
Synthesis and In Silico Docking of New Pyrazolo[4,3-e]pyrido[1,2-a]pyrimidine-based Cytotoxic Agents


 , ,
, ,  ,
,  , and
, and
Abstract
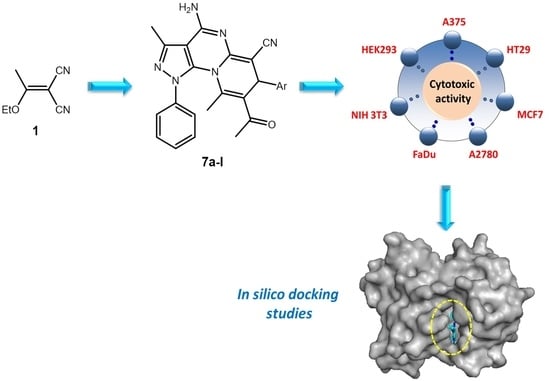
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Biological Evaluation
3. Materials and Methods
3.1. General
3.2. Synthesis
3.2.1. 5-Amino-3-methyl-1-phenyl-1H-pyrazole-4-carbonitrile (3)
3.2.2. 3-Methyl-5-{[(1E)-1-methyl-3-oxobut-1-en-1-yl]amino}-1-phenyl-1H-pyrazole-4-carbonitrile (5)
3.2.3. General Procedure for the Synthesis of Compounds 7a–l
8-Acetyl-4-amino-7-(4-chlorophenyl)-3,9-dimethyl-1-phenyl-1,7-dihydropyrazolo[4,3-e] Pyrido[1,2-a]pyrimidine-6-carbonitrile (7a)
8-Acetyl-4-amino-3,9-dimethyl-7-(4-methylphenyl)-1-phenyl-1,7-dihydropyrazolo[4,3-e] Pyrido1,2-a]pyrimidine-6-carbonitrile (7b)
8-Acetyl-4-amino-3,9-dimethyl-1,7-diphenyl-1,7-dihydropyrazolo[4,3-e]Pyrido[1,2-a]pyrimidine-6-carbonitrile (7c)
8-Acetyl-4-Amino-7-(4-methoxyphenyl)-3,9-dimethyl-1-phenyl-1,7-dihydropyrazolo[4,3-e] Pyrido[1,2-a]pyrimidine-6-carbonitrile (7d)
8-Acetyl-4-amino-7-(4-bromophenyl)-3,9-dimethyl-1-phenyl-1,7-dihydropyrazolo[4,3-e] Pyrido[1,2-a]pyrimidine-6-carbonitrile (7e)
8-Acetyl-4-amino-7-(4-fluorophenyl)-3,9-dimethyl-1-phenyl-1,7-sihydropyrazolo[4,3-e] Pyrido[1,2-a]pyrimidine-6-carbonitrile (7f)
8-Acetyl-4-amino-3,9-dimethyl-7-(4-nitrophenyl)-1-phenyl-1,7-dihydropyrazolo[4,3-e] Pyrido[1,2-a]pyrimidine-6-carbonitrile (7g)
8-Acetyl-4-amino-7-(3,4-dimethoxyphenyl)-3,9-dimethyl-1-phenyl-1,7-dihydropyrazolo[4,3-e] Pyrido[1,2-a]pyrimidine-6-carbonitrile (7h)
8-Acetyl-4-amino-7-(3,4-dichlorophenyl)-3,9-dimethyl-1-phenyl-1,7-dihydropyrazolo[4,3-e] Pyrido[1,2-a]pyrimidine-6-carbonitrile (7i)
8-Acetyl-4-amino-3,9-dimethyl-7-(5-nitro-2-thienyl)-1-phenyl-1,7-dihydropyrazolo[4,3-e] Pyrido[1,2-a]pyrimidine-6-carbonitrile (7j)
8-Acetyl-4-amino-3,9-dimethyl-7-(2-naphthyl)-1-phenyl-1,7-dihydropyrazolo[4,3-e] Pyrido[1,2-a]pyrimidine-6-carbonitrile (7k)
8-Acetyl-4-amino-3,9-dimethyl-1-phenyl-7-pyridin-3-yl-1,7-dihydropyrazolo[4,3-e] Pyrido[1,2-a]pyrimidine-6-carbonitrile (7l)
3.3. Cytotoxicity Assay (SRB Assay)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global Cancer Statistics, 2012. CA-Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef]
- Isakoff, S.J. Triple-Negative Breast Cancer Role of Specific Chemotherapy Agents. Cancer J. 2010, 16, 53–61. [Google Scholar] [CrossRef]
- Nossier, E.S.; Fahmy, H.H.; Khalifa, N.M.; El-Eraky, W.I.; Baset, M.A. Design and Synthesis of Novel Pyrazole-Substituted Different Nitrogenous Heterocyclic Ring Systems as Potential Anti-Inflammatory Agents. Molecules 2017, 22, 512. [Google Scholar] [CrossRef]
- Chekir, S.; Debbabi, M.; Regazzetti, A.; Dargere, D.; Laprevote, O.; Ben Jannet, H.; Gharbi, R. Design, synthesis and biological evaluation of novel 1,2,3-triazole linked coumarinopyrazole conjugates as potent anticholinesterase, anti-5-lipoxygenase, anti-tyrosinase and anti-cancer agents. Bioorg. Chem. 2018, 80, 189–194. [Google Scholar] [CrossRef]
- El-Naggar, M.; Hassan, A.S.; Awad, H.M.; Mady, M.F. Design, Synthesis and Antitumor Evaluation of Novel Pyrazolopyrimidines and Pyrazoloquinazolines. Molecules 2018, 23, 1249. [Google Scholar] [CrossRef] [PubMed]
- Somakala, K.; Tariq, S.; Amir, M. Synthesis, evaluation and docking of novel pyrazolo pyrimidines as potent p38 alpha MAP kinase inhibitors with improved anti-inflammatory, ulcerogenic and TNF-alpha inhibitory properties. Bioorg. Chem. 2019, 87, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Rahmouni, A.; Souiei, S.; Belkacem, M.A.; Romdhane, A.; Bouajila, J.; Ben Jannet, H. Synthesis and biological evaluation of novel pyrazolopyrimidines derivatives as anticancer and anti-5-lipoxygenase agents. Bioorg. Chem. 2016, 66, 160–168. [Google Scholar] [CrossRef]
- Rahmouni, A.; Romdhane, A.; Ben Said, A.; Majouli, K.; Ben Jannet, H. Synthesis of new pyrazole and antibacterial pyrazolopyrimidine derivatives. Turk. J. Chem. 2014, 38, 210–221. [Google Scholar] [CrossRef]
- De Vita, D.; Pandolfi, F.; Cirilli, R.; Scipione, L.; Di Santo, R.; Friggeri, L.; Mori, M.; Fiorucci, D.; Maccari, G.; Christopher, R.S.A.; et al. Discovery of in vitro antitubercular agents through in silico ligand-based approaches. Eur. J. Med. Chem. 2016, 121, 169–180. [Google Scholar] [CrossRef]
- Al-Aldiwish, W.M.; Shtewi, F.A.; Ashrif, M.M.; Ibrahim, D.M. Synthesis, biolovical activity and cytotoxicity of new fused pyrazolo[1,5-a]pyrimidine from 5-aminopyrazole incoroporated with p-chloroaniline. Am. J. Heterocycl. Chem. 2017, 3, 86–94. [Google Scholar] [CrossRef][Green Version]
- Alharthy, R.D. Design and Synthesis of Novel Pyrazolo[3,4-d]Pyrimidines: In Vitro Cytotoxic Evaluation and Free Radical Scavenging Activity Studies. Pharm. Chem. J. 2020, 54, 273–278. [Google Scholar] [CrossRef]
- Fouda, A.M.; Abbas, H.A.S.; Ahmed, E.H.; Shati, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I. Synthesis, In Vitro Antimicrobial and Cytotoxic Activities of Some New Pyrazolo[1,5-a]pyrimidine Derivatives. Molecules 2019, 24, 1080. [Google Scholar] [CrossRef]
- Husseiny, E.M. Synthesis, cytotoxicity of some pyranzoles and pyrazolo[1,5-a]pyrimidines bearing benzothiazole moiety amd investigation of their mechanism of action. Bioorg. Chem. 2020, 102, 104053. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Wang, S.B.; Wen, X.; Li, J.Z.; Quan, Z.S. 5Y Design, synthesis, and evaluation of the anticonvulsant and antidepressant activities of pyrido[2,3-d]pyrimidine derivatives. Med. Chem. Res. 2016, 25, 1287–1298. [Google Scholar] [CrossRef]
- Abdelaziz, O.A.; El Husseiny, W.M.; Selim, K.B.; Eisa, H.M. Dihydrofolate reductase inhibition effect of 5-substituted pyrido[2,3-d] pyrimidines: Synthesis, antitumor activity and molecular modeling study. Bioorg. Chem. 2019, 90, 103076. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, D.A.; Ismail, N.S.M. Design, synthesis and biological study of novel pyrido[2,3-d]pyrimidine as anti-proliferative CDK2 inhibitors. Eur. J. Med. Chem. 2011, 46, 5825–5832. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, F.I. Synthesis and antifungal activity of some new pyrido[2,3-d]pyrimidines. Eur. J. Chem. 2011, 2, 65–69. [Google Scholar] [CrossRef]
- Gfesser, G.A.; Bayburt, E.K.; Cowart, M.; Di Domenico, S.; Gomtsyan, A.; Lee, C.H.; Stewart, A.O.; Jarvis, M.F.; Kowaluk, E.A.; Bhagwat, S.S. Synthesis and structure-activity relationships of 5-heteroatom-substituted pyridopyrimidines as adenosine kinase inhibitors. Eur. J. Med. Chem. 2003, 38, 245–252. [Google Scholar] [CrossRef]
- Moreno, E.; Plano, D.; Lamberto, I.; Font, M.; Encio, I.; Palop, J.A.; Sanmartin, C. Sulfur and selenium derivatives of quinazoline and pyrido[2,3-d]pyrimidine: Synthesis and study of their potential cytotoxic activity in vitro. Eur. J. Med. Chem. 2012, 47, 283–298. [Google Scholar] [CrossRef]
- Sanmartin, C.; Echeverria, M.; Mendivil, B.; Cordeu, L.; Cubedo, E.; Garcia-Foncillas, J.; Font, M.; Palop, J.A. Synthesis and biological evaluation of new symmetrical derivatives as cytotoxic agents and apoptosis inducers. Bioorg. Med. Chem. 2005, 13, 2031–2044. [Google Scholar] [CrossRef]
- Alam, R.; Alam, A.; Panda, A.K. Design, synthesis and cytotoxicity evaluation of pyrazolyl pyrazoline and pyrazolyl aminopyrimidine derivatives as potential anticancer agents. Med. Chem. Res. 2018, 27, 560–570. [Google Scholar] [CrossRef]
- Lee, J.; Kim, K.H.; Jeong, S. Discovery of a novel class of 2-aminopyrimidines as CDK1 and CDK2 inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 4203–4205. [Google Scholar] [CrossRef]
- Sabour, R.; Harras, M.F.; Mehany, A.B.M. Design, Synthesis, Cytotoxicity Screening and Molecular Docking of New 3- Cyanopyridines as Survivin inhibitors and Apoptosis Inducers. Bioorg. Chem. 2019, 94, 103358. [Google Scholar] [CrossRef]
- Al-Refai, M.; Ibrahim, M.M.; Nurul Azmi, M.; Osman, H.; Abu Bakar, M.H.; Geyer, A. The Synthesis, Characterization, Cytotoxic Activity Assessment and Structure-Activity Relationship of 4-Aryl-6-(2,5-dichlorothiophen-3-yl)-2-methoxypyridine-3-carbonitriles. Molecules 2019, 24, 4072. [Google Scholar] [CrossRef] [PubMed]
- Ragab, F.A.; Nissan, Y.M.; Seif, E.M.; Maher, A.; Arafa, R.K. Synthesis and in vitro investigation of novel cytotoxic pyrimidine and pyrazolopyrimidne derivatives showing apoptotic effect. Bioorg. Chem. 2020, 96, 103621. [Google Scholar] [CrossRef] [PubMed]
- Haiba, M.E.; Al-Abdullah, E.S.; Ahmed, N.S.; Ghabbour, H.A.; Awad, H.M. Efficient and easy synthesis of new Benzo[h]chromene and Benzo[h] quinoline derivatives as a new class of cytotoxic agents. J. Mol. Struct. 2019, 1195, 702–711. [Google Scholar] [CrossRef]
- Debbabi, M.; Nimbarte, V.D.; Chekir, S.; Chortani, S.; Romdhane, A.; Ben Jannet, H. Design and synthesis of novel potent anticoagulant and anti-tyrosinase pyranopyrimidines and pyranotriazolopyrimidines: Insights from molecular docking and SAR analysis. Bioorg. Chem. 2019, 82, 129–138. [Google Scholar] [CrossRef]
- Horchani, M.; Della Sala, G.; Caso, A.; D’Aria, F.; Esposito, G.; Laurenzana, I.; Giancola, C.; Costantino, V.; Ben Jannet, H.; Romdhane, A. Molecular Docking and Biophysical Studies for Antiproliferative Assessment of Synthetic Pyrazolo-Pyrimidinones Tethered with Hydrazide-Hydrazones. Int. J. Mol. Sci. 2021, 22, 2742. [Google Scholar] [CrossRef]
- Romdhane, A.; Martin, M.T.; Said, A.; Jabrane, A.; Jannet, H. Synthesis of new naphto[2,1-b]pyrano[3,2-e][1,2,4]triazolo[1,5-c]pyrimidine derivatives and their evaluation as acetylcholinesterase inhibitors. J. Soc. Chim. Tun. 2012, 14, 127–131. [Google Scholar]
- Horchani, M.; Hajlaoui, A.; Harrath, A.H.; Mansour, L.; Ben Jannet, H.; Romdhane, A. New pyrazolo-triazolo-pyrimidine derivatives as antibacterial agents: Design and synthesis, molecular docking and DFT studies. J. Mol. Struct. 2020, 1199, 127007. [Google Scholar] [CrossRef]
- Aliwaini, S.; Abu Thaher, B.; Al-Masri, I.; Shurrab, N.; El-Kurdi, S.; Schollmeyer, D.; Qeshta, B.; Ghunaim, M.; Csuk, R.; Laufer, S.; et al. Design, Synthesis and Biological Evaluation of Novel Pyrazolo[1,2,4]triazolopyrimidine Derivatives as Potential Anticancer Agents. Molecules 2021, 26, 4065. [Google Scholar] [CrossRef]
- Saeed, A.; Hussain, H.; Shamraiz, U.; Rehman, N.U.; Khan, H.Y.; Badshah, A.; Heller, L.; Csuk, R.; Ali, M.; Khan, A.; et al. Synthesis of new triterpenic monomers and dimers as potential antiproliferative agents and their molecular docking studies. Eur. J. Med. Chem. 2018, 143, 948–957. [Google Scholar] [CrossRef]
- Irina, A.T.; Alexey, V.N.; Daria, V.E.; Victoria, V.G. Synthesis, cytotoxic evaluation, and molecular docking studies of the semi-synthetic “triterpenoid-steroid” hybrids. Steroids 2018, 140, 131–143. [Google Scholar]
- Ruddarraju, R.R.; Murugulla, A.C.; Kotla, R.; Tirumalasetty, M.C.B.; Wudayagiri, R.; Donthabakthuni, S.; Maroju, R.; Baburao, K.; Parasa, L.S. Design, synthesis, anticancer, antimicrobial activities and molecular docking studies of theophylline containing acetylenes and theophylline containing 1,2,3-triazoles with variant nucleoside derivatives. Eur. J. Med. Chem. 2016, 123, 379–396. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Bold, G.; Schnell, C.; Furet, P.; McSheehy, P.; Bruggen, J.; Mestan, J.; Manley, P.W.; Druckes, P.; Burglin, M.; Durler, U.; et al. Littlewood-Evans, A., A Novel Potent Oral Series of VEGFR2 Inhibitors Abrogate Tumor Growth by Inhibiting Angiogenesis. J. Med. Chem. 2016, 59, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Seto, M.; Banno, H.; Kawakita, Y.; Oorui, M.; Taniguchi, T.; Ohta, Y.; Tamura, T.; Nakayama, A.; Miki, H.; et al. Design and Synthesis of Novel Human Epidermal Growth Factor Receptor 2 (HER2)/Epidermal Growth Factor Receptor (EGFR) Dual Inhibitors Bearing a Pyrrolo[3,2-d]pyrimidine Scaffold. J. Med. Chem. 2011, 54, 8030–8050. [Google Scholar] [CrossRef] [PubMed]
- Leitans, J.; Kazaks, A.; Balode, A.; Ivanova, J.; Zalubovskis, R.; Supuran, C.T.; Tars, K. Efficient Expression and Crystallization System of Cancer-Associated Carbonic Anhydrase Isoform IX. J. Med. Chem. 2015, 58, 9004–9009. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Borland, M.; Brain, C.; Chen, C.H.T.; Cheng, H.; Chopra, R.; Chung, K.; Groarke, J.; He, G.; Hou, Y.; et al. 4-(Pyrazol-4-yl)-pyrimidines as Selective Inhibitors of Cyclin-Dependent Kinase 4/6. J. Med. Chem. 2010, 53, 7938–7957. [Google Scholar] [CrossRef]
- Gilbert, N.C.; Rui, Z.; Neau, D.B.; Waight, M.T.; Bartlett, S.G.; Boeglin, W.E.; Brash, A.R.; Newcomer, M.E. Conversion of human 5-lipoxygenase to a 15-lipoxygenase by a point mutation to mimic phosphorylation at Serine-663. FASEB J. 2012, 26, 3222–3229. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | A375 | HT29 | MCF-7 | A2780 | FaDu | NIH 3T3 | HEK293 |
|---|---|---|---|---|---|---|---|
| 3, 5 | >30 | >30 | >30 | >30 | >30 | >30 | >30 |
| 7a | 19.3 ± 3.0 | 29.9 ± 1.4 | 17.2 ± 1.4 | 18.0 ± 3.0 | >30 | 23.0 ± 1.4 | 15.8 ± 2.0 |
| 7b | 12.9 ± 1.6 | 17.5 ± 1.8 | 12.2 ± 1.3 | 14.6 ± 2.2 | 22.3 ± 2.7 | 21.3 ± 0.9 | 12.4 ± 0.9 |
| 7c | 19.3 ± 3.8 | >30 | 19.5 ± 1.1 | 25.4 ± 2.5 | >30 | >30 | 7.7 ± 1.1 |
| 7d | 21.5 ± 3.6 | 28.3 ± 3.5 | 18.4 ± 2.3 | 21.7 ± 3.9 | >30 | >30 | 16.7 ± 1.2 |
| 7e | 9.4 ± 1.2 | 13.3 ± 1.8 | 9.2 ± 0.7 | 9.1 ± 1.6 | 13.5 ± 1.5 | 12.3 ± 0.7 | 6.6 ± 0.6 |
| 7f | 18.1 ± 3.3 | 24.7 ± 4.4 | 19.2 ± 1.3 | 19.2 ± 3.9 | >30 | 24.1 ± 2.8 | 14.7 ± 1.4 |
| 7g | 16.2 ± 2.0 | 25.9 ± 3.4 | 15.7 ± 2.2 | 14.5 ± 2.2 | >30 | >30 | 17.0 ± 1.3 |
| 7h | 22.1 ± 4.9 | >30 | 25.1 ± 2.8 | 27.5 ± 8.1 | >30 | >30 | 24.0 ± 3.4 |
| 7i–l | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. |
| DX | n.d. | 0.9 ± 0.01 | 1.1 ± 0.3 | 0.01 ± 0.01 | n.d. | 0.4 ± 0.07 | n.d. |
| Compound | Free Binding Energy (Kcal·mol−1) | |||||
|---|---|---|---|---|---|---|
| DHFR (5HQY) | VEGFR2 (5EW3) | HER-2 (3RCD) | hCA-IX (5FL4) | CDK6 (3NUP) | LOX5 (3V99) | |
| 7e | −8.5 | −6.6 | −7.3 | −7.2 | −7.1 | −8.5 |
| Ref. ligand | −8.3 | −7.3 | −7.2 | −7.0 | −6.8 | −6.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horchani, M.; Heise, N.V.; Hoenke, S.; Csuk, R.; Harrath, A.H.; Ben Jannet, H.; Romdhane, A. Synthesis and In Silico Docking of New Pyrazolo[4,3-e]pyrido[1,2-a]pyrimidine-based Cytotoxic Agents. Int. J. Mol. Sci. 2021, 22, 10258. https://doi.org/10.3390/ijms221910258
Horchani M, Heise NV, Hoenke S, Csuk R, Harrath AH, Ben Jannet H, Romdhane A. Synthesis and In Silico Docking of New Pyrazolo[4,3-e]pyrido[1,2-a]pyrimidine-based Cytotoxic Agents. International Journal of Molecular Sciences. 2021; 22(19):10258. https://doi.org/10.3390/ijms221910258
Chicago/Turabian StyleHorchani, Mabrouk, Niels V. Heise, Sophie Hoenke, René Csuk, Abdel Halim Harrath, Hichem Ben Jannet, and Anis Romdhane. 2021. "Synthesis and In Silico Docking of New Pyrazolo[4,3-e]pyrido[1,2-a]pyrimidine-based Cytotoxic Agents" International Journal of Molecular Sciences 22, no. 19: 10258. https://doi.org/10.3390/ijms221910258
APA StyleHorchani, M., Heise, N. V., Hoenke, S., Csuk, R., Harrath, A. H., Ben Jannet, H., & Romdhane, A. (2021). Synthesis and In Silico Docking of New Pyrazolo[4,3-e]pyrido[1,2-a]pyrimidine-based Cytotoxic Agents. International Journal of Molecular Sciences, 22(19), 10258. https://doi.org/10.3390/ijms221910258