
Molecular Landscapes of Gastric Pre-Neoplastic and Pre-Invasive Lesions



 , , , , , and
, , , , , and 
Abstract
:1. Introduction
1.1. Epidemiology and Risk Factors
1.2. Histological and Molecular Classifications of GC
2. Precursor Lesions of Intestinal Type Adenocarcinoma and Their Molecular Alterations
2.1. Chronic and Atrophic Gastritis
2.2. Intestinal Metaplasia and the “Point of No Return”
2.3. Spasmolytic Polypeptide-Expressing Metaplasia
2.4. Dysplasia
2.5. Special Type of Gastric Adenomas
3. Precursors of Diffuse Gastric Cancer
3.1. Hereditary Diffuse Gastric Cancer
3.2. Sporadic Diffuse Gastric Cancer
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Anderson, W.F.; Camargo, M.C.; Fraumeni, J.F.; Correa, P.; Rosenberg, P.S.; Rabkin, C.S. Age-specific trends in incidence of noncardia gastric cancer in US adults. JAMA 2010, 303, 1723–1728. [Google Scholar] [CrossRef] [Green Version]
- Rubenstein, J.H.; Taylor, J.B. Meta-analysis: The association of oesophageal adenocarcinoma with symptoms of gastro-oesophageal reflux. Aliment. Pharmacol. Ther. 2010, 32, 1222–1227. [Google Scholar] [CrossRef]
- Gullo, I.; Grillo, F.; Mastracci, L.; Vanoli, A.; Carneiro, F.; Saragoni, L.; Limarzi, F.; Ferro, J.; Parente, P.; Fassan, M. Precancerous lesions of the stomach, gastric cancer and hereditary gastric cancer syndromes. Pathologica 2020, 112, 166–185. [Google Scholar] [CrossRef]
- Choi, Y.J.; Kim, N. Gastric cancer and family history. Korean J. Intern. Med. 2016, 31, 1042–1053. [Google Scholar] [CrossRef] [PubMed]
- Stolte, M.; Bayerdörffer, E.; Morgner, A.; Alpen, B.; Wündisch, T.; Thiede, C.; Neubauer, A. Helicobacter and gastric MALT lymphoma. Gut 2002, 50 (Suppl. S3), III19–III24. [Google Scholar] [CrossRef] [PubMed]
- Toh, J.W.T.; Wilson, R.B. Pathways of gastric carcinogenesis, Helicobacter pylori virulence and interactions with antioxidant systems, vitamin C and phytochemicals. Int. J. Mol. Sci. 2020, 21, 6451. [Google Scholar] [CrossRef]
- Rugge, M.; Genta, R.M.; Di Mario, F.; El-Omar, E.M.; El-Serag, H.B.; Fassan, M.; Hunt, R.H.; Kuipers, E.J.; Malfertheiner, P.; Sugano, K.; et al. Gastric cancer as preventable disease. Clin. Gastroenterol. Hepatol. 2017, 15, 1833–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, J.; Oliveira, P.; Senz, J.; São José, C.; Hansford, S.; Teles, S.P.; Ferreira, M.; Corso, G.; Pinheiro, H.; Lemos, D.; et al. Redefinition of familial intestinal gastric cancer: Clinical and genetic perspectives. J. Med. Genet. 2021, 58, 1–11. [Google Scholar] [CrossRef]
- Spoto, C.P.E.; Gullo, I.; Carneiro, F.; Montgomery, E.A.; Brosens, L.A.A. Hereditary gastrointestinal carcinomas and their precursors: An algorithm for genetic testing. Semin. Diagn. Pathol. 2018, 35, 170–183. [Google Scholar] [CrossRef]
- Van der Post, R.S.; Carneiro, F. Emerging concepts in gastric neoplasia: Heritable gastric cancers and polyposis disorders. Surg. Pathol. Clin. 2017, 10, 931–945. [Google Scholar] [CrossRef]
- Sandoval-Bórquez, A.; Saavedra, K.; Carrasco-Avino, G.; Garcia-Bloj, B.; Fry, J.; Wichmann, I.; Corvalán, A.H. Noncoding genomics in gastric cancer and the gastric precancerous cascade: Pathogenesis and biomarkers. Dis. Markers 2015, 2015, 503762. [Google Scholar] [CrossRef] [Green Version]
- Businello, G.; Galuppini, F.; Fassan, M. The impact of recent next generation sequencing and the need for a new classification in gastric cancer. Best Pract. Res. Clin. Gastroenterol. 2021, 50–51, 101730. [Google Scholar] [CrossRef] [PubMed]
- WHO. Classification of Tumours Editorial Board. Digestive System Tumours; International Agency for Research on Cancer: Lyon, France, 2019. [Google Scholar]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef]
- Graziano, F.; Humar, B.; Guilford, P. The role of the E-cadherin gene (CDH1) in diffuse gastric cancer susceptibility: From the laboratory to clinical practice. Ann. Oncol. 2003, 14, 1705–1713. [Google Scholar] [CrossRef]
- Network CGAR. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Kawazoe, A.; Lordick, F.; Janjigian, Y.Y.; Shitara, K. Biomarker-targeted therapies for advanced-stage gastric and gastro-oesophageal junction cancers: An emerging paradigm. Nat. Rev. Clin. Oncol. 2021, 18, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. ToGA Trial Investigators. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 28, 687–697. [Google Scholar] [CrossRef]
- Shitara, K.; Bang, Y.J.; Iwasa, S.; Sugimoto, N.; Ryu, M.H.; Sakai, D.; Chung, H.C.; Kawakami, H.; Yabusaki, H.; Lee, J.; et al. DESTINY-Gastric01 investigators. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N. Engl. J. Med. 2020, 18, 2419–2430. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Enzinger, P.C.; Kang, Y.K.; Yamaguchi, K.; Qin, S.; Lee, K.W.; Oh, S.C.; Li, J.; Turk, H.M.; Teixeira, A.C.; et al. Randomized double-blind placebo-controlled phase 2 study of bemarituzumab combined with modified FOLFOX6 (mFOLFOX6) in first-line (1L) treatment of advanced gastric/gastroesophageal junction adenocarcinoma (FIGHT). J. Clin. Oncol. 2021, 39 (Suppl. S3), 160. [Google Scholar] [CrossRef]
- Catenacci, D.V.; Tesfaye, A.; Tejani, M.; Cheung, E.; Eisenberg, P.; Scott, A.J.; Eng, C.; Hnatyszyn, J.; Marina, N.; Powers, J.; et al. Bemarituzumab with modified FOLFOX6 for advanced FGFR2-positive gastroesophageal cancer: FIGHT Phase III study design. Future Oncol. 2019, 15, 2073–2082. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.A.; Bang, Y.J.; Lordick, F.; Alsina, M.; Chen, M.; Hack, S.P.; Bruey, J.M.; Smith, D.; McCaffery, I.; Shames, D.S.; et al. Effect of fluorouracil, leucovorin, and oxaliplatin with or without onartuzumab in HER2-Negative, MET-positive gastroesophageal adenocarcinoma: The METGastric randomized clinical trial. JAMA Oncol. 2017, 3, 620–627. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.T.; Kim, K.; Lee, H.; Kozarewa, I.; Mortimer, P.G.S.; Odegaard, J.I.; Harrington, E.A.; Lee, J.; Lee, T.; et al. Tumor genomic profiling guides patients with metastatic gastric cancer to targeted treatment: The VIKTORY umbrella trial. Cancer Discov. 2019, 9, 1388–1405. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. REGARD Trial Investigators. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.Y.; et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Sahin, U.; Türeci, Ö.; Manikhas, G.; Lordick, F.; Rusyn, A.; Vynnychenko, I.; Dudov, A.; Bazin, I.; Bondarenko, I.; Melichar, B.; et al. FAST: A randomised phase II study of zolbetuximab (IMAB362) plus EOX versus EOX alone for first-line treatment of advanced CLDN18.2-positive gastric and gastro-oesophageal adenocarcinoma. Ann. Oncol. 2021, 32, 609–619. [Google Scholar] [CrossRef]
- Moehler, M.; Shitara, K.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; Liu, T.; et al. Nivolumab (nivo) plus chemotherapy (chemo) versus chemo as first-line (1L) treatment for advanced gastric cancer/gastroesophageal junction cancer (GC/GEJC)/esophageal adenocarcinoma (EAC): First results of the CheckMate 649 study. Ann. Oncol. 2020, 31 (Suppl. S4), S1142–S1215. [Google Scholar] [CrossRef]
- Boku, N.; Ryu, M.H.; Oh, D.; Oh, S.C.; Chung, H.C.; Lee, K.; Omori, T.; Shitara, K.; Sakuramoto, S.; Chung, I.J.; et al. Nivolumab plus chemotherapy versus chemotherapy alone in patients with previously untreated advanced or recurrent gastric/gastroesophageal junction (G/GEJ) cancer: ATTRACTION-4 (ONO-4538-37) study. Ann. Oncol. 2020, 31 (Suppl. S4), S1142–S1215. [Google Scholar] [CrossRef]
- Kang, Y.K.; Boku, N.; Satoh, T.; Ryu, M.H.; Chao, Y.; Kato, K.; Chung, H.C.; Chen, J.S.; Muro, K.; Kang, W.K.; et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 2461–2471. [Google Scholar] [CrossRef]
- Correa, P.; Haenszel, W.; Cuello, C.; Tannenbaum, S.; Archer, M. A model for gastric cancer epidemiology. Lancet 1975, 2, 58–60. [Google Scholar] [CrossRef]
- Rugge, M.; Meggio, A.; Pennelli, G.; Piscioli, F.; Giacomelli, L.; De Pretis, G.; Graham, D.Y. Gastritis staging in clinical practice: The OLGA staging system. Gut 2007, 56, 631–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa, P.; Piazuelo, M.B. The gastric precancerous cascade. J. Dig. Dis. 2012, 13, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Kawanishi, S.; Hiraku, Y.; Oikawa, S. Mechanism of guanine-specific DNA damage by oxidative stress and its role in carcinogenesis and aging. Mutat. Res. 2001, 488, 65–76. [Google Scholar] [CrossRef]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef]
- Hatakeyama, M. Structure and function of Helicobacter pylori CagA, the first-identified bacterial protein involved in human cancer. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 196–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Marusawa, H.; Matsumoto, Y.; Inuzuka, T.; Ikeda, A.; Fujii, Y.; Minamiguchi, S.; Miyamoto, S.; Kou, T.; Sakai, Y.; et al. Accumulation of somatic mutations in TP53 in gastric epithelium with Helicobacter pylori infection. Gastroenterology 2014, 147, 407–417.e3. [Google Scholar] [CrossRef]
- Matsumoto, T.; Shimizu, T.; Takai, A.; Marusawa, H. Exploring the mechanisms of gastrointestinal cancer development using deep sequencing analysis. Cancers 2015, 7, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Jiang, Q.; Lou, X.; Ji, X.; Wen, Z.; Wu, J.; Tao, H.; Jiang, T.; He, W.; Wang, C.; et al. MicroRNAs up-regulated by CagA of Helicobacter pylori induce intestinal metaplasia of gastric epithelial cells. PLoS ONE 2012, 7, e35147. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Yin, H.; Zheng, S.; Chu, A.; Li, Y.; Xing, C.; Yuan, Y.; Gong, Y. Differentially expressed mRNAs and their long noncoding RNA regulatory network with Helicobacter pylori-associated diseases including atrophic gastritis and gastric cancer. Biomed. Res. Int. 2020, 2020, 3012193. [Google Scholar] [CrossRef]
- Petkevicius, V.; Streleckiene, G.; Balciute, K.; Link, A.; Leja, M.; Malfertheiner, P.; Skieceviciene, J.; Kupcinskas, J. Association of long non-coding RNA polymorphisms with gastric cancer and atrophic gastritis. Genes 2020, 11, 1505. [Google Scholar] [CrossRef]
- Mao, Y.; Liu, R.; Zhou, H.; Yin, S.; Zhao, Q.; Ding, X.; Wang, H. Transcriptome analysis of miRNA-lncRNA-mRNA interactions in the malignant transformation process of gastric cancer initiation. Cancer Gene Ther. 2017, 24, 267–275. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, H.; Zhu, L.; Hao, B.; Zhang, W.; Hua, J.; Gu, H.; Jin, W.; Zhang, G. Helicobacter pylori infection related long noncoding RNA (lncRNA) AF147447 inhibits gastric cancer proliferation and invasion by targeting MUC2 and up-regulating miR-34c. Oncotarget 2016, 7, 82770–82782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coker, O.O.; Dai, Z.; Nie, Y.; Zhao, G.; Cao, L.; Nakatsu, G.; Wu, W.K.; Wong, S.H.; Chen, Z.; Sung, J.J.Y.; et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut 2018, 67, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gao, X.; Zeng, R.; Wu, Q.; Sun, H.; Wu, W.; Zhang, X.; Sun, G.; Yan, B.; Wu, L.; et al. Changes of the Gastric Mucosal Microbiome associated with histological stages of gastric carcinogenesis. Front. Microbiol. 2020, 11, 997. [Google Scholar] [CrossRef]
- Sung, J.J.Y.; Coker, O.O.; Chu, E.; Szeto, C.H.; Luk, S.T.Y.; Lau, H.C.H.; Yu, J. Gastric microbes associated with gastric inflammation, atrophy and intestinal metaplasia 1 year after Helicobacter pylori eradication. Gut 2020, 69, 1572–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negovan, A.; Iancu, M.; Fülöp, E.; Bănescu, C. Helicobacter pylori and cytokine gene variants as predictors of premalignant gastric lesions. World J. Gastroenterol. 2019, 25, 4105–4124. [Google Scholar] [CrossRef]
- Fu, H.; Ma, Y.; Yang, M.; Zhang, C.; Huang, H.; Xia, Y.; Lu, L.; Jin, W.; Cui, D. Persisting and increasing neutrophil infiltration associates with gastric carcinogenesis and E-cadherin downregulation. Sci. Rep. 2016, 6, 29762. [Google Scholar] [CrossRef] [Green Version]
- Coati, I.; Fassan, M.; Farinati, F.; Graham, D.Y.; Genta, R.M.; Rugge, M. Autoimmune gastritis: Pathologist’s viewpoint. World J. Gastroenterol. 2015, 21, 12179–12189. [Google Scholar] [CrossRef]
- Vannella, L.; Lahner, E.; Osborn, J.; Annibale, B. Systematic review: Gastric cancer incidence in pernicious anaemia. Aliment. Pharmacol. Ther. 2013, 37, 375–382. [Google Scholar] [CrossRef]
- Elsborg, L.; Mosbech, J. Pernicious anaemia as a risk factor in gastric cancer. Acta Med. Scand. 1979, 206, 315–318. [Google Scholar] [CrossRef]
- Rugge, M.; Fassan, M.; Pizzi, M.; Zorzetto, V.; Maddalo, G.; Realdon, S.; De Bernard, M.; Betterle, C.; Cappellesso, R.; Pennelli, G.; et al. Autoimmune gastritis: Histology phenotype and OLGA staging. Aliment. Pharmacol. Ther. 2012, 35, 1460–1466. [Google Scholar] [CrossRef] [PubMed]
- Bizzaro, N.; Antico, A.; Villalta, D. Autoimmunity and gastric cancer. Int. J. Mol. Sci. 2018, 19, 377. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.Y.; Chan, A.O.; Rashid, A.; Wong, D.K.; Seto, W.K.; Cho, C.H.; Lai, C.L.; Yuen, M.F. Interleukin-1β increases the risk of gastric cancer through induction of aberrant DNA methylation in a mouse model. Oncol. Lett. 2016, 11, 2919–2924. [Google Scholar] [CrossRef] [Green Version]
- De Vries, A.C.; van Grieken, N.C.; Looman, C.W.; Casparie, M.K.; de Vries, E.; Meijer, G.A.; Kuipers, E.J. Gastric cancer risk in patients with premalignant gastric lesions: A nationwide cohort study in the Netherlands. Gastroenterology 2008, 134, 945–952. [Google Scholar] [CrossRef]
- Li, D.; Bautista, M.C.; Jiang, S.F.; Daryani, P.; Brackett, M.; Armstrong, M.A.; Hung, Y.Y.; Postlethwaite, D.; Ladabaum, U. Risks and predictors of gastric adenocarcinoma in patients with gastric intestinal metaplasia and dysplasia: A Population-based study. Am. J. Gastroenterol. 2016, 111, 1104–1113. [Google Scholar] [CrossRef]
- Filipe, M.I.; Potet, F.; Bogomoletz, W.V.; Dawson, P.A.; Fabiani, B.; Chauveinc, P.; Fenzy, A.; Gazzard, B.; Goldfain, D.; Zeegen, R. Incomplete sulphomucin-secreting intestinal metaplasia for gastric cancer. Preliminary data from a prospective study from three centres. Gut 1985, 26, 1319–1326. [Google Scholar] [CrossRef] [Green Version]
- Yakirevich, E.; Resnick, M.B. Pathology of gastric cancer and its precursor lesions. Gastroenterol. Clin. 2013, 42, 261–284. [Google Scholar] [CrossRef] [PubMed]
- Camilo, V.; Garrido, M.; Valente, P.; Ricardo, S.; Amaral, A.L.; Barros, R.; Chaves, P.; Carneiro, F.; David, L.; Almeida, R. Differentiation reprogramming in gastric intestinal metaplasia and dysplasia: Role of SOX2 and CDX2. Histopathology 2015, 66, 343–350. [Google Scholar] [CrossRef] [PubMed]
- González, C.A.; Sanz-Anquela, J.M.; Gisbert, J.P.; Correa, P. Utility of subtyping intestinal metaplasia as marker of gastric cancer risk. A review of the evidence. Int. J. Cancer 2013, 133, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- González, C.A.; Sanz-Anquela, J.M.; Companioni, O.; Bonet, C.; Berdasco, M.; López, C.; Mendoza, J.; Martín-Arranz, M.D.; Rey, E.; Poves, E.; et al. Incomplete type of intestinal metaplasia has the highest risk to progress to gastric cancer: Results of the Spanish follow-up multicenter study. J. Gastroenterol. Hepatol. 2016, 31, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Pittayanon, R.; Rerknimitr, R.; Klaikaew, N.; Sanpavat, A.; Chaithongrat, S.; Mahachai, V.; Kullavanijaya, P.; Barkun, A. The risk of gastric cancer in patients with gastric intestinal metaplasia in 5-year follow-up. Aliment. Pharmacol. Ther. 2017, 46, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Koulis, A.; Buckle, A.; Boussioutas, A. Premalignant lesions and gastric cancer: Current understanding. World J. Gastrointest. Oncol. 2019, 11, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.K.; Ramnarayanan, K.; Zhu, F.; Srivastava, S.; Xu, C.; Tan, A.L.K.; Lee, M.; Tay, S.; Das, K.; Xing, M.; et al. Genomic and epigenomic profiling of high-risk intestinal metaplasia reveals molecular determinants of progression to gastric cancer. Cancer Cell 2018, 33, 137–150.e5. [Google Scholar] [CrossRef] [Green Version]
- Tahara, E. Genetic pathways of two types of gastric cancer. IARC Sci. Publ. 2004, 6, 327–349. [Google Scholar]
- Fassan, M.; Simbolo, M.; Bria, E.; Mafficini, A.; Pilotto, S.; Capelli, P.; Bencivenga, M.; Pecori, S.; Luchini, C.; Neves, D.; et al. High-throughput mutation profiling identifies novel molecular dysregulation in high-grade intraepithelial neoplasia and early gastric cancers. Gastric Cancer 2014, 17, 442–449. [Google Scholar] [CrossRef]
- Mu, Y.; Zhang, Q.; Mei, L.; Liu, X.; Yang, W.; Yu, J. Telomere shortening occurs early during gastrocarcinogenesis. Med. Oncol. 2012, 29, 893–898. [Google Scholar] [CrossRef]
- Lin, R.; Li, C.; Liu, Z.; Wu, R.; Lu, J. Genome-wide DNA methylation profiling identifies epigenetic signatures of gastric cardiac intestinal metaplasia. J. Transl. Med. 2020, 18, 292. [Google Scholar] [CrossRef]
- Hamamoto, T.; Yokozaki, H.; Semba, S.; Yasui, W.; Yunotani, S.; Miyazaki, K.; Tahara, E. Altered microsatellites in incomplete-type intestinal metaplasia adjacent to primary gastric cancers. J. Clin. Pathol. 1997, 50, 841–846. [Google Scholar] [CrossRef] [Green Version]
- Cortés-Márquez, A.C.; Mendoza-Elizalde, S.; Arenas-Huertero, F.; Trillo-Tinoco, J.; Valencia-Mayoral, P.; Consuelo-Sánchez, A.; Zarate-Franco, J.; Dionicio-Avendaño, A.R.; Herrera-Esquivel, J.J.; Recinos-Carrera, E.G.; et al. Differential expression of miRNA-146a and miRNA-155 in gastritis induced by Helicobacter pylori infection in paediatric patients, adults, and an animal model. BMC Infect. Dis. 2018, 18, 463. [Google Scholar] [CrossRef]
- Li, H.; Wu, Q.; Li, T.; Liu, C.; Xue, L.; Ding, J.; Shi, Y.; Fan, D. The miR-17-92 cluster as a potential biomarker for the early diagnosis of gastric cancer: Evidence and literature review. Oncotarget 2017, 8, 45060–45071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, M.; Li, L.; Zheng, W.C. Reduced miR-490-3p expression is associated with poor prognosis of Helicobacter pylori induced gastric cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3384–3388. [Google Scholar]
- Shen, J.; Xiao, Z.; Wu, W.K.; Wang, M.H.; To, K.F.; Chen, Y.; Yang, W.; Li, M.S.; Shin, V.Y.; Tong, J.H.; et al. Epigenetic silencing of miR-490-3p reactivates the chromatin remodeler SMARCD1 to promote Helicobacter pylori-induced gastric carcinogenesis. Cancer Res. 2015, 75, 754–765. [Google Scholar] [CrossRef] [Green Version]
- Min, J.; Han, T.S.; Sohn, Y.; Shimizu, T.; Choi, B.; Bae, S.W.; Hur, K.; Kong, S.H.; Suh, Y.S.; Lee, H.J.; et al. microRNA-30a arbitrates intestinal-type early gastric carcinogenesis by directly targeting ITGA2. Gastric Cancer 2020, 23, 600–613. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Wu, Y.; Wang, D.; Qin, Z.F. MicroRNA network analysis identifies key microRNAs and genes associated with precancerous lesions of gastric cancer. Genet. Mol. Res. 2014, 13, 8695–8703. [Google Scholar] [CrossRef]
- Rokkas, T.; Pistiolas, D.; Sechopoulos, P.; Robotis, I.; Margantinis, G. The long-term impact of Helicobacter pylori eradication on gastric histology: A systematic review and meta-analysis. Helicobacter 2007, 12 (Suppl. S2), 32–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, L.; Shi, R.; Huang, X.; Li, S.W.; Huang, Z.; Zhang, G. Gastric atrophy and intestinal metaplasia before and after Helicobacter pylori eradication: A meta-analysis. Digestion 2011, 83, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.M.; Kim, N.; Chang, H.; Kim, J.S.; Lee, D.H.; Jung, H.C. Follow-up study on CDX1 and CDX2 mRNA expression in noncancerous gastric mucosae after Helicobacter pylori eradication. Dig. Dis. Sci. 2016, 61, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.N.; Wang, Z.; Li, X.; Zhou, Z.G. Helicobacter pylori eradication cannot reduce the risk of gastric cancer in patients with intestinal metaplasia and dysplasia: Evidence from a meta-analysis. Gastric Cancer 2016, 19, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.M.; Kim, N.; Shin, C.M.; Lee, H.S.; Lee, D.H.; Jung, H.C.; Song, I.S. Predictive factors for improvement of atrophic gastritis and intestinal metaplasia after Helicobacter pylori eradication: A three-year follow-up study in Korea. Helicobacter 2012, 17, 86–95. [Google Scholar] [CrossRef]
- Lee, Y.C.; Chen, T.H.; Chiu, H.M.; Shun, C.T.; Chiang, H.; Liu, T.Y.; Wu, M.S.; Lin, J.T. The benefit of mass eradication of Helicobacter pylori infection: A community-based study of gastric cancer prevention. Gut 2013, 62, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Den Hollander, W.J.; Holster, I.L.; den Hoed, C.M.; Capelle, L.G.; Tang, T.J.; Anten, M.P.; Prytz-Berset, I.; Witteman, E.M.; Ter Borg, F.; Hartog, G.D.; et al. Surveillance of premalignant gastric lesions: A multicentre prospective cohort study from low incidence regions. Gut 2019, 68, 585–593. [Google Scholar] [CrossRef]
- Rugge, M.; de Boni, M.; Pennelli, G.; de Bona, M.; Giacomelli, L.; Fassan, M.; Basso, D.; Plebani, M.; Graham, D.Y. Gastritis OLGA-staging and gastric cancer risk: A twelve-year clinico-pathological follow-up study. Aliment. Pharmacol Ther. 2010, 31, 1104–1111. [Google Scholar] [CrossRef]
- Rugge, M.; Fassan, M.; Pizzi, M.; Farinati, F.; Sturniolo, G.C.; Plebani, M.; Graham, D.Y. Operative link for gastritis assessment vs. operative link on intestinal metaplasia assessment. World J. Gastroenterol. 2011, 17, 4596–4601. [Google Scholar] [CrossRef] [PubMed]
- Goldenring, J.R.; Nam, K.T.; Wang, T.C.; Mills, J.C.; Wright, N.A. Spasmolytic polypeptide-expressing metaplasia and intestinal metaplasia: Time for reevaluation of metaplasias and the origins of gastric cancer. Gastroenterology 2010, 138, 2207–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, P.H.; Lee, J.R.; Joshi, V.; Playford, R.J.; Poulsom, R.; Wright, N.A.; Goldenring, J.R. Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Lab. Investig. 1999, 79, 639–646. [Google Scholar]
- Halldórsdóttir, A.M.; Sigurdardóttrir, M.; Jónasson, J.G.; Oddsdóttir, M.; Magnússon, J.; Lee, J.R.; Goldenring, J.R. Spasmolytic polypeptide-expressing metaplasia (SPEM) associated with gastric cancer in Iceland. Dig. Dis. Sci. 2003, 48, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Goldenring, J.R.; Kaminishi, M.; Lee, J.R. Identification of spasmolytic polypeptide expressing metaplasia (SPEM) in remnant gastric cancer and surveillance postgastrectomy biopsies. Dig. Dis. Sci. 2002, 47, 573–578. [Google Scholar] [CrossRef]
- Goldenring, J.R.; Nam, K.T. Oxyntic atrophy, metaplasia, and gastric cancer. Prog. Mol. Biol. Transl. Sci. 2010, 96, 117–131. [Google Scholar]
- Petersen, C.P.; Weis, V.G.; Nam, K.T.; Sousa, J.F.; Fingleton, B.; Goldenring, J.R. Macrophages promote progression of spasmolytic polypeptide-expressing metaplasia after acute loss of parietal cells. Gastroenterology 2014, 146, 1727–1738.e8. [Google Scholar] [CrossRef] [Green Version]
- Rokutan, H.; Abe, H.; Nakamura, H.; Ushiku, T.; Arakawa, E.; Hosoda, F.; Yachida, S.; Tsuji, Y.; Fujishiro, M.; Koike, K.; et al. Initial and crucial genetic events in intestinal-type gastric intramucosal neoplasia. J. Pathol. 2019, 247, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Feng, L.; Liu, Y.; Zhou, W.X.; Ma, Y.C.; Fei, G.J.; An, N.; Li, Y.; Wu, X.; Yao, F.; et al. Differential gene expression profiling of gastric intraepithelial neoplasia and early-stage adenocarcinoma. World J. Gastroenterol. 2014, 20, 17883–17893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, M.; Tsukamoto, Y.; Uchida, T.; Ishikawa, Y.; Nagai, T.; Hijiya, N.; Nguyen, L.T.; Nakada, C.; Kuroda, A.; Okimoto, T.; et al. Genomic profiling of gastric carcinoma in situ and adenomas by array-based comparative genomic hybridization. J. Pathol. 2010, 221, 96–105. [Google Scholar] [CrossRef]
- Sugai, T.; Eizuka, M.; Arakawa, N.; Osakabe, M.; Habano, W.; Fujita, Y.; Yamamoto, E.; Yamano, H.; Endoh, M.; Matsumoto, T.; et al. Molecular profiling and comprehensive genome-wide analysis of somatic copy number alterations in gastric intramucosal neoplasias based on microsatellite status. Gastric Cancer 2018, 21, 765–775. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Abraham, S.C.; Kim, H.S.; Nam, J.H.; Choi, C.; Lee, M.C.; Park, C.S.; Juhng, S.W.; Rashid, A.; Hamilton, S.R.; et al. Inverse relationship between APC gene mutation in gastric adenomas and development of adenocarcinoma. Am. J. Pathol. 2002, 161, 611–618. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, S.; Sano, T.; Nakajima, T. Clinicopathological and molecular biological studies of gastric adenomas with special reference to p53 abnormality. Pathol. Int. 1995, 45, 51–57. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, X.; Zhang, C.; Wang, J.; Fei, G.; Di, X.; Lu, X.; Feng, L.; Cheng, S.; Yang, A. Dissecting expression profiles of gastric precancerous lesions and early gastric cancer to explore crucial molecules in intestinal-type gastric cancer tumorigenesis. J. Pathol. 2020, 251, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Yang, L.; Liang, S.; Liu, D.; Chen, X.; Ma, Z.; Zhai, S.; Li, P.; Wang, X. AEG-1 is a target of perifosine and is over-expressed in gastric dysplasia and cancers. Dig. Dis. Sci. 2013, 58, 2873–2880. [Google Scholar] [CrossRef]
- Fassan, M.; Mastracci, L.; Grillo, F.; Zagonel, V.; Bruno, S.; Battaglia, G.; Pitto, F.; Nitti, D.; Celiento, T.; Zaninotto, G.; et al. Early HER2 dysregulation in gastric and oesophageal carcinogenesis. Histopathology 2012, 61, 769–776. [Google Scholar] [CrossRef]
- Fassan, M.; Brignola, S.; Pennelli, G.; Alberti, G.; Angerilli, V.; Bressan, A.; Pellino, A.; Lanza, C.; Salmaso, R.; Lonardi, S.; et al. PD-L1 expression in gastroesophageal dysplastic lesions. Virchows Arch. 2020, 477, 151–156. [Google Scholar] [CrossRef]
- Hwang, J.; Min, B.H.; Jang, J.; Kang, S.Y.; Bae, H.; Jang, S.S.; Kim, J.I.; Kim, K.M. MicroRNA Expression profiles in gastric carcinogenesis. Sci. Rep. 2018, 8, 14393. [Google Scholar] [CrossRef]
- Zhu, M.; Zhang, N.; He, S. Similarly up-regulated microRNA-106a in matched formalin-fixed paraffin-embedded and fresh frozen samples and the dynamic changes during gastric carcinogenesis and development. Pathol. Res. Pract. 2014, 210, 909–915. [Google Scholar] [CrossRef]
- Fassan, M.; Pizzi, M.; Realdon, S.; Balistreri, M.; Guzzardo, V.; Zagonel, V.; Castoro, C.; Mastracci, L.; Farinati, F.; Nitti, D.; et al. The HER2-miR125a5p/miR125b loop in gastric and esophageal carcinogenesis. Hum. Pathol. 2013, 44, 1804–1810. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.X.; Ding, S.Z. Helicobacter pylori infection-induced H3Ser10 phosphorylation in stepwise gastric carcinogenesis and its clinical implications. Helicobacter 2018, 23, e12486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Q.; Li, G.; Wang, X.; Wang, S.; Hu, J.; Yang, L.; He, Y.; Pan, Y.; Yu, D.; Wu, Y. A decrease of histone deacetylase 6 expression caused by Helicobacter pylori infection is associated with oncogenic transformation in gastric cancer. Cell Physiol. Biochem. 2017, 42, 1326–1335. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, B.; Gao, W.; An, Y.; Dong, G.; Jia, J.; Yang, Q. Helicobacter pylori inhibits autophagic flux and promotes its intracellular survival and colonization by down-regulating SIRT1. J. Cell. Mol. Med. 2021, 25, 3348–3360. [Google Scholar] [CrossRef]
- Valente, P.; Garrido, M.; Gullo, I.; Baldaia, H.; Marques, M.; Baldaque-Silva, F.; Lopes, J.; Carneiro, F. Epithelial dysplasia of the stomach with gastric immunophenotype shows features of biological aggressiveness. Gastric Cancer 2015, 18, 720–728. [Google Scholar] [CrossRef] [Green Version]
- Sugai, T.; Uesugi, N.; Habano, W.; Sugimoto, R.; Eizuka, M.; Fujita, Y.; Osakabe, M.; Toya, Y.; Suzuki, H.; Matsumoto, T. The clinicopathological and molecular features of sporadic gastric foveolar type neoplasia. Virchows Arch. 2020, 477, 835–844. [Google Scholar] [CrossRef]
- Abraham, S.C.; Park, S.J.; Lee, J.H.; Mugartegui, L.; Wu, T.T. Genetic alterations in gastric adenomas of intestinal and foveolar phenotypes. Mod. Pathol. 2003, 16, 786–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.H.; Cho, Y.K.; Kim, S.W.; Choi, M.G.; Rhee, J.K.; Chung, Y.J.; Lee, S.H.; Kim, T.M. The chronological sequence of somatic mutations in early gastric carcinogenesis inferred from multiregion sequencing of gastric adenomas. Oncotarget 2016, 7, 39758–39767. [Google Scholar] [CrossRef] [Green Version]
- Pennelli, G.; Grillo, F.; Galuppini, F.; Ingravallo, G.; Pilozzi, E.; Rugge, M.; Fiocca, R.; Fassan, M.; Mastracci, L. Gastritis: Update on etiological features and histological practical approach. Pathologica 2020, 112, 153–165. [Google Scholar] [CrossRef]
- Matsubara, A.; Sekine, S.; Kushima, R.; Ogawa, R.; Taniguchi, H.; Tsuda, H.; Kanai, Y. Frequent GNAS and KRAS mutations in pyloric gland adenoma of the stomach and duodenum. J. Pathol. 2013, 229, 579–587. [Google Scholar] [CrossRef]
- Hashimoto, T.; Ogawa, R.; Matsubara, A.; Taniguchi, H.; Sugano, K.; Ushiama, M.; Yoshida, T.; Kanai, Y.; Sekine, S. Familial adenomatous polyposis-associated and sporadic pyloric gland adenomas of the upper gastrointestinal tract share common genetic features. Histopathology 2015, 67, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Saito, T.; Mitomi, H.; Hidaka, Y.; Murakami, T.; Nomura, R.; Watanabe, S.; Yao, T. Mutation spectrum in the Wnt/beta-catenin signaling pathway in gastric fundic gland-associated neoplasms/polyps. Virchows Arch. 2015, 467, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, F. Hereditary gastric cancer. Pathologe 2012, 33 (Suppl. S2), 231–234. [Google Scholar] [CrossRef]
- Luo, W.; Fedda, F.; Lynch, P.; Tan, D. CDH1 gene and hereditary diffuse gastric cancer syndrome: Molecular and histological alterations and implications for diagnosis and treatment. Front. Pharmacol. 2018, 9, 1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corso, G.; Pedrazzani, C.; Pinheiro, H.; Fernandes, E.; Marrelli, D.; Rinnovati, A.; Pascale, V.; Seruca, R.; Oliveira, C.; Roviello, F. E-cadherin genetic screening and clinico-pathologic characteristics of early onset gastric cancer. Eur. J. Cancer 2011, 47, 631–639. [Google Scholar] [CrossRef]
- Hansford, S.; Kaurah, P.; Li-Chang, H.; Woo, M.; Senz, J.; Pinheiro, H.; Schrader, K.A.; Schaeffer, D.F.; Shumansky, K.; Zogopoulos, G.; et al. Hereditary diffuse gastric cancer syndrome: CDH1 mutations and beyond. JAMA Oncol. 2015, 1, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Guilford, P.; Humar, B.; Blair, V. Hereditary diffuse gastric cancer: Translation of CDH1 germline mutations into clinical practice. Gastric Cancer 2010, 13, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Te Paske, I.B.A.W.; Garcia-Pelaez, J.; Sommer, A.K.; Matalonga, L.; Starzynska, T.; Jakubowska, A.; Solve-RD-GENTURIS Group; van der Post, R.S.; Lubinski, J.; Oliveira, C.; et al. A mosaic PIK3CA variant in a young adult with diffuse gastric cancer: Case report. Eur. J. Hum. Genet. 2021, 29, 1354–1358. [Google Scholar] [CrossRef]
- Van der Post, R.S.; Vogelaar, I.P.; Manders, P.; van der Kolk, L.E.; Cats, A.; van Hest, L.P.; Sijmons, R.; Aalfs, C.M.; Ausems, M.G.; Gómez García, E.B.; et al. Accuracy of hereditary diffuse gastric cancer testing criteria and outcomes in patients with a germline mutation in CDH1. Gastroenterology 2015, 149, 897–906.e19. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, F.; Huntsman, D.G.; Smyrk, T.C.; Owen, D.A.; Seruca, R.; Pharoah, P.; Caldas, C.; Sobrinho-Simões, M. Model of the early development of diffuse gastric cancer in E-cadherin mutation carriers and its implications for patient screening. J. Pathol. 2004, 203, 681–687. [Google Scholar] [CrossRef]
- Milne, A.N.; Sitarz, R.; Carvalho, R.; Carneiro, F.; Offerhaus, G.J. Early onset gastric cancer: On the road to unraveling gastric carcinogenesis. Curr. Mol. Med. 2007, 7, 15–28. [Google Scholar] [CrossRef]
- Barber, M.; Murrell, A.; Ito, Y.; Maia, A.T.; Hyland, S.; Oliveira, C.; Save, V.; Carneiro, F.; Paterson, A.L.; Grehan, N.; et al. Mechanisms and sequelae of E-cadherin silencing in hereditary diffuse gastric cancer. J. Pathol. 2008, 216, 295–306. [Google Scholar] [CrossRef]
- Grady, W.M.; Willis, J.; Guilford, P.J.; Dunbier, A.K.; Toro, T.T.; Lynch, H.; Wiesner, G.; Ferguson, K.; Eng, C.; Park, J.G.; et al. Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat. Genet. 2000, 26, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Sousa, S.; Pinheiro, H.; Karam, R.; Bordeira-Carriço, R.; Senz, J.; Kaurah, P.; Carvalho, J.; Pereira, R.; Gusmão, L.; et al. Quantification of epigenetic and genetic 2nd hits in CDH1 during hereditary diffuse gastric cancer syndrome progression. Gastroenterology 2009, 136, 2137–2148. [Google Scholar] [CrossRef]
- Humar, B.; Fukuzawa, R.; Blair, V.; Dunbier, A.; More, H.; Charlton, A.; Yang, H.K.; Kim, W.H.; Reeve, A.E.; Martin, I.; et al. Destabilized adhesion in the gastric proliferative zone and c-Src kinase activation mark the development of early diffuse gastric cancer. Cancer Res. 2007, 67, 2480–2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garziera, M.; Canzonieri, V.; Cannizzaro, R.; Geremia, S.; Caggiari, L.; De Zorzi, M.; Maiero, S.; Orzes, E.; Perin, T.; Zanussi, S.; et al. Identification and characterization of CDH1 germline variants in sporadic gastric cancer patients and in individuals at risk of gastric cancer. PLoS ONE 2013, 8, e77035. [Google Scholar]
- Group HaCC. Gastric cancer and Helicobacter pylori: A combined analysis of 12 case control studies nested within prospective cohorts. Gut 2001, 49, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.Q.; Sridhar, S.; Chen, Y.; Hunt, R.H. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998, 114, 1169–1179. [Google Scholar] [CrossRef]
- Chan, A.O.; Lam, S.K.; Wong, B.C.; Wong, W.M.; Yuen, M.F.; Yeung, Y.H.; Hui, W.M.; Rashid, A.; Kwong, Y.L. Promoter methylation of E-cadherin gene in gastric mucosa associated with Helicobacter pylori infection and in gastric cancer. Gut 2003, 52, 502–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, A.O.; Huang, C.; Hui, W.M.; Cho, C.H.; Yuen, M.F.; Lam, S.K.; Rashid, A.; Wong, B.C. Stability of E-cadherin methylation status in gastric mucosa associated with histology changes. Aliment. Pharmacol. Ther. 2006, 24, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Smyrk, T.C.; Zhang, L. Histologic and immunohistochemical differences between hereditary and sporadic diffuse gastric carcinoma. Hum. Pathol. 2018, 74, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.Y.; Kim, N.; Lee, J.; Lee, J.Y.; Hwang, Y.J.; Lee, H.S.; Yoon, H.; Shin, C.M.; Park, Y.S.; Kim, J.W.; et al. Usefulness of OLGA and OLGIM system not only for intestinal type but also for diffuse type of gastric cancer, and no interaction among the gastric cancer risk factors. Helicobacter 2018, 23, e12542. [Google Scholar] [CrossRef] [PubMed]
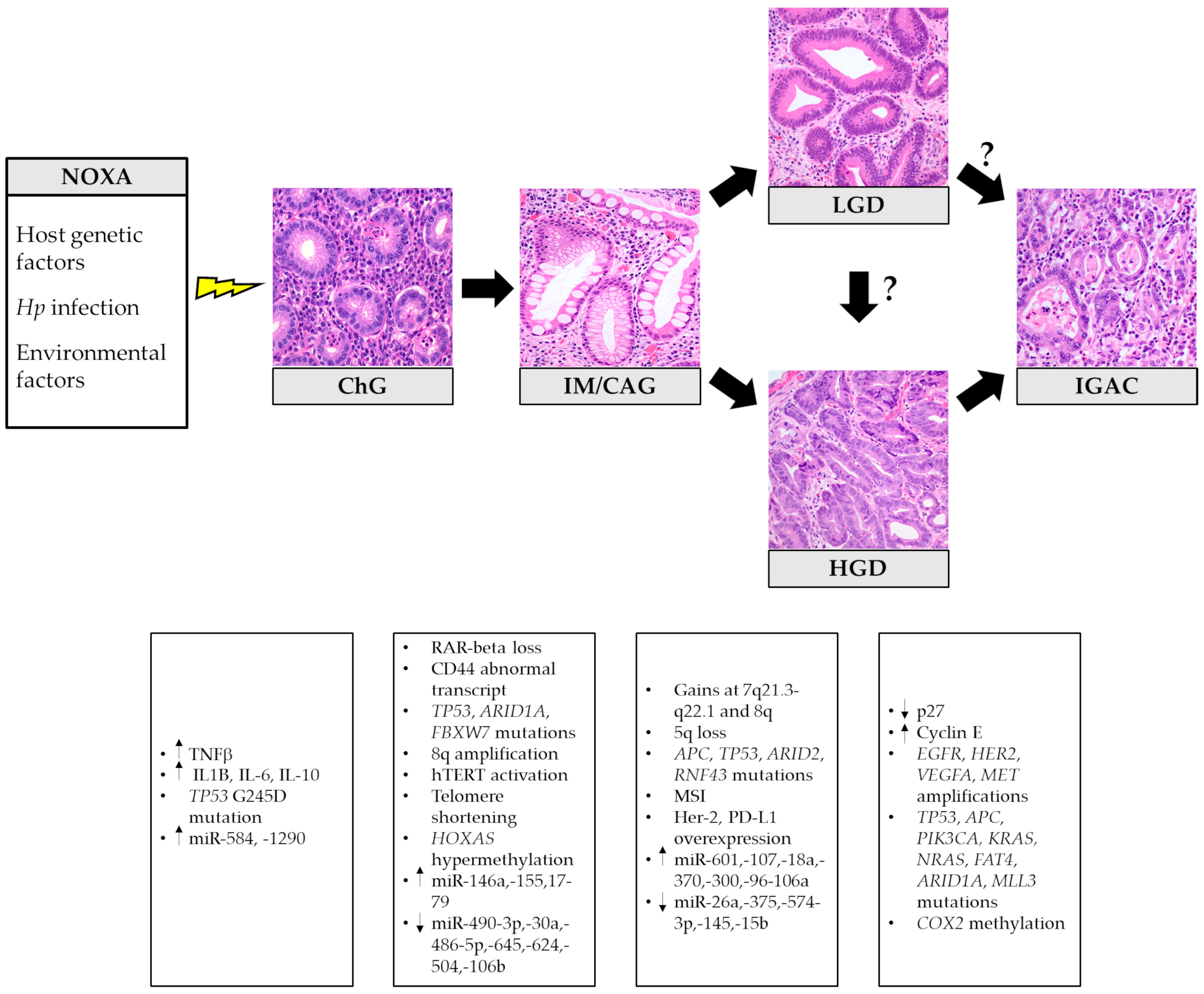

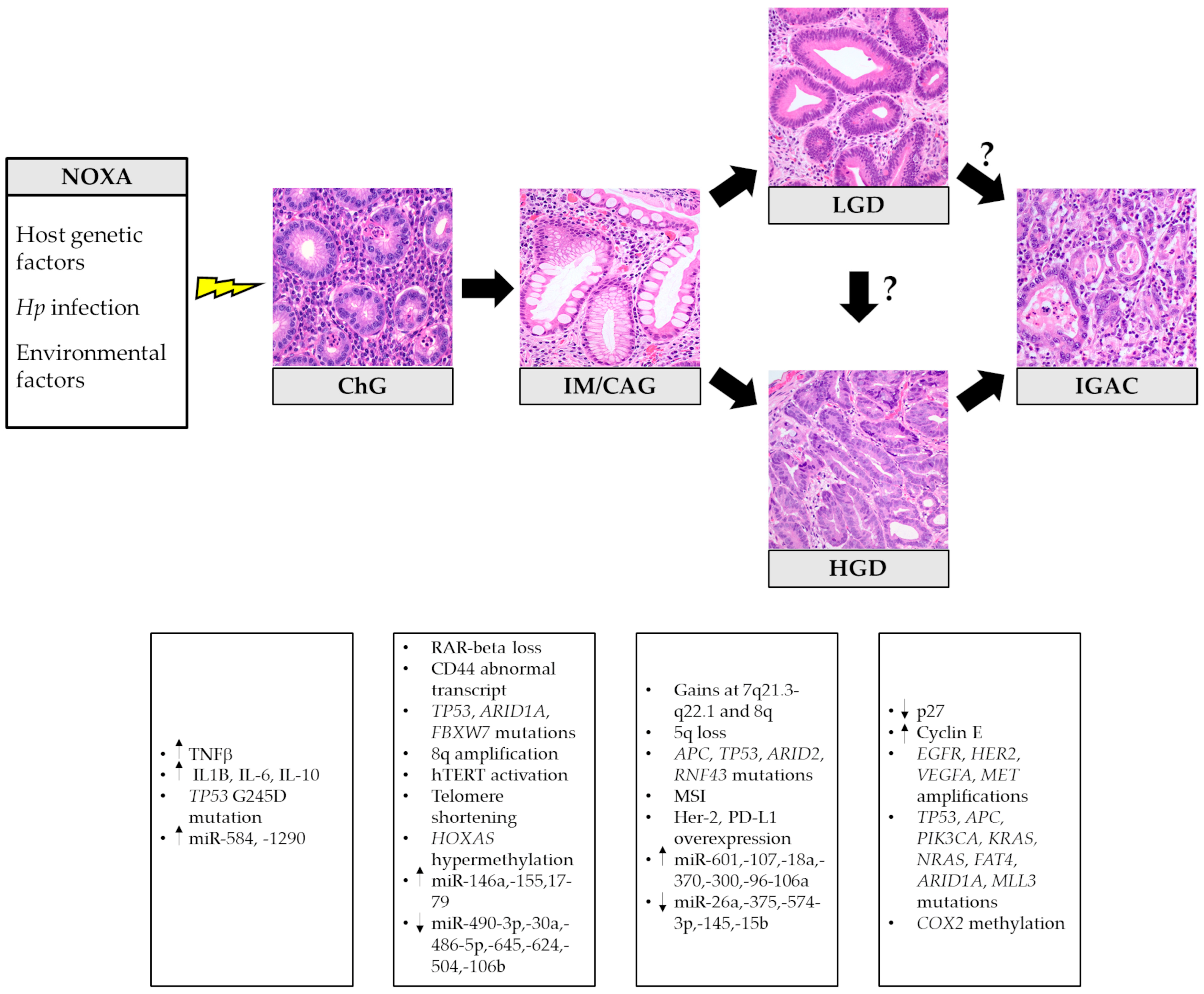{kind=link}
| TCGA | |||
|---|---|---|---|
| CIN (50%) | EBV (9%) | MSI (21%) | GS (20%) |
|
GEJ/cardia Intestinal type DNA aneuploidy Highly variable CIN TP53 mutations Amplification of TKR | Male prevalence Gastric corpus or fundus Extensive DNA promoter methylation CDKN2A promoter hypermethylation PIK3CA, ARID1A and BCOR mutations | Elderly age Gastric antrum Intestinal type Best prognosis among TCGA subtypes MLH1 promoter hypermethylation High mutational burden Possibly associated with Lynch syndrome | Younger age Distal localization Poorly cohesive histotype Worst prognosis among TCGA subtypes Low CNAs and mutational burden ARID1, RHOA and CDH1 mutations CLDN18–ARHGAP26 fusion in 15% |
| ACRG | |||
|---|---|---|---|
| MSS/TP53− (36%) | MSS/TP53+ (26%) | MSI (23%) | MMS/EMT (15%) |
| Male predominance Intestinal type Highest rate of TP53 and RHOA mutations APC, ARID1A, KRAS, PIK3CA and SMAD4 mutations | Male predominance Intestinal type EBV infection ARID1A, PIK3CA, SMAD4 and APC mutations | Gastric antrum Intestinal type Early stage at diagnosis Best prognosis among ACRG subtypes DNA methylation signature Presence of hypermutation Silencing of MLH1 ARID1A, KRAS and ALK mutations PIK3–PTEN–mTOR pathway dysregulation PD-L1 overexpression | Younger age Poorly cohesive histotype Higher frequency of peritoneal spreading Worst prognosis among ACRG subtypes Loss of CDH1 expression |
| MOLECULAR TARGET | THERAPEUTIC AGENT (Trial) | LINE OF THERAPY |
|---|---|---|
| HER2 | Trastuzumab (ToGa [20]) Trastuzumab deruxtecan (DESTINY-Gastric 01 [21]) | First line Third or later line |
| FGFR2 | Bemarituzumab (FIGHT [22,23]) | First line |
| MET | Onartuzumab (METGastric [24]) Savolitinib (VIKTORY [25]) | Second line |
| VEGF/VEGFR | Ramucirumab (RAINBOW [26]) (REGARD [27]) | Second line Second line |
| CLAUDIN 18.2 | Zolbetuximab (FAST [28]) | First line |
| PD-1/PD-L1 | Nivolumab (CHECKMATE-649 [29]) (ATTRACTION-4 [30]) (ATTRACTION-2 [31]) | First line First line Third or later line |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Businello, G.; Angerilli, V.; Parente, P.; Realdon, S.; Savarino, E.; Farinati, F.; Grillo, F.; Vanoli, A.; Galuppini, F.; Paccagnella, S.; et al. Molecular Landscapes of Gastric Pre-Neoplastic and Pre-Invasive Lesions. Int. J. Mol. Sci. 2021, 22, 9950. https://doi.org/10.3390/ijms22189950
Businello G, Angerilli V, Parente P, Realdon S, Savarino E, Farinati F, Grillo F, Vanoli A, Galuppini F, Paccagnella S, et al. Molecular Landscapes of Gastric Pre-Neoplastic and Pre-Invasive Lesions. International Journal of Molecular Sciences. 2021; 22(18):9950. https://doi.org/10.3390/ijms22189950
Chicago/Turabian StyleBusinello, Gianluca, Valentina Angerilli, Paola Parente, Stefano Realdon, Edoardo Savarino, Fabio Farinati, Federica Grillo, Alessandro Vanoli, Francesca Galuppini, Silvia Paccagnella, and et al. 2021. "Molecular Landscapes of Gastric Pre-Neoplastic and Pre-Invasive Lesions" International Journal of Molecular Sciences 22, no. 18: 9950. https://doi.org/10.3390/ijms22189950
APA StyleBusinello, G., Angerilli, V., Parente, P., Realdon, S., Savarino, E., Farinati, F., Grillo, F., Vanoli, A., Galuppini, F., Paccagnella, S., Pennelli, G., Mastracci, L., Saragoni, L., & Fassan, M. (2021). Molecular Landscapes of Gastric Pre-Neoplastic and Pre-Invasive Lesions. International Journal of Molecular Sciences, 22(18), 9950. https://doi.org/10.3390/ijms22189950