
Uridine Treatment of the First Known Case of SLC25A36 Deficiency

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
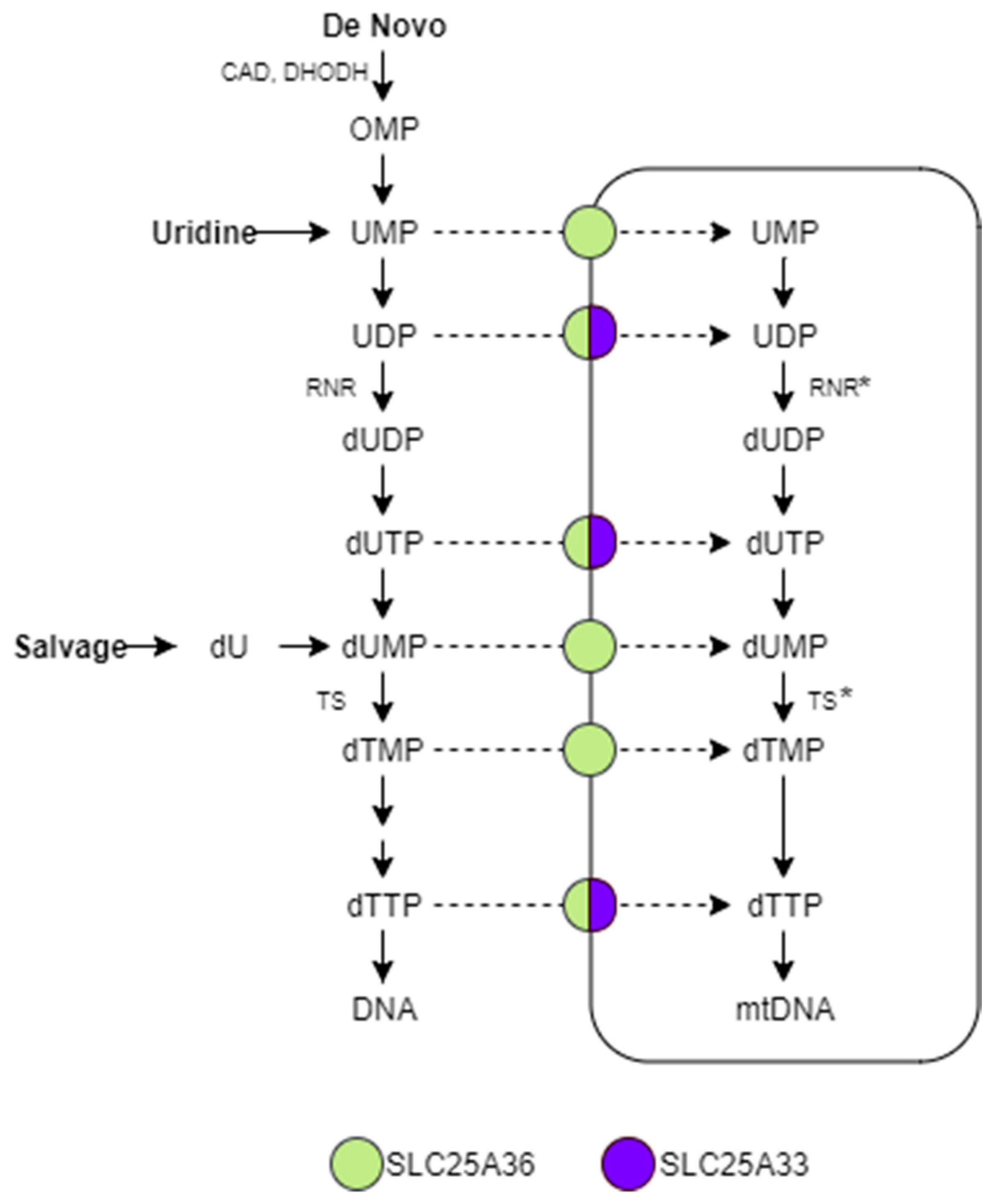
:1. Introduction
2. Results
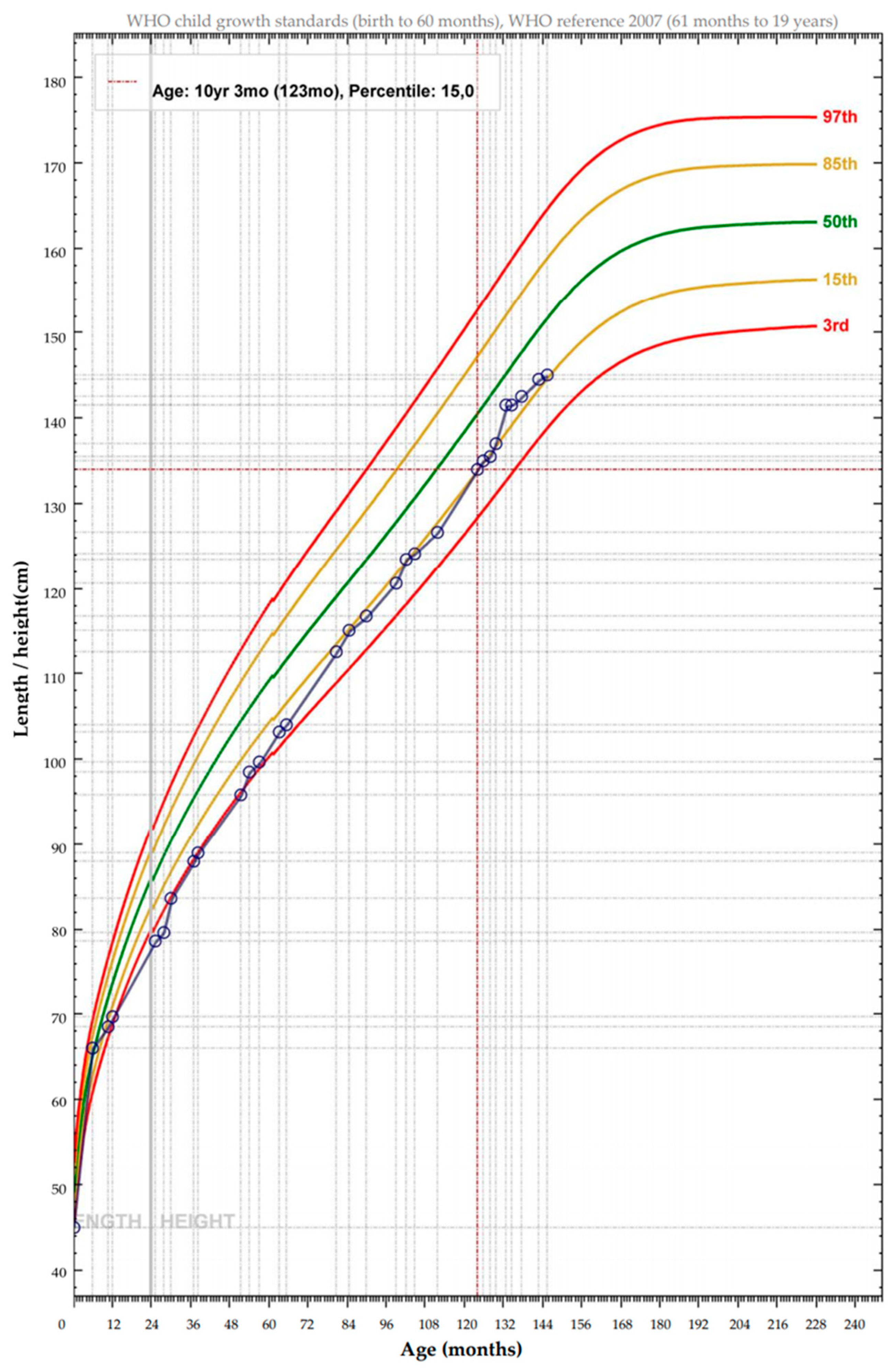
2.1. Patient and Clinical Phenotype
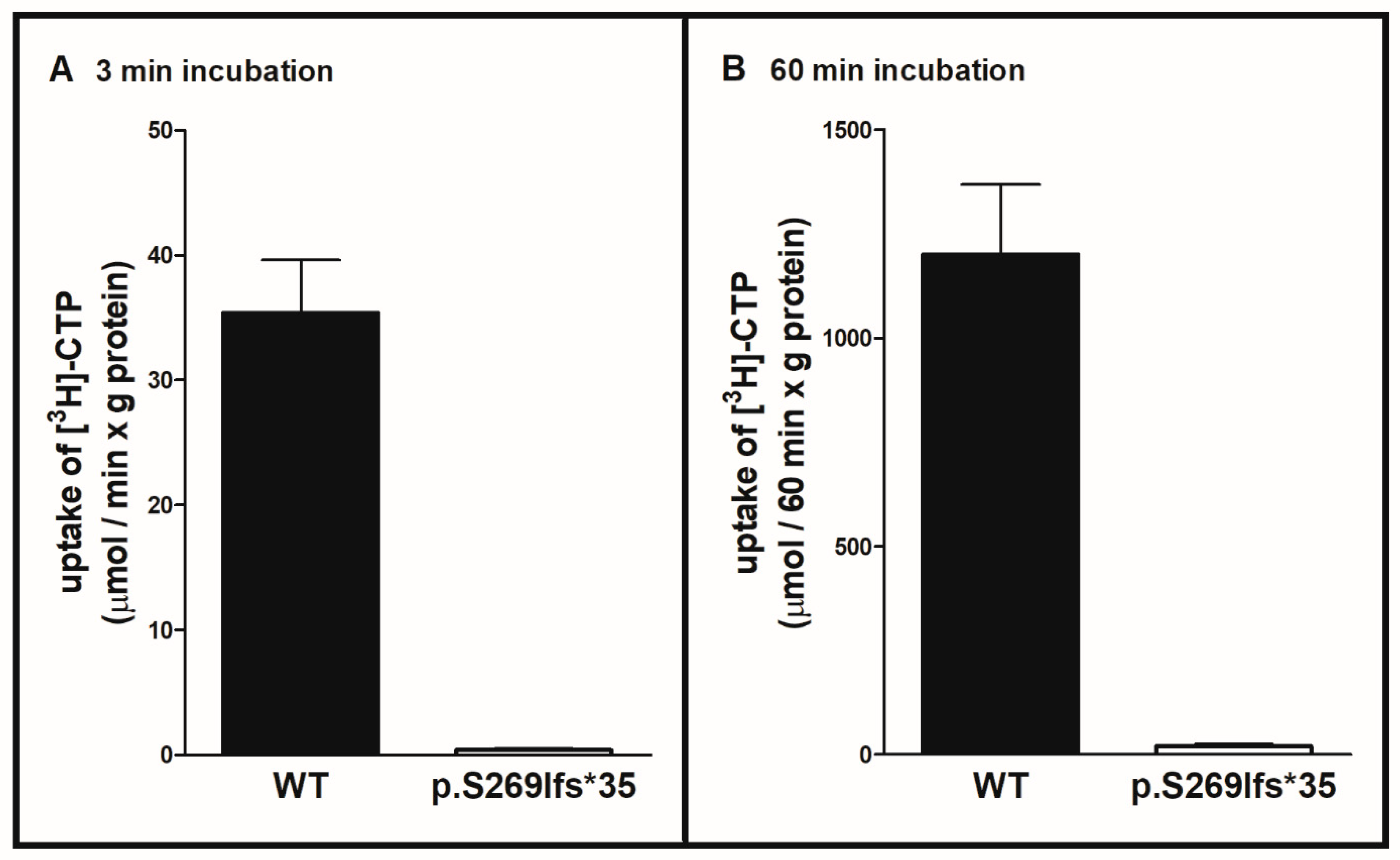
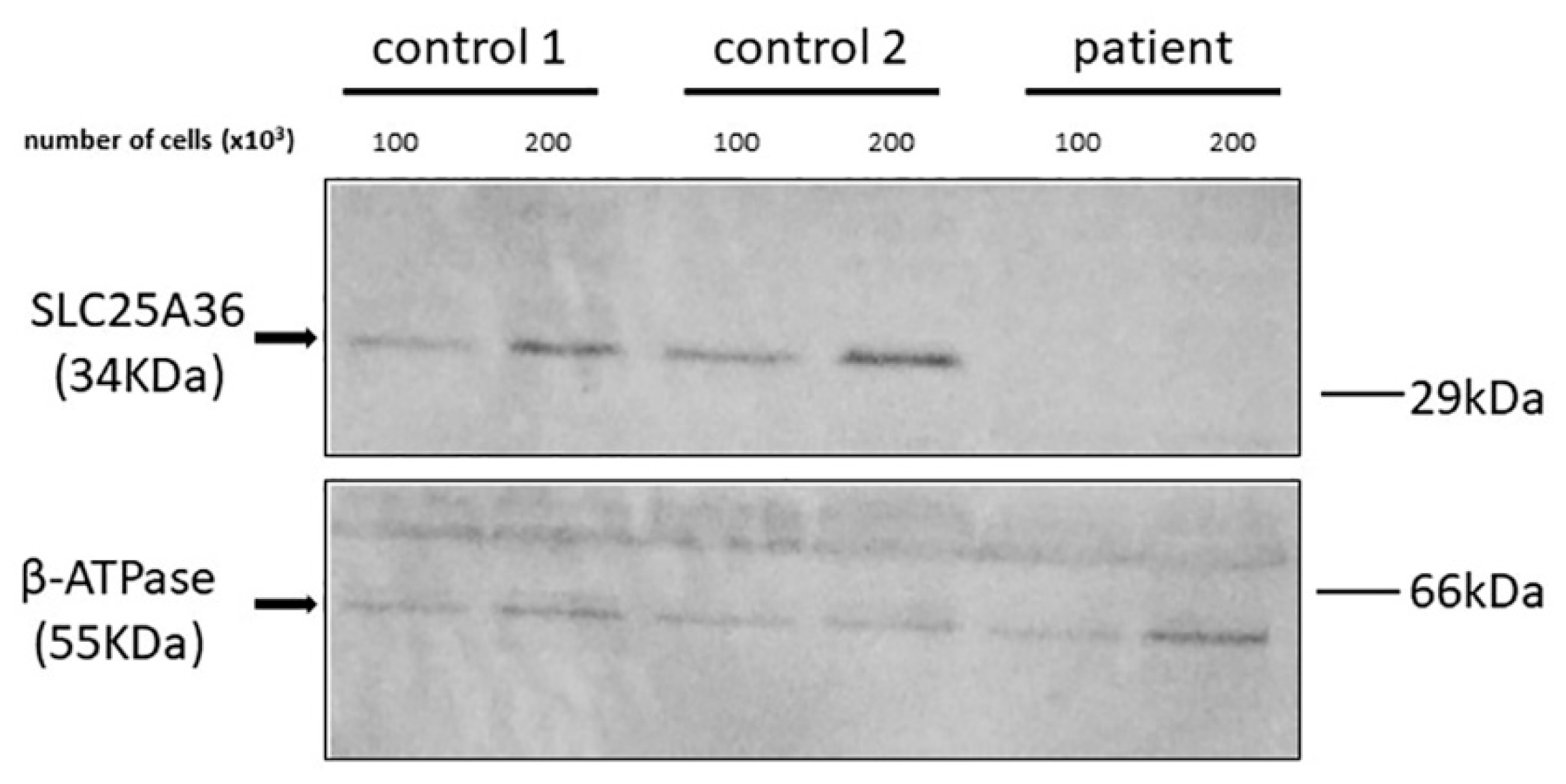
2.2. Functional Analysis of the SLC25A36 p.Ser269llefs*35 Mutant
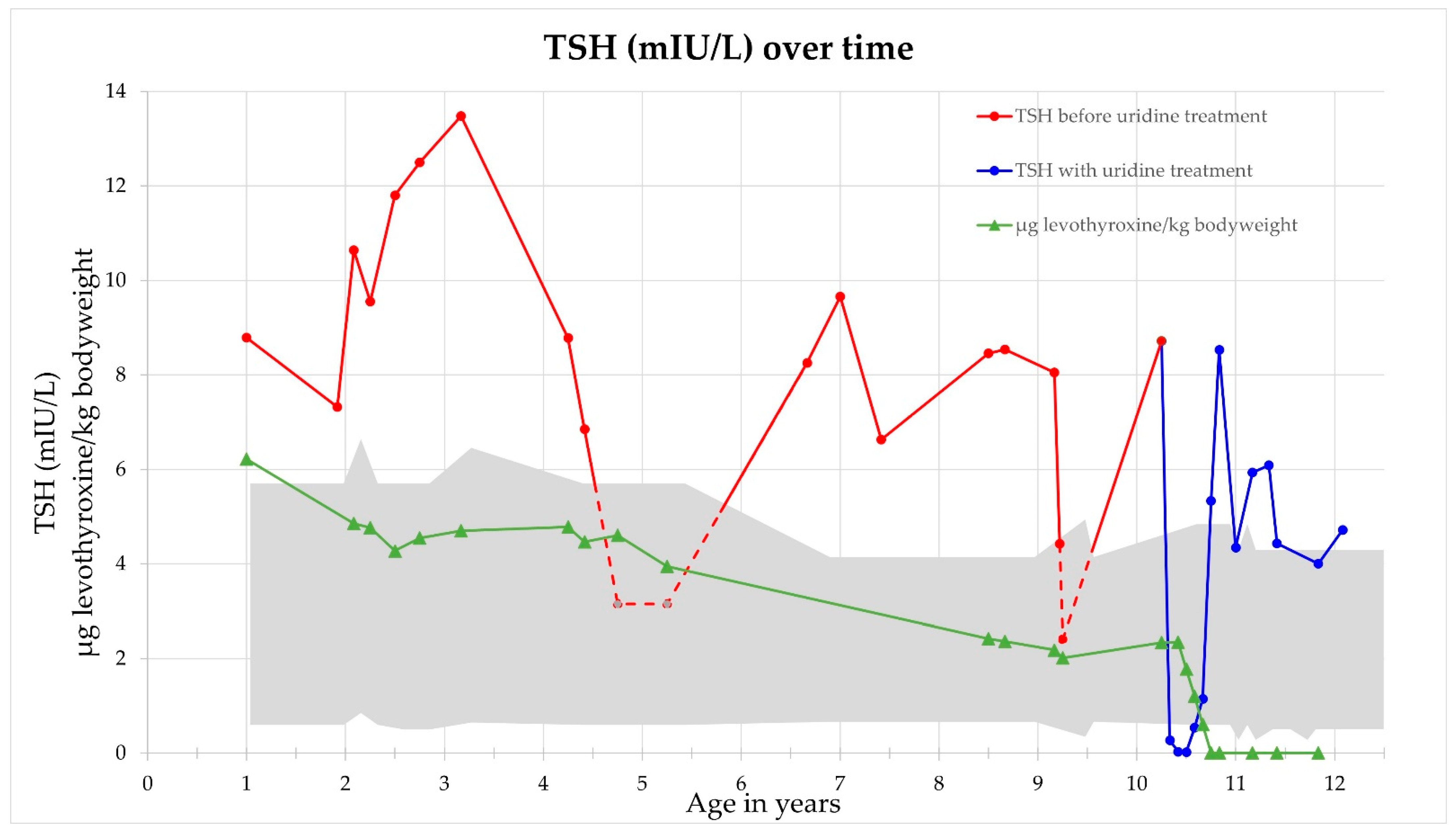
2.3. Response to Uridine Treatment
3. Discussion
4. Materials and Methods
4.1. Genetic Analysis and Clinical Diagnostics
4.2. Bacterial Expression, Reconstitution into Liposomes and Transport Assays of Recombinant WT SLC25A36 and p.Ser269llefs*35 SLC25A36 Mutant
4.3. Immunoblotting
4.4. Uridine as a Therapeutic Intervention
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| (d)NMPs | monophosphate (deoxy)nucleosides |
| (d)NTP | triphosphate (deoxy)nucleosides |
| A | adenine |
| C | cytosine |
| CAD | carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, dihydroorotase |
| CMP | cytidine-monophosphate |
| DHODH | dihydroorotate dehydrogenase |
| G | guanine |
| I | inosine |
| MCF | mitochondrial carrier family |
| mESC | mouse embryonic stem cells |
| mt | mitochondrial |
| RNR | ribonucleotide reductase |
| SLC | solute carrier superfamily |
| SLC25A36 | solute carrier family 25 member 36 |
| T | thymine |
| TMP | thymidine-monophosphate |
| TS | thymidylate synthase |
| U | uracil |
| UMP | uridine-monophosphate |
| WT | wild-type |
References
- Palmieri, F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflugers Arch. 2004, 447, 689–709. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Aspects Med. 2013, 34, 465–484. [Google Scholar] [CrossRef]
- Di Noia, M.A.; Todisco, S.; Cirigliano, A.; Rinaldi, T.; Agrimi, G.; Iacobazzi, V.; Palmieri, F. The human SLC25A33 and SLC25A36 genes of solute carrier family 25 encode two mitochondrial pyrimidine nucleotide transporters. J. Biol. Chem. 2014, 289, 33137–33148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, F. Diseases caused by defects of mitochondrial carriers: A review. Biochim. Biophys. Acta 2008, 1777, 564–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, F.; Scarcia, P.; Monné, M. Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules 2020, 10, 655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruprecht, J.J.; Kunji, E.R.S. Structural Mechanism of Transport of Mitochondrial Carriers. Annu. Rev. Biochem. 2021, 90, 535–558. [Google Scholar] [CrossRef]
- Monné, M.; Palmieri, F. Antiporters of the mitochondrial carrier family. Curr. Top. Membr. 2014, 73, 289–320. [Google Scholar] [CrossRef]
- Palmieri, F.; Bisaccia, F.; Capobianco, L.; Dolce, V.; Fiermonte, G.; Iacobazzi, V.; Zara, V. Transmembrane topology, genes, and biogenesis of the mitochondrial phosphate and oxoglutarate carriers. J. Bioenerg. Biomembr. 1993, 25, 493–501. [Google Scholar] [CrossRef]
- Palmieri, F.; Monné, M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim. Biophys. Acta 2016, 1863, 2362–2378. [Google Scholar] [CrossRef]
- Haitina, T.; Lindblom, J.; Renström, T.; Fredriksson, R. Fourteen novel human members of mitochondrial solute carrier family 25 (SLC25) widely expressed in the central nervous system. Genomics 2006, 88, 779–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, Y.; Wang, Y.; Zhong, L.; Shi, B.; Liang, H.; Han, J. Slc25a36 modulates pluripotency of mouse embryonic stem cells by regulating mitochondrial function and glutathione level. Biochem. J. 2019, 476, 1585–1604. [Google Scholar] [CrossRef]
- Grohmann, K.; Lauffer, H.; Lauenstein, P.; Hoffmann, G.F.; Seidlitz, G. Hereditary orotic aciduria with epilepsy and without megaloblastic anemia. Neuropediatrics 2015, 46, 123–125. [Google Scholar] [CrossRef]
- Koch, J.; Mayr, J.A.; Alhaddad, B.; Rauscher, C.; Bierau, J.; Kovacs-Nagy, R.; Coene, K.L.M.; Bader, I.; Holzhacker, M.; Prokisch, H.; et al. CAD mutations and uridine-responsive epileptic encephalopathy. Brain 2017, 140, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Garone, C.; Garcia-Diaz, B.; Emmanuele, V.; Lopez, L.C.; Tadesse, S.; Akman, H.O.; Tanji, K.; Quinzii, C.M.; Hirano, M. Deoxypyrimidine monophosphate bypass therapy for thymidine kinase 2 deficiency. EMBO Mol. Med. 2014, 6, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samsonoff, W.A.; Reston, J.; McKee, M.; O’Connor, B.; Galivan, J.; Maley, G.; Maley, F. Intracellular location of thymidylate synthase and its state of phosphorylation. J. Biol. Chem. 1997, 272, 13281–13285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chimploy, K.; Song, S.; Wheeler, L.J.; Mathews, C.K. Ribonucleotide reductase association with mammalian liver mitochondria. J. Biol. Chem. 2013, 288, 13145–13155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamath, V.G.; Hsiung, C.-H.; Lizenby, Z.J.; McKee, E.E. Heart mitochondrial TTP synthesis and the compartmentalization of TMP. J. Biol. Chem. 2015, 290, 2034–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, P.; Leeds, J.M.; Slabaugh, M.B.; Mathews, C.K. Ribonucleotide reductase: Evidence for specific association with HeLa cell mitochondria. Biochem. Biophys. Res. Commun. 1994, 203, 46–52. [Google Scholar] [CrossRef]
- Floyd, S.; Favre, C.; Lasorsa, F.M.; Leahy, M.; Trigiante, G.; Stroebel, P.; Marx, A.; Loughran, G.; O’Callaghan, K.; Marobbio, C.M.T.; et al. The insulin-like growth factor-I-mTOR signaling pathway induces the mitochondrial pyrimidine nucleotide carrier to promote cell growth. Mol. Biol. Cell 2007, 18, 3545–3555. [Google Scholar] [CrossRef] [Green Version]
- Cansev, M. Uridine and cytidine in the brain: Their transport and utilization. Brain Res. Rev. 2006, 52, 389–397. [Google Scholar] [CrossRef]
- Favre, C.; Zhdanov, A.; Leahy, M.; Papkovsky, D.; O’Connor, R. Mitochondrial pyrimidine nucleotide carrier (PNC1) regulates mitochondrial biogenesis and the invasive phenotype of cancer cells. Oncogene 2010, 29, 3964–3976. [Google Scholar] [CrossRef] [Green Version]
- Reunert, J.; van den Heuvel, A.; Rust, S.; Marquardt, T. Cerebro-oculo-facio-skeletal syndrome caused by the homozygous pathogenic variant Gly47Arg in ERCC2. Am. J. Med. Genet. A 2021, 185, 930–936. [Google Scholar] [CrossRef]
- Park, J.H.; Elpers, C.; Reunert, J.; McCormick, M.L.; Mohr, J.; Biskup, S.; Schwartz, O.; Rust, S.; Grüneberg, M.; Seelhöfer, A.; et al. SOD1 deficiency: A novel syndrome distinct from amyotrophic lateral sclerosis. Brain 2019, 142, 2230–2237. [Google Scholar] [CrossRef]
- Dimmock, D.; Tang, L.-Y.; Schmitt, E.S.; Wong, L.-J.C. Quantitative evaluation of the mitochondrial DNA depletion syndrome. Clin. Chem. 2010, 56, 1119–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ost, M.; Doerrier, C.; Gama-Perez, P.; Moreno-Gomez, S. Analysis of mitochondrial respiratory function in tissue biopsies and blood cells. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F.; Indiveri, C.; Bisaccia, F.; Iacobazzi, V. Mitochondrial metabolite carrier proteins: Purification, reconstitution, and transport studies. In Mitochondrial Biogenesis and Genetics; Attardi, G., Ed.; Academic Press: San Diego, CA, USA, 1995; pp. 349–369. ISBN 9780121821616. [Google Scholar]
- Indiveri, C.; Iacobazzi, V.; Giangregorio, N.; Palmieri, F. Bacterial overexpression, purification, and reconstitution of the carnitine/acylcarnitine carrier from rat liver mitochondria. Biochem. Biophys. Res. Commun. 1998, 249, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, V.; Fiermonte, G.; Longo, A.; Palmieri, F. The human gene SLC25A29, of solute carrier family 25, encodes a mitochondrial transporter of basic amino acids. J. Biol. Chem. 2014, 289, 13374–13384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marobbio, C.M.T.; Vozza, A.; Harding, M.; Bisaccia, F.; Palmieri, F.; Walker, J.E. Identification and reconstitution of the yeast mitochondrial transporter for thiamine pyrophosphate. EMBO J. 2002, 21, 5653–5661. [Google Scholar] [CrossRef] [Green Version]
- Marobbio, C.M.T.; Di Noia, M.A.; Palmieri, F. Identification of a mitochondrial transporter for pyrimidine nucleotides in Saccharomyces cerevisiae: Bacterial expression, reconstitution and functional characterization. Biochem. J. 2006, 393, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Capobianco, L.; Bisaccia, F.; Mazzeo, M.; Palmieri, F. The mitochondrial oxoglutarate carrier: Sulfhydryl reagents bind to cysteine-184, and this interaction is enhanced by substrate binding. Biochemistry 1996, 35, 8974–8980. [Google Scholar] [CrossRef] [PubMed]
- Marobbio, C.M.T.; Giannuzzi, G.; Paradies, E.; Pierri, C.L.; Palmieri, F. alpha-Isopropylmalate, a leucine biosynthesis intermediate in yeast, is transported by the mitochondrial oxalacetate carrier. J. Biol. Chem. 2008, 283, 28445–28453. [Google Scholar] [CrossRef] [Green Version]
- Monné, M.; Daddabbo, L.; Gagneul, D.; Obata, T.; Hielscher, B.; Palmieri, L.; Miniero, D.V.; Fernie, A.R.; Weber, A.P.M.; Palmieri, F. Uncoupling proteins 1 and 2 (UCP1 and UCP2) from Arabidopsis thaliana are mitochondrial transporters of aspartate, glutamate, and dicarboxylates. J. Biol. Chem. 2018, 293, 4213–4227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrimi, G.; Russo, A.; Pierri, C.L.; Palmieri, F. The peroxisomal NAD+ carrier of Arabidopsis thaliana transports coenzyme A and its derivatives. J. Bioenerg. Biomembr. 2012, 44, 333–340. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jasper, L.; Scarcia, P.; Rust, S.; Reunert, J.; Palmieri, F.; Marquardt, T. Uridine Treatment of the First Known Case of SLC25A36 Deficiency. Int. J. Mol. Sci. 2021, 22, 9929. https://doi.org/10.3390/ijms22189929
Jasper L, Scarcia P, Rust S, Reunert J, Palmieri F, Marquardt T. Uridine Treatment of the First Known Case of SLC25A36 Deficiency. International Journal of Molecular Sciences. 2021; 22(18):9929. https://doi.org/10.3390/ijms22189929
Chicago/Turabian StyleJasper, Luisa, Pasquale Scarcia, Stephan Rust, Janine Reunert, Ferdinando Palmieri, and Thorsten Marquardt. 2021. "Uridine Treatment of the First Known Case of SLC25A36 Deficiency" International Journal of Molecular Sciences 22, no. 18: 9929. https://doi.org/10.3390/ijms22189929
APA StyleJasper, L., Scarcia, P., Rust, S., Reunert, J., Palmieri, F., & Marquardt, T. (2021). Uridine Treatment of the First Known Case of SLC25A36 Deficiency. International Journal of Molecular Sciences, 22(18), 9929. https://doi.org/10.3390/ijms22189929