
The Role of Sonic Hedgehog in Human Holoprosencephaly and Short-Rib Polydactyly Syndromes


Abstract
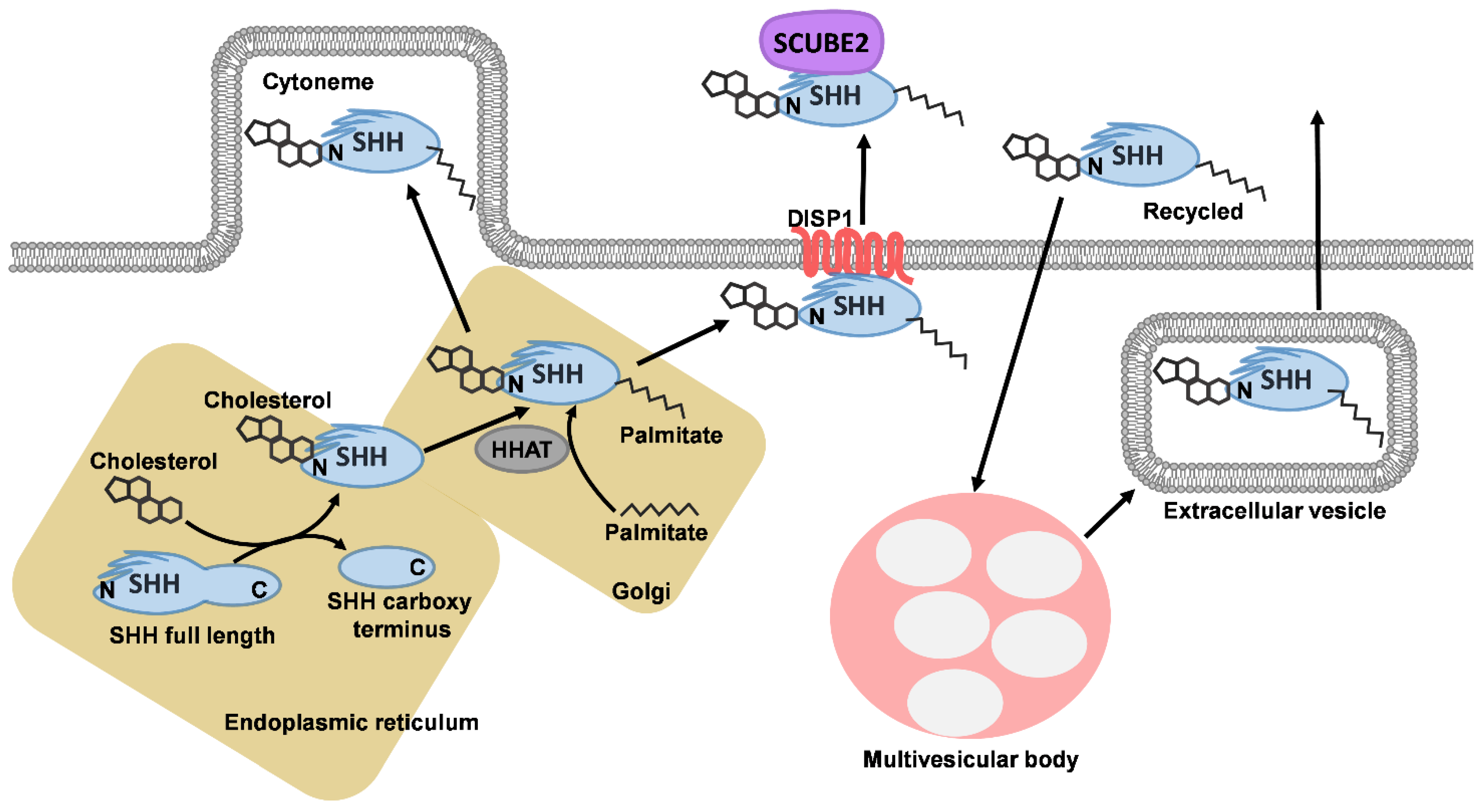
1. Sonic Hedgehog Signalling
2. Primary Cilia and Sonic Hedgehog Signalling
3. Non-Canonical Sonic Hedgehog Signalling and Modifiers of Signalling
4. Sonic Hedgehog and Holoprosencephaly
Environmental Factors and Holoprosencephaly
5. Human Ciliopathies
6. Short-Rib Polydactyly Syndromes
7. Comment
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ARL13B | ADP ribosylation factor like GRPase 13B |
| BMP | bone morphogenetic protein |
| BOC | brother of CDON |
| CDON | cell adhesion molecule-related/down regulated by oncogenes |
| COS2 | COSTAL 2 |
| DHCR7 | 7 Dehydrocholesterol reductase |
| DHH | Desert hedgehog |
| DISP1 | Dispatched 1 |
| DYNC2H1 | Dynein cytoplasmic 2 heavy chain 1 |
| EGF | Epidermal growth factor |
| FGF | Fibroblast growth factor |
| FGFR | fibroblast growth factor receptor |
| GAS1 | Growth arrest specific 1 |
| GLI | Glioma-associated (family of transcription factors) |
| GPCR | G protein coupled receptor |
| GTPase | Guanosine triphosphatase |
| HH | Hedgehog |
| HHAT | Hedgehog acyl transferase |
| HHIP | Hedgehog interacting protein |
| IFT | Intraflagellar transport |
| IGF | Insulin-like growth factor |
| IHH | Indian hedgehog |
| KIF | Kinesin family |
| LRP2 | low density lipotein (LDL) receptor-related protein 2 (also known as megalin) |
| mTOR | mammalian target of rapamycin |
| PDGFα | Platelet derived growth factor alpha |
| PKA | Protein kinase A |
| PTCH | Patched |
| SBE7 | Sonic Hedgehog brain enhancer 7 |
| SCUBE2 | signal peptide, CUB domain and EGF domain containing protein 2 |
| SHH | Sonic Hedgehog |
| SIX3 | SIX homeobox 3 |
| SLO | Smith-Lemli-Opitz |
| SMO | Smoothened |
| SRP | Short rib polydactyly |
| SRTD | Short rib thoracic dysplasia |
| SUFU | Suppressor of Fused |
| TGFβ | Transforming growth factor beta |
| TGIF | thymine guanine-interacting factor |
| VACTERL | Vertebral Anal Cardiac Tracheo Oesphageal Renal Limb |
| WDR | WD40-repeat (a series of loosely conserved structural motifs comprised of about 40 amino acids, often terminating in tryptophan (W) and aspartic acid (D)) |
| WNT | Wingless and Int-1 |
| ZIC2 | ZIC gene family 2 |
| ZPA | Zone of polarising activity |
References
- Teperino, R.; Aberger, F.; Esterbauer, H.; Riobo, N.; Pospisilik, J.A. Canonical and non-canonical Hedgehog signalling and the control of metabolism. Semin. Cell Dev. Biol. 2014, 33, 81–92. [Google Scholar] [CrossRef]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [PubMed]
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Odent, S.; Atti-Bitach, T.; Blayau, M.; Mathieu, M.; Aug, J.; Delezo de, A.L.; Gall, J.Y.; Le Marec, B.; Munnich, A.; David, V.; et al. Expression of the Sonic Hedgehog (SHH ) gene during early human development and phenotypic expression of new mutations causing holoprosencephaly. Hum. Mol. Genet. 1999, 8, 1683–1689. [Google Scholar] [CrossRef] [PubMed]
- Kopinke, D.; Norris, A.M.; Mukhopadhyay, S. Developmental and regenerative paradigms of cilia regulated hedgehog signaling. Semin. Cell Dev. Biol. 2021, 110, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Wierbowski, B.M.; Petrov, K.; Aravena, L.; Gu, G.; Xu, Y.; Salic, A. Hedgehog Pathway Activation Requires Coreceptor-Catalyzed, Lipid-Dependent Relay of the Sonic Hedgehog Ligand. Dev. Cell 2020, 55, 450–467.e8. [Google Scholar] [CrossRef]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic Hedgehog gene function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef]
- Matusek, T.; Marcetteau, J.; Therond, P.P. Functions of Wnt and Hedgehog-containing extracellular vesicles in development and disease. J. Cell Sci. 2020, 133, 18. [Google Scholar] [CrossRef]
- Kornberg, T.B.; Roy, S. Cytonemes as specialized signaling filopodia. Development 2014, 141, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Sasai, N.; Toriyama, M.; Kondo, T. Hedgehog Signal and Genetic Disorders. Front. Genet. 2019, 10, 1103. [Google Scholar] [CrossRef]
- Hall, E.T.; Cleverdon, E.R.; Ogden, S.K. Dispatching Sonic Hedgehog: Molecular Mechanisms Controlling Deployment. Trends Cell Biol. 2019, 29, 385–395. [Google Scholar]
- Andreu-Cervera, A.; Catala, M.; Schneider-Maunoury, S. Cilia, ciliopathies and hedgehog-related forebrain developmental disorders. Neurobiol. Dis. 2021, 150, 105236. [Google Scholar]
- Kong, J.H.; Siebold, C.; Rohatgi, R. Biochemical mechanisms of vertebrate hedgehog signaling. Development 2019, 146, 10. [Google Scholar] [CrossRef]
- Izzi, L.; Levesque, M.; Morin, S.; Laniel, D.; Wilkes, B.C.; Mille, F.; Krauss, R.S.; McMahon, A.P.; Allen, B.L.; Charron, F. Boc and Gas1 each form distinct Shh receptor complexes with Ptch1 and are required for Shh-mediated cell proliferation. Dev. Cell 2011, 20, 788–801. [Google Scholar] [CrossRef]
- Echevarria-Andino, M.L.; Allen, B.L. The hedgehog co-receptor BOC differentially regulates SHH signaling during craniofacial development. Development 2020, 147, 23. [Google Scholar] [CrossRef]
- Gigante, E.D.; Caspary, T. Signaling in the primary cilium through the lens of the Hedgehog pathway. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9, e377. [Google Scholar] [PubMed]
- Hasenpusch-Theil, K.; Theil, T. The Multifaceted Roles of Primary Cilia in the Development of the Cerebral Cortex. Front. Cell Dev. Biol. 2021, 9, 630161. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Christa, A.; Kur, E.; Lioubinski, O.; Bachmann, S.; Willnow, T.E.; Hammes, A. LRP2 is an auxiliary SHH receptor required to condition the forebrain ventral midline for inductive signals. Dev. Cell 2012, 22, 268–278. [Google Scholar]
- Gigante, E.D.; Long, A.B.; Ben-Ami, J.; Caspary, T. Hypomorphic Smo mutant with inefficient ciliary enrichment disrupts the highest level of vertebrate Hedgehog response. Dev. Biol. 2018, 437, 152–162. [Google Scholar] [CrossRef]
- Li, P.; Markson, J.S.; Wang, S.; Chen, S.; Vachharajani, V.; Elowitz, M.B. Morphogen gradient reconstitution reveals Hedgehog pathway design principles. Science 2018, 360, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.D.R.; Han, Y.G.; Triplett, J.W.; Farmer, W.T.; Harwell, C.C.; Ihrie, R.A. The Elegance of Sonic Hedgehog: Emerging Novel Functions for a Classic Morphogen. J. Neurosci. 2018, 38, 9338–9345. [Google Scholar] [CrossRef]
- Cederquist, G.Y.; Asciolla, J.J.; Tchieu, J.; Walsh, R.M.; Cornacchia, D.; Resh, M.D.; Studer, L. Specification of positional identity in forebrain organoids. Nat. Biotechnol. 2019, 37, 436–444. [Google Scholar] [CrossRef]
- Elliott, K.H.; Brugmann, S.A. Sending mixed signals: Cilia-dependent signaling during development and disease. Dev. Biol. 2019, 447, 28–41. [Google Scholar] [CrossRef]
- Huang, D.; Wang, Y.; Tang, J.; Luo, S. Molecular mechanisms of suppressor of fused in regulating the hedgehog signalling pathway. Oncol. Lett. 2018, 15, 6077–6086. [Google Scholar] [CrossRef]
- Doheny, D.; Manore, S.G.; Wong, G.L.; Lo, H.W. Hedgehog Signaling and Truncated GLI1 in Cancer. Cells 2020, 9, 2114. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, A.F.; Kinnebrew, M.; Kowatsch, C.; Ansell, T.B.; El Omari, K.; Bishop, B.; Pardon, E.; Schwab, R.A.; Malinauskas, T.; Qian, M.; et al. The morphogen Sonic Hedgehog inhibits its receptor Patched by a pincer grasp mechanism. Nat. Chem. Biol. 2019, 15, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Brennan, D.; Chen, X.; Cheng, L.; Mahoney, M.; Riobo, N.A. Noncanonical Hedgehog signaling. Vitam. Horm. 2012, 88, 55–72. [Google Scholar] [PubMed]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; Spohr, T. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef]
- Jeng, K.S.; Chang, C.F.; Lin, S.S. Sonic Hedgehog Signaling in Organogenesis, Tumors, and Tumor Microenvironments. Int. J. Mol. Sci. 2020, 21, 3. [Google Scholar]
- Faria, A.V.S.; Akyala, A.I.; Parikh, K.; Bruggemann, L.W.; Spek, C.A.; Cao, W.; Bruno, M.J.; Bijlsma, M.F.; Fuhler, G.M.; Peppelenbosch, M.P. Smoothened-dependent and -independent pathways in mammalian noncanonical Hedgehog signaling. J. Biol. Chem. 2019, 294, 9787–9798. [Google Scholar] [CrossRef]
- Zhang, X.M.; Ramalho-Santos, M.; McMahon, A.P. Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R symmetry by the mouse node. Cell 2001, 106, 781–792. [Google Scholar] [CrossRef]
- Caspary, T.; Garcia-Garcia, M.J.; Huangfu, D.; Eggenschwiler, J.T.; Wyler, M.R.; Rakeman, A.S.; Alcorn, H.L.; Anderson, K.V. Mouse Dispatched homolog1 is required for long-range, but not juxtacrine, Hh signaling. Curr. Biol. 2002, 12, 1628–1632. [Google Scholar] [CrossRef]
- Ho Wei, L.; Arastoo, M.; Georgiou, I.; Manning, D.R.; Riobo-Del Galdo, N.A. Activation of the Gi protein-RHOA axis by non-canonical Hedgehog signaling is independent of primary cilia. PLoS ONE 2018, 13, e0203170. [Google Scholar] [CrossRef]
- Ferent, J.; Constable, S.; Gigante, E.D.; Yam, P.T.; Mariani, L.E.; Legue, E.; Liem, K.F., Jr.; Caspary, T.; Charron, F. The Ciliary Protein Arl13b Functions Outside of the Primary Cilium in Shh-Mediated Axon Guidance. Cell Rep. 2019, 29, 3356–3366.e3. [Google Scholar] [CrossRef] [PubMed]
- Akhshi, T.; Trimble, W.S. A non-canonical Hedgehog pathway initiates ciliogenesis and autophagy. J. Cell Biol. 2021, 220, 1. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, A.; Rohatgi, R.; Siebold, C. Cholesterol access in cellular membranes controls Hedgehog signaling. Nat. Chem. Biol. 2020, 16, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Findakly, S.; Daggubati, V.; Garcia, G.; LaStella, S.A.; Choudhury, A.; Tran, C.; Li, A.; Tong, P.; Garcia, J.Q.; Puri, N.; et al. Sterol and oxysterol synthases near the ciliary base activate the Hedgehog pathway. J. Cell Biol. 2021, 220, 1. [Google Scholar] [CrossRef]
- Cruz, L.; Romero, J.A.A.; Iglesia, R.P.; Lopes, M.H. Extracellular Vesicles: Decoding a New Language for Cellular Communication in Early Embryonic Development. Front. Cell Dev. Biol. 2018, 6, 94. [Google Scholar] [CrossRef]
- Tanaka, Y.; Okada, Y.; Hirokawa, N. FGF-induced vesicular release of Sonic Hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature 2005, 435, 172–177. [Google Scholar] [CrossRef]
- Vyas, N.; Walvekar, A.; Tate, D.; Lakshmanan, V.; Bansal, D.; Lo Cicero, A.; Raposo, G.; Palakodeti, D.; Dhawan, J. Vertebrate Hedgehog is secreted on two types of extracellular vesicles with different signaling properties. Sci. Rep. 2014, 4, 7357. [Google Scholar] [CrossRef]
- Whalen, D.M.; Malinauskas, T.; Gilbert, R.J.; Siebold, C. Structural insights into proteoglycan-shaped Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 16420–16425. [Google Scholar] [CrossRef]
- Yan, D.; Lin, X. Shaping morphogen gradients by proteoglycans. Cold Spring Harb. Perspect. Biol. 2009, 1, a002493. [Google Scholar] [CrossRef]
- Roessler, E.; Belloni, E.; Gaudenz, K.; Jay, P.; Berta, P.; Scherer, S.W.; Tsui, L.C.; Muenke, M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat. Genet. 1996, 14, 357–360. [Google Scholar] [CrossRef]
- Dubourg, C.; Lazaro, L.; Pasquier, L.; Bendavid, C.; Blayau, M.; Le Duff, F.; Durou, M.R.; Odent, S.; David, V. Molecular screening of SHH, ZIC2, SIX3, and TGIF genes in patients with features of holoprosencephaly spectrum: Mutation review and genotype-phenotype correlations. Hum. Mutat. 2004, 24, 43–51. [Google Scholar] [CrossRef]
- Kruszka, P.; Muenke, M. Syndromes associated with holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Tekendo-Ngongang, C.; Muenke, M.; Kruszka, P. Holoprosencephaly Overview. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; The University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Ribeiro, L.A.; Murray, J.C.; Richieri-Costa, A. PTCH mutations in four Brazilian patients with holoprosencephaly and in one with holoprosencephaly-like features and normal MRI. Am. J. Med. Genet. A 2006, 140, 2584–2586. [Google Scholar] [CrossRef] [PubMed]
- Wotton, D.; Taniguchi, K. Functions of TGIF homeodomain proteins and their roles in normal brain development and holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Roessler, E.; Muenke, M. The molecular genetics of holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 52–61. [Google Scholar] [CrossRef]
- Dykes, I.M.; Szumska, D.; Kuncheria, L.; Puliyadi, R.; Chen, C.M.; Papanayotou, C.; Lockstone, H.; Dubourg, C.; David, V.; Schneider, J.E.; et al. A Requirement for Zic2 in the Regulation of Nodal Expression Underlies the Establishment of Left-Sided Identity. Sci. Rep. 2018, 8, 10439. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, R.J.; Godin, E.A.; O’Leary-Moore, S.K.; Parnell, S.E.; Sulik, K.K. Genesis of teratogen-induced holoprosencephaly in mice. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 29–42. [Google Scholar] [CrossRef]
- Sagai, T.; Amano, T.; Maeno, A.; Ajima, R.; Shiroishi, T. SHH signaling mediated by a prechordal and brain enhancer controls forebrain organization. Proc. Natl. Acad. Sci. USA 2019, 116, 23636–23642. [Google Scholar] [CrossRef]
- Solomon, B.D.; Mercier, S.; Velez, J.I.; Pineda-Alvarez, D.E.; Wyllie, A.; Zhou, N.; Dubourg, C.; David, V.; Odent, S.; Roessler, E.; et al. Analysis of genotype-phenotype correlations in human holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 133–141. [Google Scholar] [CrossRef]
- Sun, M.R.; Chung, H.M.; Matsuk, V.; Fink, D.M.; Stebbins, M.J.; Palecek, S.P.; Shusta, E.V.; Lipinski, R.J. Sonic Hedgehog Signaling in Cranial Neural Crest Cells Regulates Microvascular Morphogenesis in Facial Development. Front. Cell Dev. Biol. 2020, 8, 590539. [Google Scholar] [CrossRef]
- Suzuki, A.; Sangani, D.R.; Ansari, A.; Iwata, J. Molecular mechanisms of midfacial developmental defects. Dev. Dyn. 2016, 245, 276–293. [Google Scholar] [CrossRef]
- Dubourg, C.; Bendavid, C.; Pasquier, L.; Henry, C.; Odent, S.; David, V. Holoprosencephaly. Orphanet J. Rare Dis. 2007, 2, 8. [Google Scholar] [CrossRef]
- Tichy, J.; Zinke, J.; Bunz, B.; Meyermann, R.; Harter, P.N.; Mittelbronn, M. Expression Profile of Sonic Hedgehog Pathway Members in the Developing Human Fetal Brain. Biomed. Res. Int. 2015, 2015, 494269. [Google Scholar] [CrossRef]
- Memi, F.; Zecevic, N.; Radonjic, N. Multiple roles of Sonic Hedgehog in the developing human cortex are suggested by its widespread distribution. Brain Struct. Funct. 2018, 223, 2361–2375. [Google Scholar] [CrossRef]
- Reilly, S.K.; Yin, J.; Ayoub, A.E.; Emera, D.; Leng, J.; Cotney, J.; Sarro, R.; Rakic, P.; Noonan, J.P. Evolutionary genomics. Evolutionary changes in promoter and enhancer activity during human corticogenesis. Science 2015, 347, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.D.; Bear, K.A.; Wyllie, A.; Keaton, A.A.; Dubourg, C.; David, V.; Mercier, S.; Odent, S.; Hehr, U.; Paulussen, A.; et al. Genotypic and phenotypic analysis of 396 individuals with mutations in Sonic Hedgehog. J. Med. Genet. 2012, 49, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Hadley, D.W.; Kruszka, P.; Muenke, M. Challenging issues arising in counseling families experiencing holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Ming, J.E.; Muenke, M. Multiple hits during early embryonic development: Digenic diseases and holoprosencephaly. Am. J. Hum. Genet. 2002, 71, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Oliver, G. Pathogenesis of holoprosencephaly. J. Clin. Investig. 2009, 119, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.D.; Lacbawan, F.; Jain, M.; Domene, S.; Roessler, E.; Moore, C.; Dobyns, W.B.; Muenke, M. A novel SIX3 mutation segregates with holoprosencephaly in a large family. Am. J. Med. Genet. A 2009, 149A, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.K.; Stearns, T. Hedgehog signaling and the primary cilium: Implications for spatial and temporal constraints on signaling. Development 2021, 148, 9. [Google Scholar] [CrossRef] [PubMed]
- Petrov, K.; de Almeida Magalhaes, T.; Salic, A. Mechanism and ultrasensitivity in Hedgehog signaling revealed by Patched1 disease mutations. Proc. Natl. Acad. Sci. USA 2021, 118, 6. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef]
- Seppala, M.; Depew, M.J.; Martinelli, D.C.; Fan, C.M.; Sharpe, P.T.; Cobourne, M.T. Gas1 is a modifier for holoprosencephaly and genetically interacts with Sonic Hedgehog. J. Clin. Investig. 2007, 117, 1575–1584. [Google Scholar] [CrossRef]
- Muenke, M.; Cohen, M.M., Jr. Genetic approaches to understanding brain development: Holoprosencephaly as a model. Ment. Retard. Dev. Disabil. Res. Rev. 2000, 6, 15–21. [Google Scholar] [CrossRef]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176–1187.e14. [Google Scholar] [CrossRef]
- Weaver, D.D.; Solomon, B.D.; Akin-Samson, K.; Kelley, R.I.; Muenke, M. Cyclopia (synophthalmia) in Smith-Lemli-Opitz syndrome: First reported case and consideration of mechanism. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154, 142–145. [Google Scholar] [CrossRef]
- Hong, M.; Christ, A.; Christa, A.; Willnow, T.E.; Krauss, R.S. Cdon mutation and fetal alcohol converge on Nodal signaling in a mouse model of holoprosencephaly. eLife 2020, 9, e60351. [Google Scholar] [CrossRef] [PubMed]
- Kietzman, H.W.; Everson, J.L.; Sulik, K.K.; Lipinski, R.J. The teratogenic effects of prenatal ethanol exposure are exacerbated by Sonic Hedgehog or GLI2 haploinsufficiency in the mouse. PLoS ONE 2014, 9, e89448. [Google Scholar]
- Addissie, Y.A.; Troia, A.; Wong, Z.C.; Everson, J.L.; Kozel, B.A.; Muenke, M.; Lipinski, R.J.; Malecki, K.M.C.; Kruszka, P. Identifying environmental risk factors and gene-environment interactions in holoprosencephaly. Birth Defects Res. 2021, 113, 63–76. [Google Scholar] [CrossRef]
- Fitriasari, S.; Trainor, P.A. Diabetes, Oxidative Stress, and DNA Damage Modulate Cranial Neural Crest Cell Development and the Phenotype Variability of Craniofacial Disorders. Front. Cell Dev. Biol. 2021, 9, 644410. [Google Scholar] [CrossRef] [PubMed]
- Beames, T.G.; Lipinski, R.J. Gene-environment interactions: Aligning birth defects research with complex etiology. Development 2020, 147, 21. [Google Scholar] [CrossRef]
- Solomon, B.D. The etiology of VACTERL association: Current knowledge and hypotheses. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Pelizzo, G.; Chiricosta, L.; Mazzon, E.; Zuccotti, G.V.; Avanzini, M.A.; Croce, S.; Lima, M.; Bramanti, P.; Calcaterra, V. Discovering Genotype Variants in an Infant with VACTERL through Clinical Exome Sequencing: A Support for Personalized Risk Assessment and Disease Prevention. Pediatr. Rep. 2021, 13, 6. [Google Scholar] [CrossRef]
- Engelke, M.F.; Waas, B.; Kearns, S.E.; Suber, A.; Boss, A.; Allen, B.L.; Verhey, K.J. Acute Inhibition of Heterotrimeric Kinesin-2 Function Reveals Mechanisms of Intraflagellar Transport in Mammalian Cilia. Curr. Biol. 2019, 29, 1137–1148.e4. [Google Scholar] [CrossRef] [PubMed]
- Revenkova, E.; Liu, Q.; Gusella, G.L.; Iomini, C. The Joubert syndrome protein ARL13B binds tubulin to maintain uniform distribution of proteins along the ciliary membrane. J. Cell Sci. 2018, 131, 9. [Google Scholar] [CrossRef] [PubMed]
- Gigante, E.D.; Taylor, M.R.; Ivanova, A.A.; Kahn, R.A.; Caspary, T. ARL13B regulates Sonic Hedgehog signaling from outside primary cilia. eLife 2020, 9, e50434. [Google Scholar] [CrossRef]
- Wang, W.; Jack, B.M.; Wang, H.H.; Kavanaugh, M.A.; Maser, R.L.; Tran, P.V. Intraflagellar Transport Proteins as Regulators of Primary Cilia Length. Front. Cell Dev. Biol. 2021, 9, 661350. [Google Scholar] [CrossRef]
- May, S.R.; Ashique, A.M.; Karlen, M.; Wang, B.; Shen, Y.; Zarbalis, K.; Reiter, J.; Ericson, J.; Peterson, A.S. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 2005, 287, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Ocbina, P.J.; Anderson, K.V. Intraflagellar transport, cilia, and mammalian Hedgehog signaling: Analysis in mouse embryonic fibroblasts. Dev. Dyn. 2008, 237, 2030–2038. [Google Scholar] [CrossRef] [PubMed]
- Hasenpusch-Theil, K.; Laclef, C.; Colligan, M.; Fitzgerald, E.; Howe, K.; Carroll, E.; Abrams, S.R.; Reiter, J.F.; Schneider-Maunoury, S.; Theil, T. A transient role of the ciliary gene Inpp5e in controlling direct versus indirect neurogenesis in cortical development. eLife 2020, 9, e58162. [Google Scholar] [CrossRef] [PubMed]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the Primary Cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yuan, X.; Yang, S. IFT80 is essential for chondrocyte differentiation by regulating Hedgehog and Wnt signaling pathways. Exp. Cell Res. 2013, 319, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhu, Q.; Cheng, J.; Wu, Y.; Fan, M.; Zhang, J.; Wu, H. Opposite regulation of Wnt/beta-catenin and Shh signaling pathways by Rack1 controls mammalian cerebellar development. Proc. Natl. Acad. Sci. USA 2019, 116, 4661–4670. [Google Scholar] [CrossRef] [PubMed]
- Liem, K.F., Jr.; Jessell, T.M.; Briscoe, J. Regulation of the neural patterning activity of Sonic Hedgehog by secreted BMP inhibitors expressed by notochord and somites. Development 2000, 127, 4855–4866. [Google Scholar] [CrossRef] [PubMed]
- Brodski, C.; Blaess, S.; Partanen, J.; Prakash, N. Crosstalk of Intercellular Signaling Pathways in the Generation of Midbrain Dopaminergic Neurons In Vivo and from Stem Cells. J. Dev. Biol. 2019, 7, 3. [Google Scholar] [CrossRef]
- Christ, A.; Herzog, K.; Willnow, T.E. LRP2, an auxiliary receptor that controls Sonic Hedgehog signaling in development and disease. Dev. Dyn. 2016, 245, 569–579. [Google Scholar] [CrossRef]
- Tickle, C.; Towers, M. Sonic Hedgehog Signaling in Limb Development. Front. Cell Dev. Biol. 2017, 5, 14. [Google Scholar] [CrossRef]
- McQueen, C.; Towers, M. Establishing the pattern of the vertebrate limb. Development 2020, 147, 17. [Google Scholar] [CrossRef]
- Loo, C.K.; Pereira, T.N.; Ramsing, M.; Vogel, I.; Petersen, O.B.; Ramm, G.A. Mechanism of pancreatic and liver malformations in human fetuses with short-rib polydactyly syndrome. Birth Defects Res. A Clin. Mol. Teratol. 2016, 106, 549–562. [Google Scholar] [CrossRef]
- Clotman, F.; Libbrecht, L.; Killingsworth, M.C.; Loo, C.C.; Roskams, T.; Lemaigre, F.P. Lack of cilia and differentiation defects in the liver of human foetuses with the Meckel syndrome. Liver Int. 2008, 28, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Lam, W.L.; Oh, J.D.H.; Johnson, E.J.; Pertinez, S.P.; Stephens, C.; Davey, M.G. Experimental evidence that preaxial polydactyly and forearm radial deficiencies may share a common developmental origin. J. Hand Surg. Eur. Vol. 2019, 44, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. A 2019, 179, 2393–2419. [Google Scholar] [CrossRef] [PubMed]
- Rix, S.; Calmont, A.; Scambler, P.J.; Beales, P.L. An Ift80 mouse model of short rib polydactyly syndromes shows defects in hedgehog signalling without loss or malformation of cilia. Hum. Mol. Genet. 2011, 20, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Taylor, S.P.; Nevarez, L.; Lachman, R.S.; Nickerson, D.A.; Bamshad, M.; University of Washington Center for Mendelian Genomics, C.; Krakow, D.; Cohn, D.H. IFT52 mutations destabilize anterograde complex assembly, disrupt ciliogenesis and result in short rib polydactyly syndrome. Hum. Mol. Genet. 2016, 25, 4012–4020. [Google Scholar] [CrossRef] [PubMed]
- Mill, P.; Lockhart, P.J.; Fitzpatrick, E.; Mountford, H.S.; Hall, E.A.; Reijns, M.A.; Keighren, M.; Bahlo, M.; Bromhead, C.J.; Budd, P.; et al. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am. J. Hum. Genet. 2011, 88, 508–515. [Google Scholar] [CrossRef]
- Sasai, N.; Briscoe, J. Primary cilia and graded Sonic Hedgehog signaling. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 753–772. [Google Scholar] [CrossRef] [PubMed]
- Goetz, S.C.; Ocbina, P.J.; Anderson, K.V. The primary cilium as a Hedgehog signal transduction machine. Methods Cell Biol. 2009, 94, 199–222. [Google Scholar] [PubMed]
- Hwang, S.H.; White, K.A.; Somatilaka, B.N.; Shelton, J.M.; Richardson, J.A.; Mukhopadhyay, S. The G protein-coupled receptor Gpr161 regulates forelimb formation, limb patterning and skeletal morphogenesis in a primary cilium-dependent manner. Development 2018, 145, 1. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Boutaud, L.; Reilly, M.L.; Benmerah, A. Cilia in hereditary cerebral anomalies. Biol. Cell 2019, 111, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Higginbotham, H.; Li, J.; Nichols, J.; Hirt, J.; Ghukasyan, V.; Anton, E.S. Developmental disruptions underlying brain abnormalities in ciliopathies. Nat. Commun. 2015, 6, 7857. [Google Scholar] [CrossRef] [PubMed]
- Arellano, J.I.; Guadiana, S.M.; Breunig, J.J.; Rakic, P.; Sarkisian, M.R. Development and distribution of neuronal cilia in mouse neocortex. J. Comp. Neurol. 2012, 520, 848–873. [Google Scholar] [CrossRef]
- Valenza, F.; Cittaro, D.; Stupka, E.; Biancolini, D.; Patricelli, M.G.; Bonanomi, D.; Lazarevic, D. A novel truncating variant of GLI2 associated with Culler-Jones syndrome impairs Hedgehog signalling. PLoS ONE 2019, 14, e0210097. [Google Scholar] [CrossRef]
- Bear, K.A.; Solomon, B.D. GLI2 mutations typically result in pituitary anomalies with or without postaxial polydactyly. Am. J. Med. Genet. A 2015, 167, 2491–2492. [Google Scholar] [CrossRef]
- Elizabeth, M.S.M.; Verkerk, A.; Hokken-Koelega, A.C.S.; Verlouw, J.A.M.; Argente, J.; Pfaeffle, R.; Visser, T.J.; Peeters, R.P.; De Graaff, L.C.G. Unique near-complete deletion of GLI2 in a patient with combined pituitary hormone deficiency and post-axial polydactyly. Growth Horm. IGF Res. 2020, 50, 35–41. [Google Scholar] [CrossRef]
- Brooks, E.R.; Islam, M.T.; Anderson, K.V.; Zallen, J.A. Sonic Hedgehog signaling directs patterned cell remodeling during cranial neural tube closure. eLife 2020, 9, e60234. [Google Scholar] [CrossRef] [PubMed]
- Le, T.L.; Sribudiani, Y.; Dong, X.; Huber, C.; Kois, C.; Baujat, G.; Gordon, C.T.; Mayne, V.; Galmiche, L.; Serre, V.; et al. Bi-allelic Variations of SMO in Humans Cause a Broad Spectrum of Developmental Anomalies Due to Abnormal Hedgehog Signaling. Am. J. Hum. Genet. 2020, 106, 779–792. [Google Scholar] [CrossRef]
- Hong, M.; Srivastava, K.; Kim, S.; Allen, B.L.; Leahy, D.J.; Hu, P.; Roessler, E.; Krauss, R.S.; Muenke, M. BOC is a modifier gene in holoprosencephaly. Hum. Mutat. 2017, 38, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Cordero, D.R.; Bendavid, C.; Shanske, A.L.; Haddad, B.R.; Muenke, M. Holoprosencephaly-Polydactyly syndrome: In search of an etiology. Eur. J. Med. Genet. 2008, 51, 106–112. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sereke, S.G.; Oriekot, A.; Bongomin, F. Overlapping holoprosencephaly-polydactyl syndrome and asphyxiating thoracic dystrophy, an incidental finding in late prenatal ultrasound: A rare case report. Clin. Case Rep. 2021, 9, 1577–1582. [Google Scholar] [CrossRef]
- Shou, Y.; Liang, F.; Xu, S.; Li, X. The Application of Brain Organoids: From Neuronal Development to Neurological Diseases. Front. Cell Dev. Biol. 2020, 8, 579659. [Google Scholar] [CrossRef]
- Shin, J.O.; Song, J.; Choi, H.S.; Lee, J.; Lee, K.; Ko, H.W.; Bok, J. Activation of Sonic Hedgehog signaling by a Smoothened agonist restores congenital defects in mouse models of endocrine-cerebro-osteodysplasia syndrome. EBioMedicine 2019, 49, 305–317. [Google Scholar] [CrossRef]
- Fish, E.W.; Parnell, S.E.; Sulik, K.K.; Baker, L.K.; Murdaugh, L.B.; Lamson, D.; Williams, K.P. Preaxial polydactyly following early gestational exposure to the smoothened agonist, SAG, in C57BL/6J mice. Birth Defects Res. 2017, 109, 49–54. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Basset-Seguin, N.; Dreno, B.; Garbe, C.; Gutzmer, R.; Hauschild, A.; Krattinger, R.; Lear, J.T.; Malvehy, J.; et al. Sonidegib and vismodegib in the treatment of patients with locally advanced basal cell carcinoma: A joint expert opinion. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 1944–1956. [Google Scholar] [CrossRef]
- Kish, T.; Corry, L. Sonidegib (Odomzo) for the Systemic Treatment of Adults with Recurrent, Locally Advanced Basal Cell Skin Cancer. Pharm. Ther. 2016, 41, 322–325. [Google Scholar]
- Fu, J.; Rodova, M.; Roy, S.K.; Sharma, J.; Singh, K.P.; Srivastava, R.K.; Shankar, S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013, 330, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Matevossian, A.; Resh, M.D. Hedgehog Acyltransferase as a target in estrogen receptor positive, HER2 amplified, and tamoxifen resistant breast cancer cells. Mol. Cancer 2015, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Tolani, B.; Hoang, N.T.; Acevedo, L.A.; Giroux Leprieur, E.; Li, H.; He, B.; Jablons, D.M. Preclinical characterization of therapeutic antibodies targeted at the carboxy-terminus of Sonic Hedgehog. Oncotarget 2018, 9, 14311–14323. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Syndromes | Genetic Abnormality | Clinical Features |
|---|---|---|
| Chromosomal | Trisomy 13 (almost 50% of all cases holoprosencephaly) | Holoprosencephaly in 8–39%, cleft lip and palate, rocker bottom feet, postaxial polydactyly |
| Trisomy 18, 21, 22 | Holoprosencephaly infrequent | |
| triploidy | Holoprosencephaly rare, syndactyly, lethal in 1st or 2nd trimester | |
| 13q deletion | ZIC2 | Holoprosencephaly, kidney, heart, eye, facial and limb anomalies |
| 18p deletion | TGIF1 | Holoprosencephaly rare |
| Smith Lemli Opitz | DHCR7 | Holoprosencephaly about 5% |
| Hartsfield | FGFR1 | Holoprosencephaly occasional, ectrodactyly |
| Kallman syndrome 2 | FGFR1 | Cleft lip and palate, hypogonadism and holoprosencephaly occasional |
| Steinfeld | CDON | Microform holoprosencephaly |
| Culler Jones | GLI2 | Hypopituitarism, post axial polydactyly, holoprosencephaly unusual |
| Non-syndromic causes of Holoprosencephaly | Estimated incidence | |
| SHH | 5–6% | |
| ZIC2 | 5% | |
| SIX3 | 3% | |
| TGIF1 | <1% | |
| Less common | ||
| CNOT1 | <1.5% | |
| DISP1 | <1.2% | |
| FGF8 | <2.2% | |
| FGFR1 | 1.2% | |
| DLL1 | <1% | |
| PPP1R12A, RAD21, SMC1A, SMC3, STAG2, STIL, PTCH1, CDON | Rare causes of holoprosencephaly | |
| Forms of Holoprosencephaly (in Decreasing Order of Severity) | Clinical Features |
|---|---|
| Alobar holoprosencephaly (most severe) | one cerebral lobe with fused deep gray nuclei, absent corpus callosum absent olfactory bulbs and tracts cyclopia, proboscis, synophthalmia, hypotelorism |
| Semilobar holoprosencephaly | fused left and right frontal parietal lobes, incomplete separation of cerebral hemispheres, varying separation of the deep gray nuclei. Absent or hypoplastic olfactory bulbs and tracts, absent corpus callosum, varying non separation of deep gray nuclei. midline cleft lip and flat nose. |
| Lobar holoprosencephaly | Fully developed cerebral hemispheres but frontal lobes are joined anteriorly in the midline, normal or hypoplastic corpus callosum separated deep gray nuclei midline cleft lip and flat nose. |
| Middle interhemispheric variant | failure of separation of posterior frontal and parietal lobes incomplete formation of the corpus callosum, with normal callosal genu and splenium but absence of the callosal body. |
| Agenesis of corpus callosum | Part or all of the corpus callosum can be absent |
| Microform holoprosencephaly | facial midline anomalies with normal brain structure or microcephaly. |
| Arhinencephaly | absent olfactory bulbs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loo, C.K.C.; Pearen, M.A.; Ramm, G.A. The Role of Sonic Hedgehog in Human Holoprosencephaly and Short-Rib Polydactyly Syndromes. Int. J. Mol. Sci. 2021, 22, 9854. https://doi.org/10.3390/ijms22189854
Loo CKC, Pearen MA, Ramm GA. The Role of Sonic Hedgehog in Human Holoprosencephaly and Short-Rib Polydactyly Syndromes. International Journal of Molecular Sciences. 2021; 22(18):9854. https://doi.org/10.3390/ijms22189854
Chicago/Turabian StyleLoo, Christine K. C., Michael A. Pearen, and Grant A. Ramm. 2021. "The Role of Sonic Hedgehog in Human Holoprosencephaly and Short-Rib Polydactyly Syndromes" International Journal of Molecular Sciences 22, no. 18: 9854. https://doi.org/10.3390/ijms22189854
APA StyleLoo, C. K. C., Pearen, M. A., & Ramm, G. A. (2021). The Role of Sonic Hedgehog in Human Holoprosencephaly and Short-Rib Polydactyly Syndromes. International Journal of Molecular Sciences, 22(18), 9854. https://doi.org/10.3390/ijms22189854