj. 2013, 1830, 4719–4733. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Freitas, D.; Campos, D.; Gomes, J.; Pinto, F.; Macedo, J.A.; Matos, R.; Mereiter, S.; Pinto, M.T.; Polonia, A.; Gartner, F.; et al. O-glycans truncation modulates gastric cancer cell signaling and transcription leading to a more aggressive phenotype. EBioMedicine 2019, 40, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Remigante, A.; Civello, D.A.; Bernardinelli, E.; Szabó, Z.; Morabito, R.; Marino, A.; Sarikas, A.; Patsch, W.; Paulmichi, M.; et al. O-GlcNAcylation Suppresses the Ion Current IClswell by Preventing the Binding of the Protein ICln to α-Integrin. Front. Cell Dev. Biol. 2020, 8, 1–23. [Google Scholar] [CrossRef]
- Pirillo, A.; Svecla, M.; Catapano, A.L.; Holleboom, A.G.; Norata, G.D. Impact of protein glycosylation on lipoprotein metabolism and atherosclerosis. Cardiovasc. Res. 2021, 117, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Holleboom, A.G.; Karlsson, H.; Lin, R.S.; Beres, T.M.; Sierts, J.A.; Herman, D.S.; Stroes, E.S.G.; Aerts, J.M.; Kastelein, J.J.P.; Motazacker, M.M.; et al. Heterozygosity for a loss-of-function mutation in GALNT2 improves plasma triglyceride clearance in man. Cell Metab. 2011, 14, 811–818. [Google Scholar] [CrossRef]
- Khetarpal, S.A.; Schjoldager, K.T.; Christoffersen, C.; Raghavan, A.; Edmondson, A.C.; Reutter, H.M.; Ahmed, B.; Ouazzani, R.; Peloso, G.M.; Vitali, C.; et al. Loss of Function of GALNT2 Lowers High-Density Lipoproteins in Humans, Nonhuman Primates, and Rodents. Cell Metab. 2016, 24, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Shrikhande, G.V.; Scali, S.T.; da Silva, C.G.; Damrauer, S.M.; Csizmadia, E.; Putheti, P.; Matthey, M.; Arjoon, R.; Patel, R.; Siracuse, J.J.; et al. O-Glycosylation regulates ubiquitination and degradation of the anti-inflammatory protein A20 to accelerate atherosclerosis in diabetic ApoE-null mice. PLoS ONE 2010, 5, e14240. [Google Scholar] [CrossRef]
- Liu, Y.-W.; Huang, M.-S.; Hsu, L.-W.; Chang, H.-Y.; Lee, C.-H.; Lee, C.-Y.; Chen, D.P.; Li, Y.H.; Chao, T.H.; Su, P.F.; et al. Genetic risk model for instent restenosis of second and third generation drug eluting stents. IScience. 2021, 24. in press. [Google Scholar] [CrossRef]
- Adhikara, I.M.; Yagi, K.; Mayasari, D.S.; Suzuki, Y.; Ikeda, K.; Ryanto, G.R.T.; Sasaki, N.; Rikitake, Y.; Nadanaka, S.; Kitagawa, H.; et al. Chondroitin sulfate N-acetylgalactosaminyltransferase-2 impacts foam cell formation and atherosclerosis by altering macrophage glycosaminoglycan chain. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1076–1091. [Google Scholar] [CrossRef]
- Kato, K.; Jeanneau, C.; Tarp, M.A.; Benet-Pagès, A.; Lorenz-Depiereux, B.; Bennett, E.P.; Mandel, U.; Strom, T.M.; Clausen, H. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis: Secretion of fibroblast growth factor 23 requires O-glycosylation. J. Biol. Chem. 2006, 281, 18370–18377. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, C.A.; Kawane, T.; Moriishi, T.; Purushothaman, A.; Miyazaki, T.; Komori, H.; Mori, M.; Qin, X.; Hashimoto, A.; Sugahara, K.; et al. Overexpression of Galnt3 in chondrocytes resulted in dwarfism due to the increase of mucin-type O-glycans and reduction of glycosaminoglycans. J. Biol. Chem. 2014, 289, 26584–26596. [Google Scholar] [CrossRef]
- Andrés-Bergós, J.; Tardio, L.; Larranaga-Vera, A.; Gómez, R.; Herrero-Beaumont, G.; Largo, R. The increase in O-linked N-acetylglucosamine protein modification stimulates chondrogenic differentiation both in vitro and in vivo. J. Biol. Chem. 2012, 287, 33615–33628. [Google Scholar] [CrossRef] [PubMed]
- Siddals, K.W.; Allen, J.; Sinha, S.; Canfield, A.E.; Kalra, P.A.; Martin Gibson, J. Apposite insulin-like growth factor (IGF) receptor glycosylation is critical to the maintenance of vascular smooth muscle phenotype in the presence of factors promoting osteogenic differentiation and mineralization. J. Biol. Chem. 2011, 286, 16623–16630. [Google Scholar] [CrossRef] [PubMed]
- Hang, Q.; Zhou, Y.; Hou, S.; Zhang, D.; Yang, X.; Chen, J.; Ben, Z.; Cheng, C.; Shen, A. Asparagine-linked glycosylation of bone morphogenetic protein-2 is required for secretion and osteoblast differentiation. Glycobiology 2014, 24, 292–304. [Google Scholar] [CrossRef]
- Wen, X.; Liu, A.; Yu, C.; Wang, L.; Zhou, M.; Wang, N.; Fang, M.; Wang, W.; Lin, H. Inhibiting post-translational core fucosylation prevents vascular calcification in the model of uremia. Int. J. Biochem. Cell Biol. 2016, 79, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Heath, J.M.; Sun, Y.; Yuan, K.; Bradley, W.E.; Litovsky, S.; Dell’Italia, L.J.; Catham, J.J.; Wu, H.; Chen, Y. Activation of AKT by O-linked N-Acetylglucosamine induces vascular calcification in diabetes mellitus. Circ. Res. 2014, 114, 1094–1102. [Google Scholar] [CrossRef]
- Xu, T.H.; Sheng, Z.; Li, Y.; Qiu, X.; Tian, B.; Yao, L. OGT knockdown counteracts high phosphate-induced vascular calcification in chronic kidney disease through autophagy activation by downregulating YAP. Life Sci. 2020, 261, 118121. [Google Scholar] [CrossRef]
- de las Rivas, M.; Paul Daniel, E.J.; Narimatsu, Y.; Compañón, I.; Kato, K.; Hermosilla, P.; Thureau, A.; Ceballos-Laita, L.; Coelho, H.; Bernado, P.; et al. Molecular basis for fibroblast growth factor 23 O-glycosylation by GalNAc-T3. Nat. Chem. Biol. 2020, 16, 351–360. [Google Scholar] [CrossRef]
- Sun, Y.; Byon, C.H.; Yuan, K.; Chen, J.; Mao, X.; Heath, J.M.; Javed, A.; Zhang, K.; Anderson, P.G.; Chen, Y. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ. Res. 2012, 111, 543–552. [Google Scholar] [CrossRef]
- Lin, M.E.; Chen, T.; Leaf, E.M.; Speer, M.Y.; Giachelli, C.M. Runx2 Expression in Smooth Muscle Cells Is Required for Arterial Medial Calcification in Mice. Am. J. Pathol. 2015, 185, 1958–1969. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Shigematsu, T.; Hatamura, I.; Saji, F.; Mune, S.; Kunimoto, K.; Hanba, Y.; Shiizaki, K.; Sakaguchi, T.; Negi, S. Reduced expression of perlecan in the aorta of secondary hyperparathyroidism model rats with medial calcification. Ren. Fail. 2010, 32, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Purnomo, E.; Emoto, N.; Nugrahaningsih, D.A.A.; Nakayama, K.; Yagi, K.; Heiden, S.; Nadanaka, S.; Kitagawa, H.; Hirata, K. Glycosaminoglycan overproduction in the aorta increases aortic calcification in murine chronic kidney disease. J. Am. Heart Assoc. 2013, 2, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Liang, Q.; Chen, Y.; Yang, P.; Liu, X.; Li, Y.; Feng, S.; Wu, J.; Liu, W.; Tang, J.; et al. Hyaluronan negatively regulates vascular calcification involving BMP2 signaling. Lab Investig. 2018, 98, 1320–1332. [Google Scholar] [CrossRef]
- Borland, S.J.; Morris, T.G.; Borland, S.C.; Morgan, M.R.; Francis, S.E.; Merry, C.L.R.; Canfield, A.E. Regulation of vascular smooth muscle cell calcification by syndecan-4/FGF-2/PKCα signalling and cross-talk with TGFβ. Cardiovasc. Res. 2017, 113, 1639–1652. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.Y.; Giachelli, C.M. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 2008, 199, 271–277. [Google Scholar] [CrossRef]
- Rahman, M.S.; Akhtar, N.; Jamil, H.M.; Banik, R.S.; Asaduzzaman, S.M. TGF-β/BMP signaling and other molecular events: Regulation of osteoblastogenesis and bone formation. Bone Res. 2015, 3, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Van De Watering, F.C.J.; Van Den Beucken, J.J.J.P.; Van Der Woning, S.P.; Briest, A.; Eek, A.; Qureshi, H.; Winnubst, L.; Boerman, O.C.; Jansen, J.A. Non-glycosylated BMP-2 can induce ectopic bone formation at lower concentrations compared to glycosylated BMP-2. J. Control. Release 2012, 159, 69–77. [Google Scholar] [CrossRef]
- Lowery, J.W.; Amich, J.M.; Andonian, A.; Rosen, V. N-linked glycosylation of the bone morphogenetic protein receptor type 2 (BMPR2) enhances ligand binding. Cell Mol. Life Sci. 2014, 71, 3165–3172. [Google Scholar] [CrossRef][Green Version]
- Sako, D.; Grinberg, A.V.; Liu, J.; Davies, M.V.; Castonguay, R.; Maniatis, S.; Andreucci, A.J.; Pobre, E.G.; Tomkinson, K.N.; Monell, T.E.; et al. Characterization of the ligand binding functionality of the extracellular domain of activin receptor type IIB. J. Biol. Chem. 2010, 285, 21037–21048. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Into, T.; Lowenstein, C.J.; Matsushita, K. Nitric oxide regulates vascular calcification by interfering with TGF-β signalling. Cardiovasc. Res. 2008, 77, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Sha, X.; Brunner, A.M.; Purchio, A.F.; Gentry, L.E. Transforming growth factor β1, Importance of glycosylaytion and acidic proteases for processing and secretion. Mol. Endocrinol. 1989, 3, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Misawa, M.; Matsuzaki, T.; Sakurai, T.; Muramatsu, T.; Sato, M. A novel glycosylation signal regulates transforming growth factor receptors as evidenced by endo-Galactosidase C expression in rodent cells. Glycobiology 2011, 21, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Franc, V.; Heck, A.J.R. Similar Albeit Not the Same: In-Depth Analysis of Proteoforms of Human Serum, Bovine Serum, and Recombinant Human Fetuin. J. Proteome Res. 2018, 17, 2861–2869. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Ford, M.L.; Tomlinson, L.A.; Rajkumar, C.; McMahon, L.P.; Holt, S.G. Phosphorylated fetuin-A-containing calciprotein particles are associated with aortic stiffness and a procalcific milieu in patients with pre-dialysis CKD. Nephrol. Dial. Transpl. 2012, 27, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Bellia, C.; Agnello, L.; Lo Sasso, B.; Milano, S.; Bivona, G.; Scazzone, C.; Pivetti, A.; Novo, G.; Palermo, C.; Bonomo, V.; et al. Fetuin-A is Associated to Serum Calcium and AHSG T256S Genotype but Not to Coronary Artery Calcification. Biochem. Genet. 2016, 54, 222–231. [Google Scholar] [CrossRef]
- Mohammadi-Noori, E.; Salehi, N.; Mozafari, H.; Elieh Ali Komi, D.; Saidi, M.; Bahrehmand, F.; Vaisi-Raygani, A.; Elahirad, S.; Moini, A.; Kiani, A. Association of AHSG gene polymorphisms with serum Fetuin-A levels in individuals with cardiovascular calcification in west of Iran. Mol. Biol. Rep. 2020, 47, 1809–1820. [Google Scholar] [CrossRef]
- Lin, Y.H.; Zhu, J.; Meijer, S.; Franc, V.; Heck, A.J.R. Glycoproteogenomics: A frequent gene polymorphism affects the glycosylation pattern of the human serum fetuin/α-2-HS-Glycoprotein. Mol. Cell Proteom. 2019, 18, 1479–1490. [Google Scholar] [CrossRef]
- Nielsen, M.I.; Stegmayr, J.; Grant, O.C.; Yang, Z.; Nilsson, U.J.; Boos, I.; Carlsson, M.C.; Woods, R.J.; Unverzagt, C.; Leffler, H.; et al. Galectin binding to cells and glycoproteins with genetically modified glycosylation reveals galectin–glycan specificities in a natural context. J. Biol. Chem. 2018, 293, 20249–20262. [Google Scholar] [CrossRef]
- Chen, P.W.; Hsu, L.W.; Chang, H.Y.; Huang, T.C.; Yu, J.R.; Liao, H.Y.; Lee, C.H.; Liu, P.Y. Elevated platelet galectin-3 and rho-associated protein kinase activity are associated with hemodialysis arteriovenous shunt dysfunction among subjects with diabetes mellitus. Biomed. Res. Int. 2019. [Google Scholar] [CrossRef]
- Iacobini, C.; Fantauzzi, C.B.; Pugliese, G.; Menini, S. Role of galectin-3 in bone cell differentiation, bone pathophysiology and vascular osteogenesis. Int. J. Mol. Sci. 2017, 18, 2481. [Google Scholar] [CrossRef]
- Zhang, Q.; Yin, K.; Ni, Z. Galectin-3 and abdominal aortic calcification in patients on hemodialysis. Vasc. Med. 2020, 25, 575–576. [Google Scholar] [CrossRef]
- Ibarrola, J.; Martínez-Martínez, E.; Sádaba, J.R.; Arrieta, V.; García-Peña, A.; Álvarez, V.; Fernandez-Celiz, A.; Gainza, A.; Rossignol, P.; Ramos, V.C.; et al. Beneficial effects of galectin-3 blockade in vascular and aortic valve alterations in an experimental pressure overload model. Int. J. Mol. Sci. 2017, 18, 1664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qiao, Y.; Wu, Q.; Chen, Y.; Zou, S.; Liu, X.; Zhu, G.; Zhao, Y.; Chen, Y.; Yu, Y.; et al. The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chennupati, R.; Jin, Y.J.; Li, R.; Wang, S.P.; Günther, S.; Offermanns, S. YAP/TAZ Are Required to Suppress Osteogenic Differentiation of Vascular Smooth Muscle Cells. IScience 2020, 23, 101860. [Google Scholar] [CrossRef] [PubMed]
- Uemura, M.; Nagasawa, A.; Terai, K. Yap/Taz transcriptional activity in endothelial cells promotes intramembranous ossification via the BMP pathway. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.H.; Du, Y.; Sheng, Z.; Li, Y.; Qiu, X.; Tian, B.; Yao, L. OGT-Mediated KEAP1 Glycosylation Accelerates NRF2 Degradation Leading to High Phosphate-Induced Vascular Calcification in Chronic Kidney Disease. Front. Physiol. 2020, 11, 1092. [Google Scholar] [CrossRef]
- Dai, X.Y.; Zhao, M.M.; Cai, Y.; Guan, Q.C.; Zhao, Y.; Guan, Y.; Kong, W.; Zhu, W.G.; Xu, M.J.; Wang, X. Phosphate-induced autophagy counteracts vascular calcification by reducing matrix vesicle release. Kidney Int. 2013, 83, 1042–1051. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Han, Y.; Chen, Y.; Liu, L.; Lang, J.; Yang, C.; Luo, H.; Ning, J. Advanced glycation end-products suppress autophagy by AMPK/mTOR signaling pathway to promote vascular calcification. Mol. Cell Biochem. 2020, 471, 91–100. [Google Scholar] [CrossRef]
- Thompson, B.; Towler, D.A. Arterial calcification and bone physiology: Role of the bone-vascular axis. Nat. Rev. Endocrinol. 2012, 8, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Quarles, L.D. How fibroblast growth factor 23 works. J. Am. Soc. Nephrol. 2007, 18, 1637–1647. [Google Scholar] [CrossRef]
- Scialla, J.J.; Lau, W.L.; Reilly, M.P.; Isakova, T.; Yang, H.Y.; Crouthamel, M.H.; Chavkin, N.W.; Rahman, M.; Wahl, P.; Amaral, A.P.; et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int. 2013, 83, 1159–1168. [Google Scholar] [CrossRef]
- Sprecher, E. Familial tumoral calcinosis: From characterization of a rare phenotype to the pathogenesis of ectopic calcification. J. Investig. Dermatol. 2010, 130, 652–660. [Google Scholar] [CrossRef]
- Duncan, E.L.; Danoy, P.; Kemp, J.P.; Leo, P.J.; McCloskey, E.; Nicholson, G.C.; Eastell, R.; Prince, R.L.; Eisman, J.A.; Jones, G.; et al. Genome-wide association study using extreme truncate selection identifies novel genes affecting bone mineral density and fracture risk. PLoS Genet. 2011, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, L.; Liabeuf, S.; Renard, C.; Lenglet, A.; Lemke, H.D.; Choukroun, G.; Drueke, T.B.; Massy, Z.A. FGF23 is independently associated with vascular calcification but not bone mineral density in patients at various CKD stages. Osteoporos. Int. 2012, 23, 2017–2025. [Google Scholar] [CrossRef]
- Freedman, B.I.; Divers, J.; Russell, G.B.; Palmer, N.D.; Bowden, D.W.; Carr, J.J.; Wagenknecht, L.E.; Hightower, R.C.; Xu, J.; Smith, S.C.; et al. Plasma FGF23 and Calcified Atherosclerotic Plaque in African Americans with Type 2 Diabetes Mellitus. Am. J. Nephrol. 2015, 42, 391–401. [Google Scholar] [CrossRef]
- Chen, Y.X.; Huang, C.; Duan, Z.B.; Xu, C.Y.; Chen, Y. Klotho/FGF23 axis mediates high phosphate-induced vascular calcification in vascular smooth muscle cells via Wnt7b/β-catenin pathway. Kaohsiung J. Med. Sci. 2019, 35, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, K.; Olauson, H.; Amin, R.; Ponnusamy, A.; Goetz, R.; Taylor, R.F.; Mohammadi, M.; Canfield, A.; Kublickiene, K.; Larsson, T.E. Arterial Klotho Expression and FGF23 Effects on Vascular Calcification and Function. PLoS ONE 2013, 8, e60658. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, Y.H.; Song, M.; An, S.H.; Byun, H.Y.; Heo, K.; Lim, S.; Oh, Y.S.; Ryu, S.H.; Suh, P.G. O-GlcNAc modification modulates the expression of osteocalcin via OSE2 and Runx2. Biochem. Biophys. Res. Commun. 2007, 362, 325–329. [Google Scholar] [CrossRef]
- Jonason, J.H.; Xiao, G.; Zhang, M.; Xing, L.; Chen, D. Post-translational regulation of Runx2 in bone and cartilage. J. Dent. Res. 2009, 88, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Nagel, A.K.; Ball, L.E. O-GlcNAc modification of the runt-related transcription factor 2 (Runx2) links osteogenesis and nutrient metabolism in bone marrow mesenchymal stem cells. Mol. Cell Proteom. 2014, 13, 3381–3395. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N. A Role for Proteoglycan in Vascular Disease. Matrix Biol. 2018, 71–72, 396–420. [Google Scholar] [CrossRef]
- Wight, T.N.; Merrilees, M.J. Proteoglycans in atherosclerosis and restenosis: Key roles for versican. Circ. Res. 2004, 94, 1158–1167. [Google Scholar] [CrossRef]
- Little, P.J.; Tannock, L.; Olin, K.L.; Chait, A.; Wight, T.N. Proteoglycans synthesized by arterial smooth muscle cells in the presence of transforming growth factor-β1 exhibit increased binding to LDLs. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 55–60. [Google Scholar] [CrossRef]
- Nagy, N.; Kuipers, H.F.; Frymoyer, A.R.; Ishak, H.D.; Bollyky, J.B.; Wight, T.N.; Bollyky, P.L. 4-Methylumbelliferone treatment and hyaluronan inhibition as a therapeutic strategy in inflammation, autoimmunity, and cancer. Front. Immunol. 2015, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Cui, W.; Liu, B.; Hu, H.; Liu, J.; Xie, R.; Yang, X.; Gu, G.; Zhang, J.; Zheng, H. Atorvastatin protects vascular smooth muscle cells from TGF-β1-stimulated calcification by inducing autophagy via suppression of the β-catenin pathway. Cell Physiol. Biochem. 2014, 33, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.; Wakabayashi, M.; Hata, K.; Matsubara, T.; Honma, S.; Wakisaka, S.; Kiyonari, H.; Shioi, G.; Yamaguchi, A.; Tsumaki, N.; et al. Osterix regulates calcification and degradation of chondrogenic matrices through matrix metalloproteinase 13 (MMP13) expression in association with transcription factor Runx2 during endochondral ossification. J. Biol. Chem. 2012, 287, 33179–33190. [Google Scholar] [CrossRef]
- Kiyono, K.; Suzuki, H.I.; Matsuyama, H.; Morishita, Y.; Komuro, A.; Kano, M.R.; Sugimoto, K.; Miyazono, K. Autophagy is activated by TGF-β and potentiates TGF-β-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 2009, 69, 8844–8852. [Google Scholar] [CrossRef]
- Su, Z.; Zong, P.; Chen, J.; Yang, S.; Shen, Y.; Lu, Y.; Yang, C.; Kong, X.; Sheng, Y.; Sun, W. Celastrol attenuates arterial and valvular calcification via inhibiting BMP2/Smad1/5 signalling. J. Cell. Mol. Med. 2020, 24, 12476–12490. [Google Scholar] [CrossRef] [PubMed]


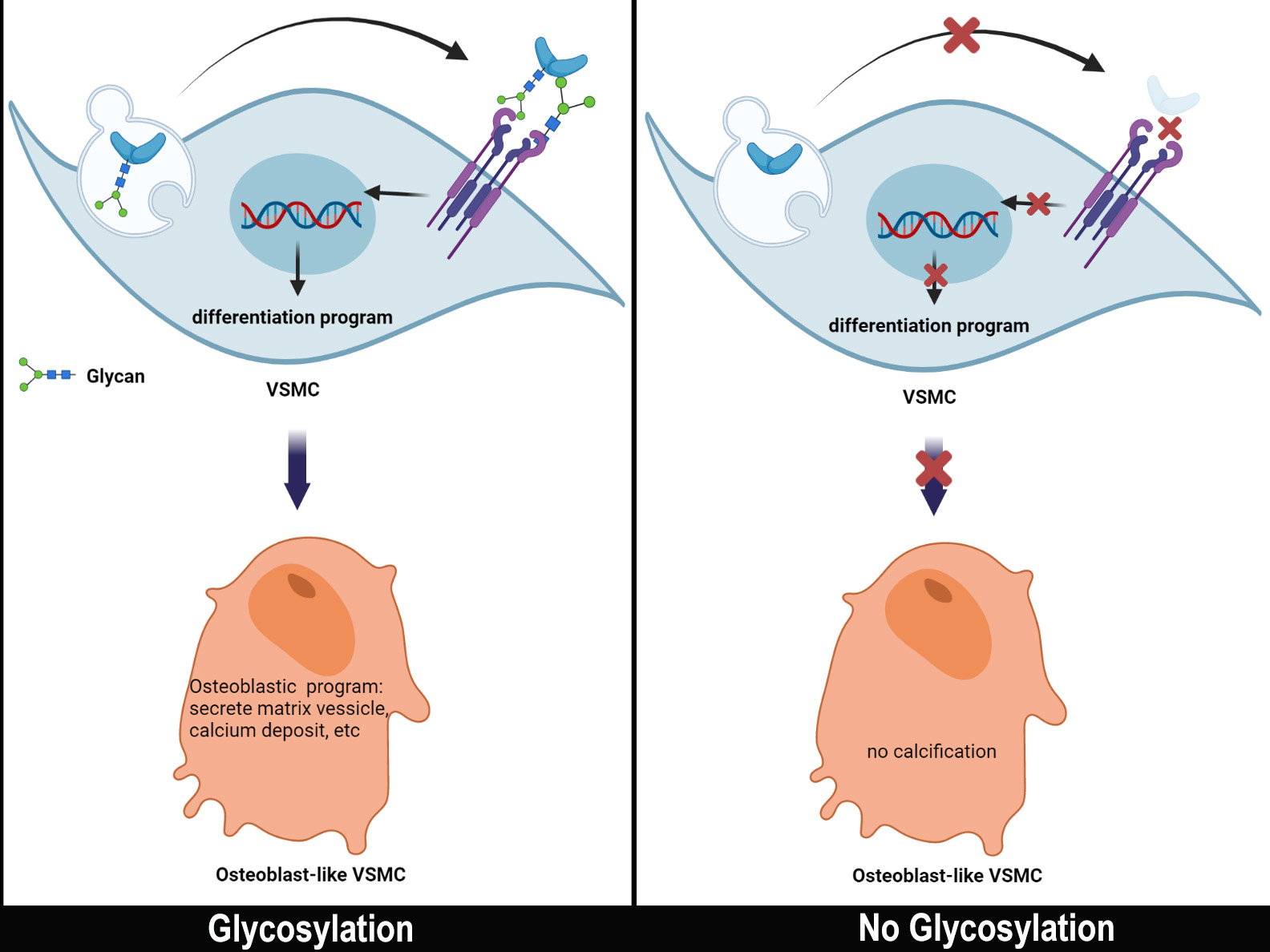{kind=link}
{kind=link}
| Enzyme | Glycosylation Type | Glycosylated Protein | Effect on VC | Reference |
|---|---|---|---|---|
| N/A | N-glycosylation | IGFR1 | Decrease | [76] |
| N/A | N-glycosylation | BMP2, BMP2R | Increase | [77] |
| N/A | N-glycosylation | Fetuin-A | Decrease | |
| N/A | N-glycosylation | TGFβR | Increase | [78] |
| OGT | O-glycosylation (GlcNac) | AKT | Increase | [79] |
| OGT | O-glicosylation (GlcNac) | YAP | Increase | [80] |
| GALNT3 | O-glycosylation (GalNac) | FGF23 | Increase | [81] |
| OGT, OGA | O-glycosylation | Runx2 | Increase | [82,83] |
| EXT1, EXTL2 | Mixed | Proteoglycan | Various | [84,85,86,87] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masbuchin, A.N.; Rohman, M.S.; Liu, P.-Y. Role of Glycosylation in Vascular Calcification. Int. J. Mol. Sci. 2021, 22, 9829. https://doi.org/10.3390/ijms22189829
Masbuchin AN, Rohman MS, Liu P-Y. Role of Glycosylation in Vascular Calcification. International Journal of Molecular Sciences. 2021; 22(18):9829. https://doi.org/10.3390/ijms22189829
Chicago/Turabian StyleMasbuchin, Ainun Nizar, Mohammad Saifur Rohman, and Ping-Yen Liu. 2021. "Role of Glycosylation in Vascular Calcification" International Journal of Molecular Sciences 22, no. 18: 9829. https://doi.org/10.3390/ijms22189829
APA StyleMasbuchin, A. N., Rohman, M. S., & Liu, P.-Y. (2021). Role of Glycosylation in Vascular Calcification. International Journal of Molecular Sciences, 22(18), 9829. https://doi.org/10.3390/ijms22189829