
Tagging and Capturing of Lentiviral Vectors Using Short RNAs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Design of RNA Scaffolds Capable of Binding to a Bacteriophage Lambda N Protein
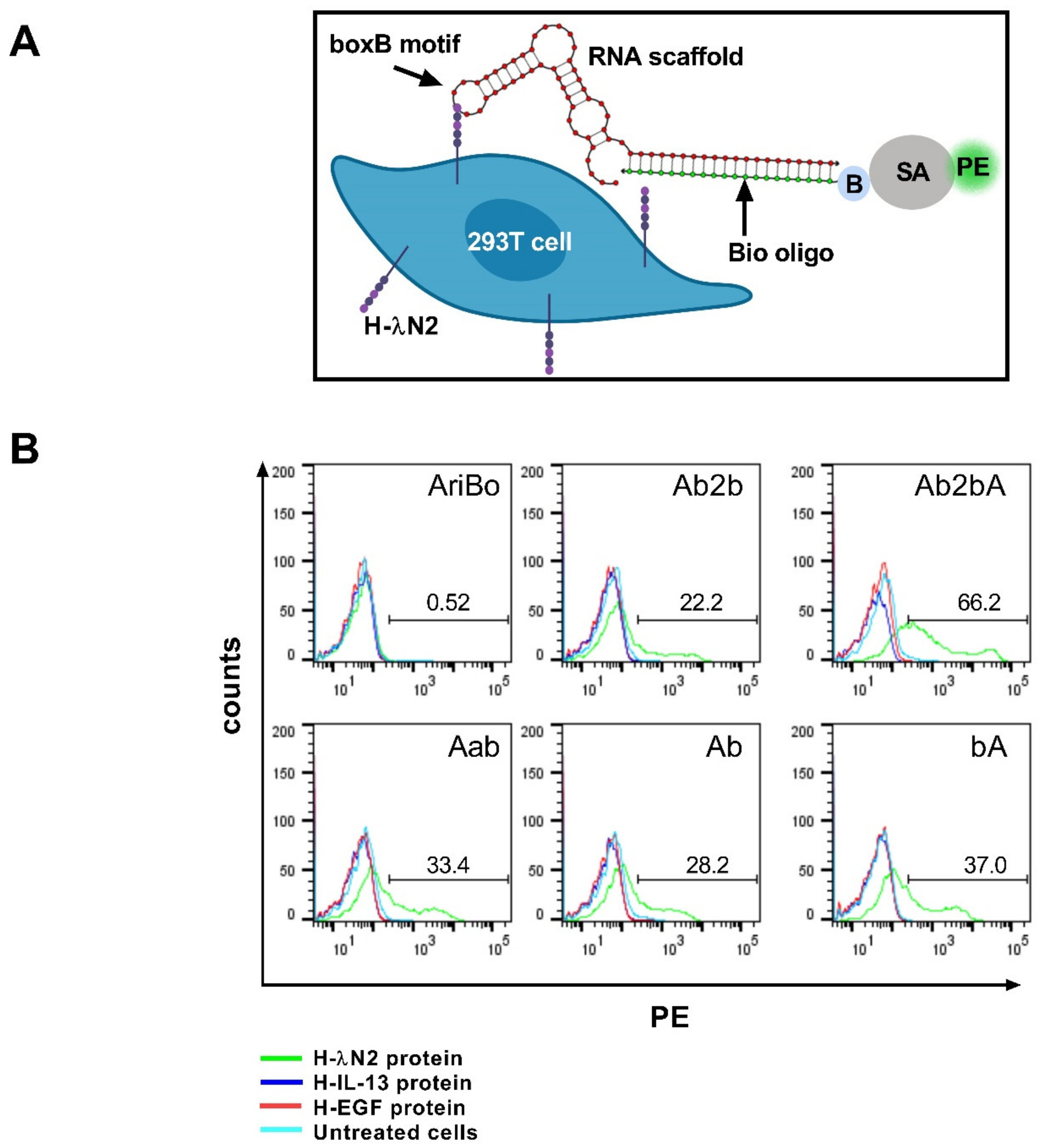
2.2. Testing the Ability of RNA Scaffolds to Bind to the H-λN2 Domain Displayed on Transfected Cells
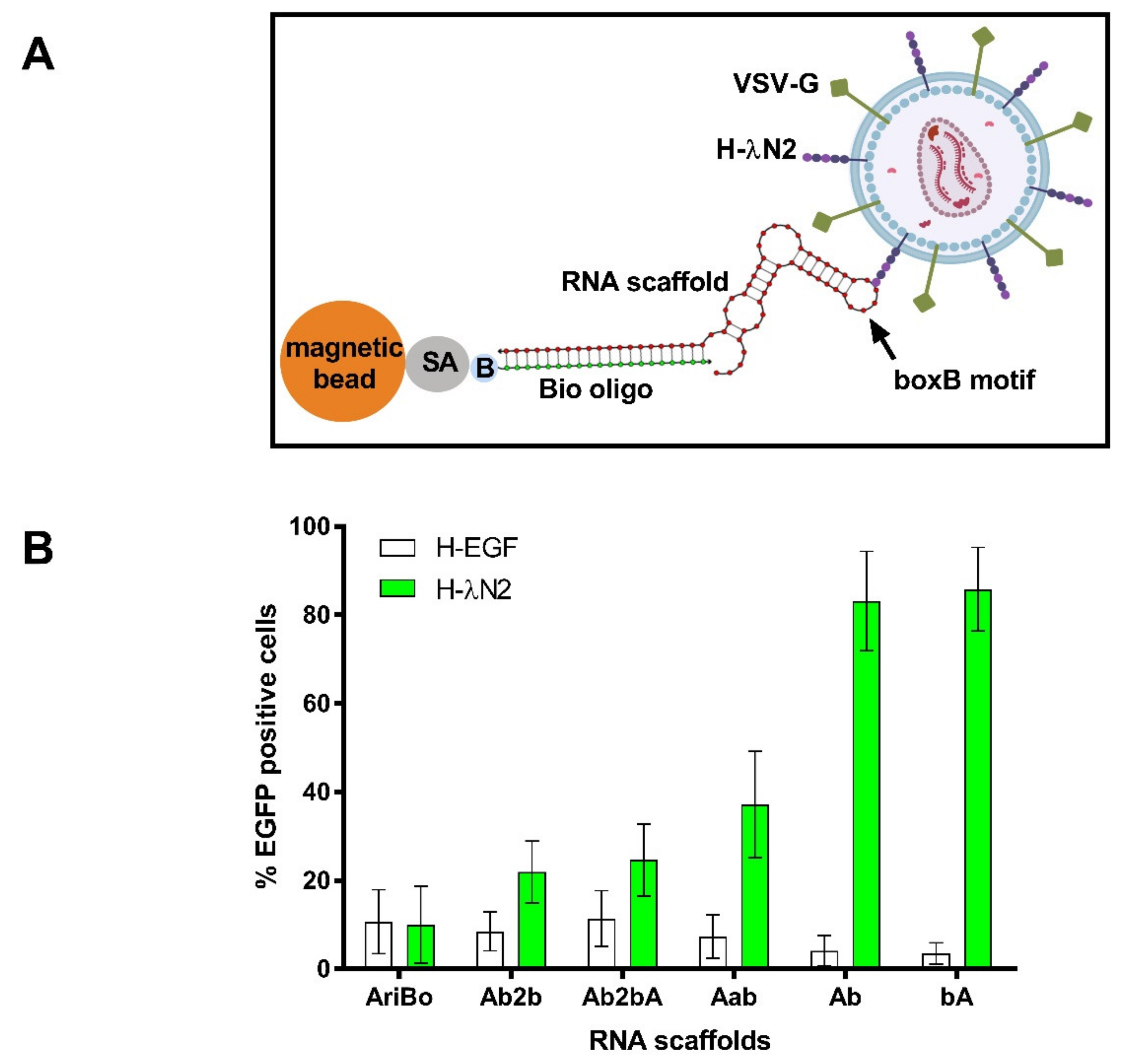
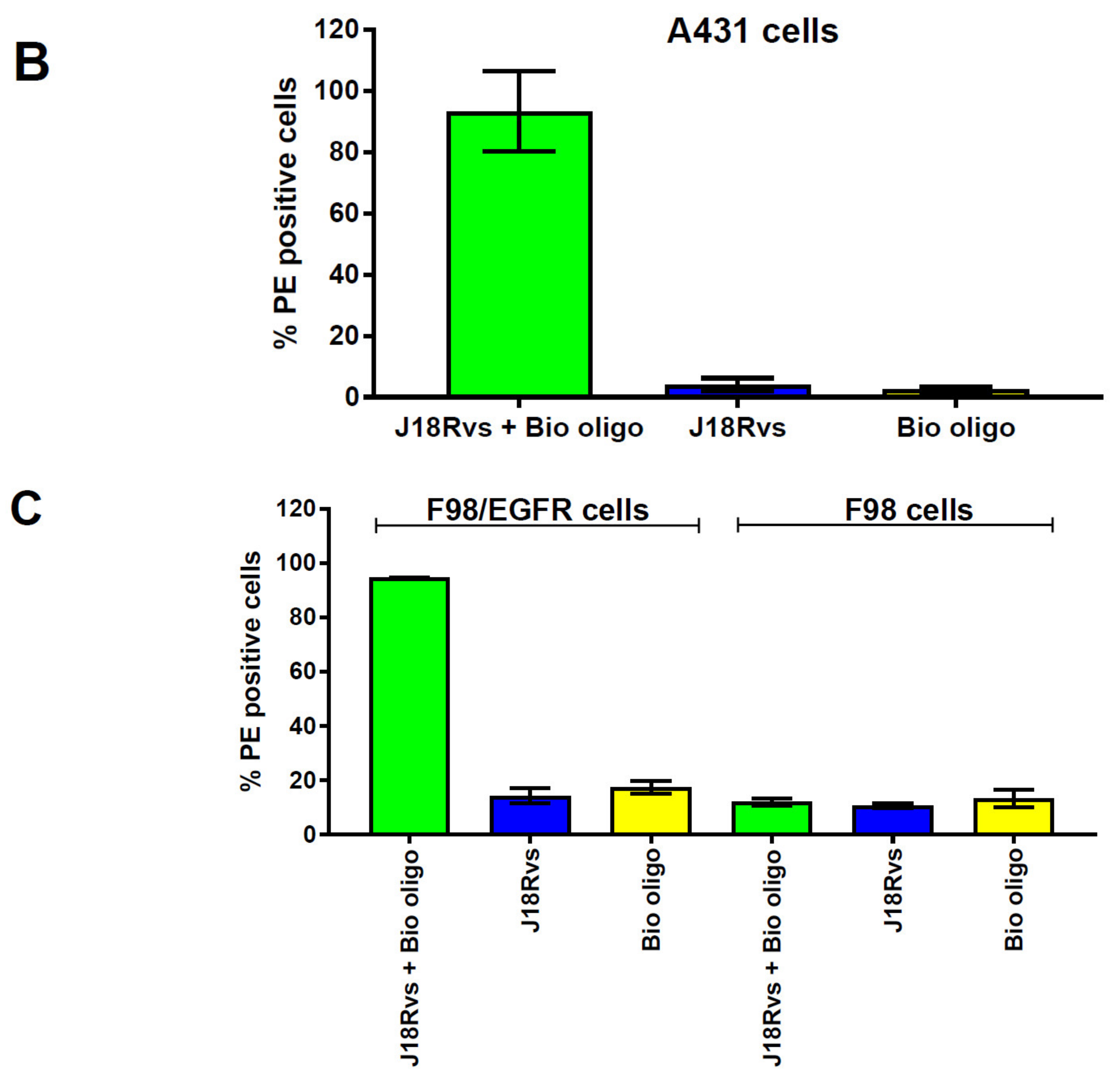
2.3. Testing the Capacity of RNA Scaffolds to Bind to the H-λN2 Protein Displayed on Lentiviral Vector Particles
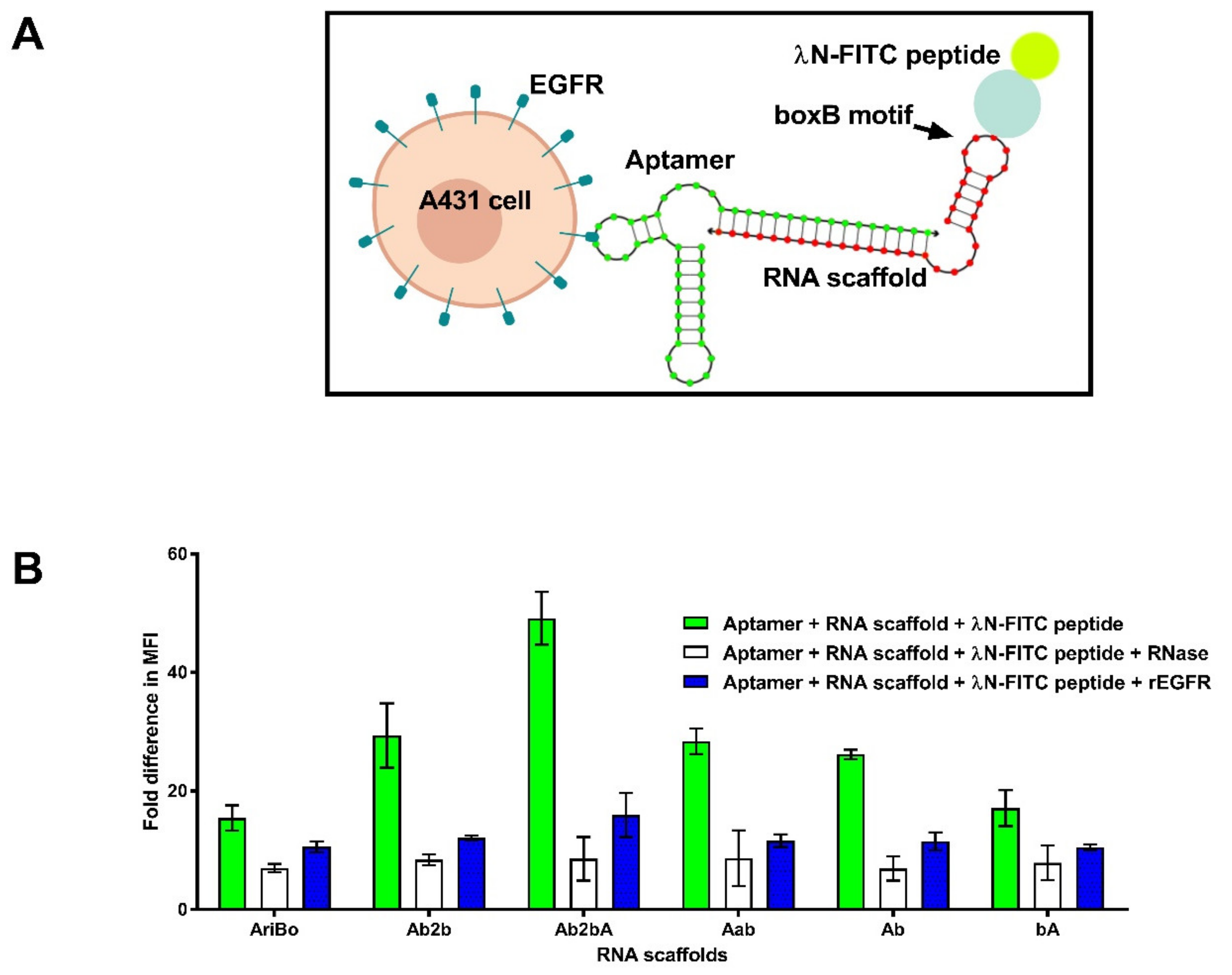
2.4. RNA Scaffold Binding to RNA Aptamers
2.5. Assessing the Functionality of RNA Aptamer/Scaffold Complexes
3. Discussion
4. Materials and Methods
4.1. Plasmid Constructs
4.2. Cells
4.3. Lentiviral Vector Production
4.4. Production of Aptamer and Scaffold RNAs
4.5. Binding of J18 and J18 Rvs Aptamers to Cells
4.6. RNA Scaffold Binding to Cells Displaying the H-λN2 Protein
4.7. Binding of RNA Scaffolds to the J18 Rvs Aptamer and the λN Peptide
4.8. Scaffold Binding to Lentiviral Vectors Displaying the H-λN2 Protein
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EGFR | Epidermal growth factor receptor |
| EGF | Epidermal growth factor |
| IL-13 | Interleukin-13 |
| LV | Lentiviral |
| MV | Measles virus |
| SA | Streptavidin |
| PE | Phycoerythrin |
| VSV-G | Vesicular stomatitis virus G protein |
References
- Sakuma, T.; Barry, M.A.; Ikeda, Y. Lentiviral vectors: Basic to translational. Biochem. J. 2012, 443, 603–618. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L. Genetic engineering of hematopoiesis: Current stage of clinical translation and future perspectives. EMBO Mol. Med. 2019, 11, e9958. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, X.; Han, W.; Zhang, Y. Tisagenlecleucel, an approved anti-CD19 chimeric antigen receptor T-cell therapy for the treatment of leukemia. Drugs Today 2017, 53, 597–608. [Google Scholar] [CrossRef]
- Schuessler-Lenz, M.; Enzmann, H.; Vamvakas, S. Regulators’ advice can make a difference: European medicines agency approval of zynteglo for beta thalassemia. Clin. Pharmacol. Ther. 2020, 107, 492–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, J.L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Cavalieri, V.; Baiamonte, E.; Iacono, M.L. Non-primate lentiviral vectors and their applications in gene therapy for ocular disorders. Viruses 2018, 10, 316. [Google Scholar] [CrossRef] [Green Version]
- Somaiah, N.; Block, M.S.; Kim, J.W.; Shapiro, G.I.; Do, K.T.; Hwu, P.; Eder, J.P.; Jones, R.L.; Lu, H.; Ter Meulen, J.H.; et al. First-in-class, first-in-human study evaluating LV305, a dendritic-cell tropic lentiviral vector, in sarcoma and other solid tumors expressing NY-ESO-1. Clin. Cancer Res. 2019, 25, 5808–5817. [Google Scholar] [CrossRef] [Green Version]
- Capasso, C.; Hirvinen, M.; Cerullo, V. Beyond gene delivery: Strategies to engineer the surfaces of viral vectors. Biomedicines 2013, 1, 3–16. [Google Scholar] [CrossRef]
- Metzner, C.; Kochan, F.; Dangerfield, J.A. Postexit surface engineering of retroviral/lentiviral vectors. BioMed Res. Int. 2013, 2013, 253521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lévy, C.; Verhoeyen, E.; Cosset, F.-L. Surface engineering of lentiviral vectors for gene transfer into gene therapy target cells. Curr. Opin. Pharmacol. 2015, 24, 79–85. [Google Scholar] [CrossRef]
- Frecha, C.; Szecsi, J.; Cosset, F.-L.; Verhoeyen, E. Strategies for targeting lentiviral vectors. Curr. Gene Ther. 2008, 8, 449–460. [Google Scholar] [CrossRef]
- Cronin, J.; Zhang, X.-Y.; Reiser, J. Altering the tropism of lentiviral vectors through pseudotyping. Curr. Gene Ther. 2005, 5, 387–398. [Google Scholar] [CrossRef]
- Bischof, D.; Cornetta, K. Flexibility in cell targeting by pseudotyping lentiviral vectors. Methods Mol. Biol. 2010, 614, 53–68. [Google Scholar] [PubMed]
- Gutierrez-Guerrero, A.; Cosset, F.-L.; Verhoeyen, E. Lentiviral vector pseudotypes: Precious tools to improve gene modification of hematopoietic cells for research and gene therapy. Viruses 2020, 12, 1016. [Google Scholar] [CrossRef]
- Buchholz, C.J.; Friedel, T.; Büning, H. Surface-engineered viral vectors for selective and cell type-specific gene delivery. Trends Biotechnol. 2015, 33, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.M.; Buchholz, C.J. Surface-engineered lentiviral vectors for selective gene transfer into sub-types of lymphocytes. Mol. Ther. Methods Clin. Dev. 2019, 12, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funke, S.; Maisner, A.; Mühlebach, M.D.; Koehl, U.; Grez, M.; Cattaneo, R.; Cichutek, K.; Buchholz, C.J. Targeted cell entry of lentiviral vectors. Mol. Ther. 2008, 16, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.; Marino, M.P.; Suzuki, A.; Joshi, B.; Husain, S.R.; Maisner, A.; Galanis, E.; Puri, R.K.; Reiser, J. Specific targeting of human interleukin (IL)-13 receptor alpha2-positive cells with lentiviral vectors displaying IL-13. Hum. Gene Ther. Methods 2012, 23, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Anliker, B.; Abel, T.; Kneissl, S.; Hlavaty, J.; Caputi, A.; Brynza, J.; Schneider, I.C.; Münch, R.C.; Petznek, H.; Kontermann, R.E.; et al. Specific gene transfer to neurons, endothelial cells and hematopoietic progenitors with lentiviral vectors. Nat. Methods 2010, 7, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Münch, R.C.; Mühlebach, M.D.; Schaser, T.; Kneissl, S.; Jost, C.; Plückthun, A.; Cichutek, K.; Buchholz, C.J. DARPins: An efficient targeting domain for lentiviral vectors. Mol. Ther. 2011, 19, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.H.; Schaffer, D.V. Selection of novel vesicular stomatitis virus glycoprotein variants from a peptide insertion library for enhanced purification of retroviral and lentiviral vectors. J. Virol. 2006, 80, 3285–3292. [Google Scholar] [CrossRef] [Green Version]
- Nesbeth, D.; Williams, S.L.; Chan, L.; Brain, T.; Slater, N.K.; Farzaneh, F.; Darling, D. Metabolic biotinylation of lentiviral pseudotypes for scalable paramagnetic microparticle-dependent manipulation. Mol. Ther. 2006, 13, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Annoni, A.; Gregori, S.; Naldini, L.; Cantore, A. Modulation of immune responses in lentiviral vector-mediated gene transfer. Cell Immunol. 2019, 342, 103802. [Google Scholar] [CrossRef]
- DePolo, N.J.; Reed, J.D.; Sheridan, P.L.; Townsend, K.; Sauter, S.L.; Jolly, D.J.; Dubensky, T.W. VSV-G pseudotyped lentiviral vector particles produced in human cells are inactivated by human serum. Mol. Ther. 2000, 2, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Munis, A.M.; Mattiuzzo, G.; Bentley, E.M.; Collins, M.K.; Eyles, J.E.; Takeuchi, Y. Use of heterologous vesiculovirus G proteins circumvents the humoral anti-envelope immunity in lentivector-based In Vivo gene delivery. Mol. Ther. Nucleic Acids 2019, 17, 126–137. [Google Scholar] [CrossRef] [Green Version]
- Schauber-Plewa, C.; Simmons, A.; Tuerk, M.J.; Pacheco, C.D.; Veres, G. Complement regulatory proteins are incorporated into lentiviral vectors and protect particles against complement inactivation. Gene Ther. 2005, 12, 238–245. [Google Scholar] [CrossRef] [Green Version]
- Croyle, M.A.; Callahan, S.M.; Auricchio, A.; Schumer, G.; Linse, K.D.; Wilson, J.M.; Brunner, L.J.; Kobinger, G.P. PEGylation of a vesicular stomatitis virus G pseudotyped lentivirus vector prevents inactivation in serum. J. Virol. 2004, 78, 912–921. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.; Yan, M.; Lu, Y.; Chen, I.S. Retargeting vesicular stomatitis virus glycoprotein pseudotyped lentiviral vectors with enhanced stability by In Situ synthesized polymer shell. Hum. Gene Ther. Methods 2013, 24, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keryer-Bibens, C.; Barreau, C.; Osborne, H.B. Tethering of proteins to RNAs by bacteriophage proteins. Biol. Cell 2008, 100, 125–138. [Google Scholar] [CrossRef] [Green Version]
- Baron-Benhamou, J.; Gehring, N.H.; Kulozik, A.E.; Hentze, M.W. Using the lambdaN peptide to tether proteins to RNAs. Methods Mol. Biol. 2004, 257, 135–154. [Google Scholar] [PubMed]
- Di Tomasso, G.; Lampron, P.; Dagenais, P.; Omichinski, J.G.; Legault, P. The ARiBo tag: A reliable tool for affinity purification of RNAs under native conditions. Nucleic Acids Res. 2011, 39, e18. [Google Scholar] [CrossRef] [PubMed]
- Austin, R.J.; Xia, T.; Ren, J.; Takahashi, T.T.; Roberts, R.W. Differential modes of recognition in N peptide-box B complexes. Biochemistry 2003, 42, 14957–14967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, R.J.; Xia, T.; Ren, J.; Takahashi, T.T.; Roberts, R.W. Designed arginine-rich RNA-binding peptides with picomolar affinity. J. Am. Chem. Soc. 2002, 124, 10966–10967. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Peng, K.-W.; Vongpunsawad, S.; Harvey, M.; Mizuguchi, H.; Hayakawa, T.; Cattaneo, R.; Russell, S.J. Antibody-targeted cell fusion. Nat. Biotechnol. 2004, 22, 331–336. [Google Scholar] [CrossRef]
- Zadeh, J.N.; Steenberg, C.D.; Bois, J.; Wolfe, B.R.; Pierce, M.B.; Khan, A.R.; Dirks, R.M.; Pierce, N.A. Nupack: Analysis and design of nucleic acid systems. J. Comput. Chem. 2010, 32, 170–173. [Google Scholar] [CrossRef]
- Li, N.; Ebright, J.N.; Stovall, G.M.; Chen, X.; Nguyen, H.H.; Singh, A.; Syrett, A.; Ellington, A.D. Technical and biological issues relevant to cell typing with aptamers. J. Proteome Res. 2009, 8, 2438–2448. [Google Scholar] [CrossRef]
- Li, N.; Larson, T.; Nguyen, H.H.; Sokolov, K.V.; Ellington, A.D. Directed evolution of gold nanoparticle delivery to cells. Chem. Commun. 2010, 46, 392–394. [Google Scholar] [CrossRef] [Green Version]
- Afonin, K.A.; Viard, M.; Koyfman, A.Y.; Martins, A.N.; Kasprzak, W.K.; Panigaj, M.; Desai, R.; Santhanam, A.; Grabow, W.W.; Jaeger, L.; et al. Multifunctional RNA nanoparticles. Nano Lett. 2014, 14, 5662–5671. [Google Scholar] [CrossRef] [Green Version]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H.; Parks, W.P. In Vitro cultivation of human tumors: Establishment of cell lines derived from a series of solid tumors. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Tahiri-Alaoui, A.; Frigotto, L.; Manville, N.; Ibrahim, J.; Romby, P.; James, W. High affinity nucleic acid aptamers for streptavidin incorporated into bi-specific capture ligands. Nucleic Acids Res. 2002, 30, e45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, R.F.; Yang, W.; Adams, D.M.; Rotaru, J.H.; Shukla, S.; Sekido, M.; Tjarks, W.; Fenstermaker, R.A.; Ciesielski, M.; Nawrocky, M.M.; et al. Molecular targeting of the epidermal growth factor receptor for neutron capture therapy of gliomas. Cancer Res. 2002, 62, 3159–3166. [Google Scholar]
- Marino, M.P.; Panigaj, M.; Ou, W.; Manirarora, J.; Wei, C.-H.; Reiser, J. A scalable method to concentrate lentiviral vectors pseudotyped with measles virus glycoproteins. Gene Ther. 2015, 22, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M. Aptamers targeting cell surface proteins. Biochimie 2018, 145, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Cerchia, L.; de Franciscis, V. Targeting cancer cells with nucleic acid aptamers. Trends Biotechnol. 2010, 28, 517–525. [Google Scholar] [CrossRef]
- Tan, W.; Wang, H.; Chen, Y.; Zhang, X.; Zhu, H.; Yang, C.; Yang, R.; Liu, C. Molecular aptamers for drug delivery. Trends Biotechnol. 2011, 29, 634–640. [Google Scholar] [CrossRef] [Green Version]
- Panigaj, M.; Reiser, J. Aptamer guided delivery of nucleic acid-based nanoparticles. DNA RNA Nanotechnol. 2016, 2, 42. [Google Scholar] [CrossRef]
- Baron-Benhamou, J.; Gehring, N.H.; Kulozik, A.E.; Hentze, M.W.; Schoenberg, D.R. Using the λN peptide to tether proteins to RNAs. In mRNA Processing and Metabolism; Schoenberg, D.R., Ed.; Humana Press: Totowa, NJ, USA, 2004; Volume 257, pp. 135–154. [Google Scholar]
- Bardwell, V.J.; Wickens, M. Purification of RNA and RNA-protein complexes by an R17 coat protein affinity method. Nucleic Acids Res. 1990, 18, 6587–6594. [Google Scholar] [CrossRef] [Green Version]
- Bos, T.J.; Nussbacher, J.K.; Aigner, S.; Yeo, G.W. Tethered function assays as tools to elucidate the molecular roles of RNA-binding proteins. Adv. Exp. Med. Biol. 2016, 907, 61–88. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.Y.; Kutner, R.H.; Bialkowska, A.; Marino, M.P.; Klimstra, W.B.; Reiser, J. Cell-specific targeting of lentiviral vectors mediated by fusion proteins derived from Sindbis virus, vesicular stomatitis virus, or avian sarcoma/leukosis virus. Retrovirology 2010, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leber, M.F.; Neault, S.; Jirovec, E.; Barkley, R.; Said, A.; Bell, J.C.; Ungerechts, G. Engineering and combining oncolytic measles virus for cancer therapy. Cytokine Growth Factor Rev. 2020, 56, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Argaw, T.; Marino, M.P.; Timmons, A.; Eldridge, L.; Takeda, K.; Li, P.; Kwilas, A.; Ou, W.; Reiser, J. In vivo targeting of lentiviral vectors pseudotyped with the Tupaia paramyxovirus H glycoprotein bearing a cell-specific ligand. Mol. Ther. Methods Clin. Dev. 2021, 21, 670–680. [Google Scholar] [CrossRef]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune responses to viral gene therapy vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Bruno, J.G. Potential inherent stimulation of the innate immune system by nucleic acid aptamers and possible corrective approaches. Pharmaceuticals 2018, 11, 62. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, H.; Kutner, R.H.; Bazan, N.G.; Reiser, J. Simplified lentivirus vector production in protein-free media using polyethylenimine-mediated transfection. J. Virol. Methods 2009, 157, 113–121. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; La Russa, V.F.; Reiser, J. Transduction of bone-marrow-derived mesenchymal stem cells by using lentivirus vectors pseudotyped with modified RD114 envelope glycoproteins. J. Virol. 2004, 78, 1219–1229. [Google Scholar] [CrossRef] [Green Version]
- Kutner, R.H.; Zhang, X.Y.; Reiser, J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc. 2009, 4, 495–505. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panigaj, M.; Marino, M.P.; Reiser, J. Tagging and Capturing of Lentiviral Vectors Using Short RNAs. Int. J. Mol. Sci. 2021, 22, 10263. https://doi.org/10.3390/ijms221910263
Panigaj M, Marino MP, Reiser J. Tagging and Capturing of Lentiviral Vectors Using Short RNAs. International Journal of Molecular Sciences. 2021; 22(19):10263. https://doi.org/10.3390/ijms221910263
Chicago/Turabian StylePanigaj, Martin, Michael P. Marino, and Jakob Reiser. 2021. "Tagging and Capturing of Lentiviral Vectors Using Short RNAs" International Journal of Molecular Sciences 22, no. 19: 10263. https://doi.org/10.3390/ijms221910263
APA StylePanigaj, M., Marino, M. P., & Reiser, J. (2021). Tagging and Capturing of Lentiviral Vectors Using Short RNAs. International Journal of Molecular Sciences, 22(19), 10263. https://doi.org/10.3390/ijms221910263