
Inhibition of APE1/Ref-1 for Neovascular Eye Diseases: From Biology to Therapy

 ,
, 
Abstract
:1. Introduction
2. APE1/Ref-1
2.1. APE/Ref-1 Functions
2.2. Structure of APE1/Ref-1
2.3. APE1/Ref-1 Expression
3. Targeting APE1/Ref-1 as a Therapeutic Approach
3.1. APE1/Ref-1 Redox Inhibitors
3.2. Specificity of APE1/Ref-1 Redox Inhibitors
3.3. APE1/Ref-1 in Cancer
3.4. APE1/Ref-1 in Neuronal Diseases
3.5. APE1/Ref-1 in Other Diseases
4. Neovascular Eye Diseases
4.1. Age-Related Macular Degeneration
4.2. Diabetic Retinopathy and Diabetic Macular Edema
4.3. Retinopathy of Prematurity
4.4. Shortcomings of Current Therapies
5. APE1/Ref-1 and Angiogenesis in Neovascular Eye Disease
5.1. Role of HIF-1
5.2. Role of VEGF
5.3. Suppression of Angiogenesis In Vitro with APE1/Ref-1 Inhibitors
5.4. Suppression of Angiogenesis In Vivo with APE1/Ref-1 Inhibitors
6. APE1/Ref-1 and Inflammation in Neovascular Eye Disease
6.1. Role of NF-κB
6.2. Role of STAT3
7. APE1/Ref-1 and Oxidative Stress in Neovascular Eye Disease
7.1. Role of Nrf2
7.2. Role of HO-1
8. APE1/Ref-1 and Cell-Cycle Control in Neovascular Eye Disease
9. Clinical Relevance and Significance
10. Outstanding Research Questions
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| AMD | Age-related macular degeneration |
| AP-1 | Activator protein 1 |
| APE1/Ref-1 | Apurinic/apyrimidinic endonuclease 1/reduction-oxidation factor 1 |
| APX2009 | (E)-N,N-diethyl-2-((3-methoxy-1,4-dihydronaphthalen-2-yl)methylene)pentanamide |
| APX2014 | (E)-N-methoxy-2-((3-methoxy-1,4-dioxo-1,4-dihydronaphthalen-2-yl)methylene)pentanamide |
| APX3330 | (2E)-2-[(4,5-dimethoxy-2-methyl-3,6-dioxo-1,4-cyclohexadien-1-yl)methyelene]-undecanoic acid |
| ARE | Antioxidant response element |
| ARPE-19 | Adult retinal pigment epithelium-19 |
| AUC | Area under the curve |
| BER | Base-excision repair |
| BMI | Body mass index |
| CA9 | Carbonic anhydrase 9 |
| CBF/NF-Y | CCAAT-binding factor |
| CCL20 | C-C motif chemokine ligand 20 |
| CI | Cell index |
| CIPN | Chemotherapy-induced peripheral neuropathy |
| CNS | Central nervous system |
| CREB | cAMP-responsive element (CRE)-binding protein |
| CXCR4 | CXC motif chemokine receptor 4 |
| DME | Diabetic macular edema |
| DR | Diabetic retinopathy |
| ECFCs | Erythroid-colony-forming unit cells |
| ECM | Extracellular matrix |
| EMSA | Electrophoretic mobility shift assay |
| ERK | Extracellular-signal-regulated kinase |
| HEY-C2 | Ovarian cancer cell line |
| HIF-1 | Hypoxia-inducible factor 1 |
| HIF-1α | Hypoxia-inducible factor 1 α subunit |
| HIF-1β | Hypoxia-inducible factor 1 β subunit |
| HO-1 | Heme oxygenase 1 |
| HRECs | Human retinal microvascular endothelial cells |
| HSF-1 | Heat shock factor 1 |
| HUVEC | Human umbilical vein endothelial cells |
| ICAM-1 | Intracellular adhesion molecule 1 |
| IBD | Inflammatory bowel disease |
| IKK | IκB kinase |
| IL-1β | Interleukin 1β |
| IL-6 | Interleukin 6 |
| IL-8 | Interleukin 8 |
| IP | Intraperitoneal |
| IVT | Intravitreal |
| JAK/STAT | Janus kinase/signal transducer and activator of transcription |
| LC-MS/MS | Liquid chromatography with tandem mass spectrometry |
| L-CNV | Laser-induced choroidal neovascularization |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MPP+ | 1-methyl-4-phenylpyridinium |
| mtDNA | Mitochondrial DNA |
| MTF-1 | Metal regulatory transcription factor 1 |
| NF-κB | Nuclear factor κ light-chain-enhancer of activated B cells |
| NPDR | Non-proliferative diabetic retinopathy |
| NPM1 | Nucleophosmin 1 |
| Nrf1 | Nuclear factor erythroid 2-related factor 1 |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| nvAMD | Neovascular age-related macular degeneration |
| OCT | Optical coherence tomography |
| oxLDL | Oxidized low density lipoprotein |
| PBPK | Physiological-based pharmacokinetic |
| PD | Parkinson’s disease |
| PDAC | Pancreatic ductal adenocarcinoma |
| PDGF-B | Platelet-derived growth factor subunit B |
| PDR | Proliferative diabetic retinopathy |
| PI3K/Akt | Phosphoinositide-3-kinase |
| PEBP-2 | Polyomavirus enhancer-binding protein 2 |
| Rf/6a | Macaque choroidal endothelial cell-like cell line |
| ROP | Retinopathy of prematurity |
| ROS | Reactive oxygen species |
| RPE | Retinal pigment epithelium |
| RVECs | Retinal vascular endothelial cells |
| SKOV-3X | Human ovarian cancer cell line |
| STAT3 | Signal transducer and activator of transcription 3 |
| TF(s) | Transcription factor(s) |
| TNF-α | Tumor necrosis factor alpha |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| VEGF | Vascular endothelial growth factor |
| VE-PTP | Vascular endothelial protein tyrosine phosphatase |
| VHL | Von Hippel–Lindau |
| YY1 | Ying yang-1 |
References
- Cabral, T.; Mello, L.G.M.; Lima, L.H.; Polido, J.; Regatieri, C.V.; Belfort, R., Jr.; Mahajan, V.B. Retinal and choroidal angiogenesis: A review of new targets. Int. J. Retin. Vitr. 2017, 3, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.; Wang, C.C.; Adamis, A.P. Ocular neovascularization: An epidemiologic review. Surv. Ophthalmol. 1998, 43, 245–269. [Google Scholar] [CrossRef]
- Dreyfuss, J.L.; Giordano, R.J.; Regatieri, C.V. Ocular angiogenesis. J. Ophthalmol. 2015, 2015, 892043. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; He, M.; Congdon, N. The worldwide epidemic of diabetic retinopathy. Indian J. Ophthalmol. 2012, 60, 428–431. [Google Scholar]
- Blencowe, H.; Lawn, J.E.; Vazquez, T.; Fielder, A.; Gilbert, C. Preterm-associated visual impairment and estimates of retinopathy of prematurity at regional and global levels for 2010. Pediatr. Res. 2013, 74, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solebo, A.L.; Teoh, L.; Rahi, J. Epidemiology of blindness in children. Arch. Dis Child. 2017, 102, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhao, J.; Sun, X. Resistance to anti-VEGF therapy in neovascular age-related macular degeneration: A comprehensive review. Drug Des. Devel. Ther. 2016, 10, 1857–1867. [Google Scholar]
- Kelley, M.R.; Georgiadis, M.M.; Fishel, M.L. APE1/Ref-1 role in redox signaling: Translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr. Mol. Pharmacol. 2012, 5, 36–53. [Google Scholar] [CrossRef]
- Li, M.; Wilson, D.M., 3rd. Human apurinic/apyrimidinic endonuclease 1. Antioxid. Redox Signal. 2014, 20, 678–707. [Google Scholar] [CrossRef] [Green Version]
- Kane, C.M.; Linn, S. Purification and characterization of an apurinic/apyrimidinic endonuclease from HeLa cells. J. Biol. Chem. 1981, 256, 3405–3414. [Google Scholar] [CrossRef]
- Grafstrom, R.H.; Shaper, N.L.; Grossman, L. Human placental apurinic/apyrimidinic endonuclease. Mechanism of action. J. Biol. Chem. 1982, 257, 13459–13464. [Google Scholar] [CrossRef]
- Shaper, N.L.; Grafstrom, R.H.; Grossman, L. Human placental apurinic/apyrimidinic endonuclease. Its isolation and characterization. J. Biol. Chem. 1982, 257, 13455–13458. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Curran, T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992, 11, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Caston, R.A.; Gampala, S.; Armstrong, L.; Messmann, R.A.; Fishel, M.L.; Kelley, M.R. The multifunctional APE1 DNA repair-redox signaling protein as a drug target in human disease. Drug Discov. Today 2021, 26, 218–228. [Google Scholar] [CrossRef]
- Georgiadis, M.M.; Luo, M.; Gaur, R.K.; Delaplane, S.; Li, X.; Kelley, M.R. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat. Res. 2008, 643, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Shah, F.; Logsdon, D.; Messmann, R.A.; Fehrenbacher, J.C.; Fishel, M.L.; Kelley, M.R. Exploiting the Ref-1-APE1 node in cancer signaling and other diseases: From bench to clinic. NPJ Precis. Oncol. 2017, 1, 19. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Luo, M.; Marasco, D.; Logsdon, D.; LaFavers, K.A.; Chen, Q.; Reed, A.; Kelley, M.R.; Gross, M.L.; Georgiadis, M.M. Inhibition of apurinic/apyrimidinic endonuclease I’s redox activity revisited. Biochemistry 2013, 52, 2955–2966. [Google Scholar] [CrossRef]
- Gorman, M.A.; Morera, S.; Rothwell, D.G.; de La Fortelle, E.; Mol, C.D.; Tainer, J.A.; Hickson, I.D.; Freemont, P.S. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 1997, 16, 6548–6558. [Google Scholar] [CrossRef] [Green Version]
- Xanthoudakis, S.; Miao, G.G.; Curran, T. The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc. Natl. Acad. Sci. USA 1994, 91, 23–27. [Google Scholar] [CrossRef] [Green Version]
- Evans, A.R.; Limp-Foster, M.; Kelley, M.R. Going APE over ref-1. Mutat Res. 2000, 461, 83–108. [Google Scholar] [CrossRef]
- Izumi, T.; Mitra, S. Deletion analysis of human AP-endonuclease: Minimum sequence required for the endonuclease activity. Carcinogenesis 1998, 19, 525–527. [Google Scholar] [CrossRef]
- Vascotto, C.; Fantini, D.; Romanello, M.; Cesaratto, L.; Deganuto, M.; Leonardi, A.; Radicella, J.P.; Kelley, M.R.; D’Ambrosio, C.; Scaloni, A.; et al. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol. Cell Biol. 2009, 29, 1834–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Chen, Q.; Georgiadis, M.M. High-resolution crystal structures reveal plasticity in the metal binding site of apurinic/apyrimidinic endonuclease I. Biochemistry 2014, 53, 6520–6529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acid. Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhang, J.; He, H.; Su, D.; Chen, Q.; Gross, M.L.; Kelley, M.R.; Georgiadis, M.M. Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease. Biochemistry 2012, 51, 695–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.R.; Joo, H.K.; Jeon, B.H. The biological role of apurinic/apyrimidinic endonuclease1/redox factor-1 as a therapeutic target for vascular inflammation and as a serologic biomarker. Biomedicines 2020, 8, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, X.; Zhou, T.; Kelley, M.R.; Edwards, P.; Gao, H.; Qiao, X. Inhibition of APE1/Ref-1 redox activity rescues human retinal pigment epithelial cells from oxidative stress and reduces choroidal neovascularization. Redox Biol. 2014, 2, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Chiarini, L.B.; Freitas, F.G.; Petrs-Silva, H.; Linden, R. Evidence that the bifunctional redox factor / AP endonuclease Ref-1 is an anti-apoptotic protein associated with differentiation in the developing retina. Cell Death Differ. 2000, 7, 272–281. [Google Scholar] [CrossRef] [Green Version]
- Jiang, A.; Gao, H.; Kelley, M.R.; Qiao, X. Inhibition of APE1/Ref-1 redox activity with APX3330 blocks retinal angiogenesis in vitro and in vivo. Vis. Res. 2011, 51, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Joo, H.K.; Lee, E.O.; Park, M.S.; Cho, H.S.; Kim, S.; Jin, H.; Jeong, J.O.; Kim, C.S.; Jeon, B.H. Plasma APE1/Ref-1 correlates with atherosclerotic inflammation in ApoE(−/−) mice. Biomedicines 2020, 8, 366. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.A.; Lim, B.K.; Seo, H.J.; Kim, S.K.; Ahn, K.T.; Jeon, B.H.; Jeong, J.O. Elevation of serum APE1/Ref-1 in experimental murine myocarditis. Int. J. Mol. Sci. 2017, 18, 2664. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Joo, H.K.; Lee, E.O.; Cho, H.S.; Choi, S.; Kim, C.S.; Jeon, B.H. ATP binding cassette transporter A1 is involved in extracellular secretion of acetylated APE1/Ref-1. Int. J. Mol. Sci. 2019, 20, 3178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eads, J.R.; Krishnamurthi, S.S.; Saltzman, J.; Bokar, J.A.; Savvides, P.; Meropol, N.J.; Gibbons, J.; Koon, H.; Sharma, N.; Rogers, L.; et al. Phase I clinical trial of temozolomide and methoxyamine (TRC-102), an inhibitor of base excision repair, in patients with advanced solid tumors. Invest. New Drugs 2021, 39, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Poletto, M.; Malfatti, M.C.; Dorjsuren, D.; Scognamiglio, P.L.; Marasco, D.; Vascotto, C.; Jadhav, A.; Maloney, D.J.; Wilson, D.M., III; Simeonov, A.; et al. Inhibitors of the apurinic/apyrimidinic endonuclease 1 (APE1)/nucleophosmin (NPM1) interaction that display anti-tumor properties. Mol. Carcinog. 2016, 55, 688–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.; Joo, H.K.; Jeon, B.H. Dynamic regulation of APE1/Ref-1 as a therapeutic target protein. Chonnam. Med. J. 2016, 52, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, A.A.; Jiang, Y.; Luo, M.; Reed, A.M.; Shahda, S.; He, Y.; Maitra, A.; Kelley, M.R.; Fishel, M.L. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS ONE 2012, 7, e47462. [Google Scholar] [CrossRef]
- Fishel, M.L.; Colvin, E.S.; Luo, M.; Kelley, M.R.; Robertson, K.A. Inhibition of the redox function of APE1/Ref-1 in myeloid leukemia cell lines results in a hypersensitive response to retinoic acid-induced differentiation and apoptosis. Exp. Hematol. 2010, 38, 1178–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishel, M.L.; Wu, X.; Devlin, C.M.; Logsdon, D.P.; Jiang, Y.; Luo, M.; He, Y.; Yu, Z.; Tong, Y.; Lipking, K.P.; et al. Apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/Ref-1) redox function negatively regulates NRF2. J. Biol. Chem. 2015, 290, 3057–3068. [Google Scholar] [CrossRef] [Green Version]
- Fishel, M.L.; Jiang, Y.; Rajeshkumar, N.V.; Scandura, G.; Sinn, A.L.; He, Y.; Shen, C.; Jones, D.R.; Pollok, K.E.; Ivan, M.; et al. Impact of APE1/Ref-1 redox inhibition on pancreatic tumor growth. Mol. Cancer Ther. 2011, 10, 1698–1708. [Google Scholar] [CrossRef] [Green Version]
- Su, D.; Delaplane, S.; Luo, M.; Rempel, D.L.; Vu, B.; Kelley, M.R.; Gross, M.L.; Georgiadis, M.M. Interactions of apurinic/apyrimidinic endonuclease with a redox inhibitor: Evidence for an alternate conformation of the enzyme. Biochemistry 2011, 50, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Delaplane, S.; Jiang, A.; Reed, A.; He, Y.; Fishel, M.; Nyland, R.L., II; Borch, R.F.; Qiao, X.; Georgiadis, M.M.; et al. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: Small-molecule inhibition of the redox function of Ape1. Antioxid. Redox Signal. 2008, 10, 1853–1867. [Google Scholar] [CrossRef] [PubMed]
- Jedinak, A.; Dudhgaonkar, S.; Kelley, M.R.; Sliva, D. Apurinic/apyrimidinic endonuclease 1 regulates inflammatory response in macrophages. Anticancer Res. 2011, 31, 379–385. [Google Scholar] [PubMed]
- Sardar Pasha, S.P.B.; Sishtla, K.; Sulaiman, R.S.; Park, B.; Shetty, T.; Shah, F.; Fishel, M.L.; Wikel, J.H.; Kelley, M.R.; Corson, T.W. Ref-1/APE1 inhibition with novel small molecules blocks ocular neovascularization. J. Pharmacol. Exp. Ther. 2018, 367, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Kelley, M.R.; Wikel, J.H.; Guo, C.; Pollok, K.E.; Bailey, B.J.; Wireman, R.; Fishel, M.L.; Vasko, M.R. Identification and characterization of new chemical entities targeting apurinic/apyrimidinic endonuclease 1 for the prevention of chemotherapy-induced peripheral neuropathy. J. Pharmacol. Exp. Ther. 2016, 359, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Kelley, M.R.; Luo, M.; Reed, A.; Su, D.; Delaplane, S.; Borch, R.F.; Nyland, R.L., II; Gross, M.L.; Georgiadis, M.M. Functional analysis of novel analogues of E3330 that block the redox signaling activity of the multifunctional AP endonuclease/redox signaling enzyme APE1/Ref-1. Antioxid. Redox Signal. 2011, 14, 1387–1401. [Google Scholar] [CrossRef]
- Laev, S.S.; Salakhutdinov, N.F.; Lavrik, O.I. Inhibitors of nuclease and redox activity of apurinic/apyrimidinic endonuclease 1/redox effector factor 1 (APE1/Ref-1). Bioorg. Med. Chem. 2017, 25, 2531–2544. [Google Scholar] [CrossRef]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The essential medicinal chemistry of curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- Nyland, R.L.; Luo, M.; Kelley, M.R.; Borch, R.F. Design and synthesis of novel quinone inhibitors targeted to the redox function of apurinic/apyrimidinic endonuclease 1/redox enhancing factor-1 (Ape1/ref-1). J. Med. Chem. 2010, 53, 1200–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logsdon, D.P.; Shah, F.; Carta, F.; Supuran, C.T.; Kamocka, M.; Jacobsen, M.H.; Sandusky, G.E.; Kelley, M.R.; Fishel, M.L. Blocking HIF signaling via novel inhibitors of CA9 and APE1/Ref-1 dramatically affects pancreatic cancer cell survival. Sci. Rep. 2018, 8, 13759. [Google Scholar] [CrossRef] [PubMed]
- Gampala, S.; Shah, F.; Lu, X.; Moon, H.R.; Babb, O.; Umesh Ganesh, N.; Sandusky, G.; Hulsey, E.; Armstrong, L.; Mosley, A.L.; et al. Ref-1 redox activity alters cancer cell metabolism in pancreatic cancer: Exploiting this novel finding as a potential target. J. Exp. Clin. Cancer Res. 2021, 40, 251. [Google Scholar] [CrossRef] [PubMed]
- Hiramoto, M.; Shimizu, N.; Sugimoto, K.; Tang, J.; Kawakami, Y.; Ito, M.; Aizawa, S.; Tanaka, H.; Makino, I.; Handa, H. Nuclear targeted suppression of NF-kappa B activity by the novel quinone derivative E3330. J. Immunol. 1998, 160, 810–819. [Google Scholar] [PubMed]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanuthala, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell 2018, 23, 833–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Dai, N.; Wang, D.; Zhong, Z. Distinct APE1 activities affect the regulation of VEGF transcription under hypoxic conditions. Comput. Struct. Biotechnol. J. 2019, 17, 324–332. [Google Scholar] [CrossRef]
- Li, Y.; Liu, X.; Zhou, T.; Kelley, M.R.; Edwards, P.A.; Gao, H.; Qiao, X. Suppression of choroidal neovascularization through inhibition of APE1/Ref-1 redox activity. Invest. Ophthalmol. Vis. Sci. 2014, 55, 4461–4469. [Google Scholar] [CrossRef] [PubMed]
- Shah, F.; Goossens, E.; Atallah, N.M.; Grimard, M.; Kelley, M.R.; Fishel, M.L. APE1/Ref-1 knockdown in pancreatic ductal adenocarcinoma—Characterizing gene expression changes and identifying novel pathways using single-cell RNA sequencing. Mol. Oncol. 2017, 11, 1711–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIlwain, D.W.; Fishel, M.L.; Boos, A.; Kelley, M.R.; Jerde, T.J. APE1/Ref-1 redox-specific inhibition decreases survivin protein levels and induces cell cycle arrest in prostate cancer cells. Oncotarget 2018, 9, 10962–10977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Poornima, V.; Michael, C.; Alex, Z.; Peng, Y.; Ruizhuo, N.; Xiaoxi, Q.; Kelley, M.R.; Jieli, C. APX3330 promotes neurorestorative effects after stroke in type one diabetic rats. Aging Dis. 2018, 9, 453–466. [Google Scholar]
- Bhat, A.A.; Lu, H.; Soutto, M.; Capobianco, A.; Rai, P.; Zaika, A.; El-Rifai, W. Exposure of Barrett’s and esophageal adenocarcinoma cells to bile acids activates EGFR-STAT3 signaling axis via induction of APE1. Oncogene 2018, 37, 6011–6024. [Google Scholar] [CrossRef] [PubMed]
- Cesaratto, L.; Codarin, E.; Vascotto, C.; Leonardi, A.; Kelley, M.R.; Tiribelli, C.; Tell, G. Specific inhibition of the redox activity of APE1/Ref-1 by E3330 blocks TNF-alpha-induced activation of IL-8 production in liver cancer cell lines. PLoS ONE 2013, 8, e70909. [Google Scholar] [CrossRef]
- Sriramajayam, K.; Peng, D.; Lu, H.; Zhou, S.; Bhat, N.; McDonald, O.G.; Que, J.; Zaika, A.; El-Rifai, W. Activation of NRF2 by APE1/REF1 is redox-dependent in Barrett’s related esophageal adenocarcinoma cells. Redox Biol. 2021, 43, 101970. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.R.; Shahda, S.; Lakhani, N.J.; O’Neil, B.; Chu, L.; Anderson, A.K.; Wan, J.; Mosley, A.L.; Liu, H.; Messmann, R.A. A phase I study targeting the APE1/Ref-1 DNA repair-redox signaling protein with the APX3330 inhibitor [abstract]. In Proceedings of the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics, Boston, MA, USA, 26–30 October 2019. [Google Scholar]
- Shimizu, N.; Sugimoto, K.; Tang, J.; Nishi, T.; Sato, I.; Hiramoto, M.; Aizawa, S.; Hatakeyama, M.; Ohba, R.; Hatori, H.; et al. High-performance affinity beads for identifying drug receptors. Nat. Biotechnol. 2000, 18, 877–881. [Google Scholar] [CrossRef]
- Codrich, M.; Comelli, M.; Malfatti, M.C.; Mio, C.; Ayyildiz, D.; Zhang, C.; Kelley, M.R.; Terrosu, G.; Pucillo, C.E.M.; Tell, G. Inhibition of APE1-endonuclease activity affects cell metabolism in colon cancer cells via a p53-dependent pathway. DNA Repair 2019, 82, 102675. [Google Scholar] [CrossRef] [PubMed]
- Bhakat, K.K.; Mantha, A.K.; Mitra, S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid. Redox Signal. 2009, 11, 621–638. [Google Scholar] [CrossRef] [Green Version]
- Bobola, M.S.; Blank, A.; Berger, M.S.; Stevens, B.A.; Silber, J.R. Apurinic/apyrimidinic endonuclease activity is elevated in human adult gliomas. Clin. Cancer Res. 2001, 7, 3510–3518. [Google Scholar] [PubMed]
- Carrero, P.; Okamoto, K.; Coumailleau, P.; O’Brien, S.; Tanaka, H.; Poellinger, L. Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1alpha. Mol. Cell Biol. 2000, 20, 402–415. [Google Scholar] [CrossRef] [Green Version]
- Kelley, M.R.; Cheng, L.; Foster, R.; Tritt, R.; Jiang, J.; Broshears, J.; Koch, M. Elevated and altered expression of the multifunctional DNA base excision repair and redox enzyme Ape1/Ref-1 in prostate cancer. Clin. Cancer Res. 2001, 7, 824–830. [Google Scholar]
- Koukourakis, M.I.; Giatromanolaki, A.; Kakolyris, S.; Sivridis, E.; Georgoulias, V.; Funtzilas, G.; Hickson, I.D.; Gatter, K.C.; Harris, A.L. Nuclear expression of human apurinic/apyrimidinic endonuclease (HAP1/Ref-1) in head-and-neck cancer is associated with resistance to chemoradiotherapy and poor outcome. Int. J. Radiat. Oncol. Biol. Phys. 2001, 50, 27–36. [Google Scholar] [CrossRef]
- Puglisi, F.; Aprile, G.; Minisini, A.M.; Barbone, F.; Cataldi, P.; Tell, G.; Kelley, M.R.; Damante, G.; Beltrami, C.A.; Di Loreto, C. Prognostic significance of Ape1/Ref-1 subcellular localization in non-small cell lung carcinomas. Anticancer Res. 2001, 21, 4041–4049. [Google Scholar] [PubMed]
- Robertson, K.A.; Bullock, H.A.; Xu, Y.; Tritt, R.; Zimmerman, E.; Ulbright, T.M.; Foster, R.S.; Einhorn, L.H.; Kelley, M.R. Altered expression of Ape1/Ref-1 in germ cell tumors and overexpression in NT2 cells confers resistance to bleomycin and radiation. Cancer Res. 2001, 61, 2220–2225. [Google Scholar] [PubMed]
- Fishel, M.L.; Kelley, M.R. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Mol. Aspects Med. 2007, 28, 375–395. [Google Scholar] [CrossRef]
- Kelley, M.R.; Logsdon, D.; Fishel, M.L. Targeting DNA repair pathways for cancer treatment: What’s new? Future Oncol. 2014, 10, 1215–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logsdon, D.P.; Grimard, M.; Luo, M.; Shahda, S.; Jiang, Y.; Tong, Y.; Yu, Z.; Zyromski, N.; Schipani, E.; Carta, F.; et al. Regulation of HIF1alpha under hypoxia by APE1/Ref-1 impacts CA9 expression: Dual targeting in patient-derived 3D pancreatic cancer models. Mol. Cancer Ther. 2016, 15, 2722–2732. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zhou, S.; Sandusky, G.E.; Kelley, M.R.; Fishel, M.L. Reduced expression of DNA repair and redox signaling protein APE1/Ref-1 impairs human pancreatic cancer cell survival, proliferation, and cell cycle progression. Cancer Invest. 2010, 28, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Sharbeen, G.; McCarroll, J.; Goldstein, D.; Phillips, P.A. Exploiting base excision repair to improve therapeutic approaches for pancreatic cancer. Front. Nutr. 2015, 2, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gampala, S.; Shah, F.; Zhang, C.; Rhodes, S.D.; Babb, O.; Grimard, M.; Wireman, R.S.; Rad, E.; Calver, B.; Bai, R.Y.; et al. Exploring transcriptional regulators Ref-1 and STAT3 as therapeutic targets in malignant peripheral nerve sheath tumours. Br. J. Cancer 2021, 124, 1566–1580. [Google Scholar] [CrossRef] [PubMed]
- Heisel, C.; Yousif, J.; Mijiti, M.; Charizanis, K.; Brigell, M.; Corson, T.W.; Kelley, M.R. APE1/Ref-1 as a novel target for retinal diseases. Cell. Signal. 2021, 2, 133–138. [Google Scholar]
- Zou, G.M.; Karikari, C.; Kabe, Y.; Handa, H.; Anders, R.A.; Maitra, A. The Ape-1/Ref-1 redox antagonist E3330 inhibits the growth of tumor endothelium and endothelial progenitor cells: Therapeutic implications in tumor angiogenesis. J. Cell Physiol. 2009, 219, 209–218. [Google Scholar] [CrossRef]
- Lee, Y.R.; Park, M.S.; Joo, H.K.; Kim, K.M.; Kim, J.; Jeon, B.H.; Choi, S. Therapeutic positioning of secretory acetylated APE1/Ref-1 requirement for suppression of tumor growth in triple-negative breast cancer in vivo. Sci. Rep. 2018, 8, 8701. [Google Scholar] [CrossRef] [Green Version]
- Shahda, S.; Lakhani, N.J.; O’Neil, B.; Rasco, D.W.; Wan, J.; Mosley, A.L.; Liu, H.; Kelley, M.R.; Messmann, R.A. A phase I study of the APE1 protein inhibitor APX3330 in patients with advanced solid tumors. J. Clin. Oncol. 2019, 37, 3097. [Google Scholar] [CrossRef]
- Davydov, V.; Hansen, L.A.; Shackelford, D.A. Is DNA repair compromised in Alzheimer’s disease? Neurobiol. Aging 2003, 24, 953–968. [Google Scholar] [CrossRef]
- Marcon, G.; Tell, G.; Perrone, L.; Garbelli, R.; Quadrifoglio, F.; Tagliavini, F.; Giaccone, G. APE1/Ref-1 in Alzheimer’s disease: An immunohistochemical study. Neurosci. Lett. 2009, 466, 124–127. [Google Scholar] [CrossRef]
- Tan, Z.; Sun, N.; Schreiber, S.S. Immunohistochemical localization of redox factor-1 (Ref-1) in Alzheimer’s hippocampus. Neuroreport 1998, 9, 2749–2752. [Google Scholar] [CrossRef]
- Shaikh, A.Y.; Martin, L.J. DNA base-excision repair enzyme apurinic/apyrimidinic endonuclease/redox factor-1 is increased and competent in the brain and spinal cord of individuals with amyotrophic lateral sclerosis. Neuromol. Med. 2002, 2, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Mu, S.; Yang, Q.; Guo, S.; Chen, X.; Guo, H. Ape1 protects against MPP+-induced neurotoxicity through ERK1/2 signaling in PC12 cells. Neuroreport 2017, 28, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Leak, R.K.; Li, P.; Zhang, F.; Sulaiman, H.H.; Weng, Z.; Wang, G.; Stetler, R.A.; Shi, Y.; Cao, G.; Gao, Y.; et al. Apurinic/apyrimidinic endonuclease 1 upregulation reduces oxidative DNA damage and protects hippocampal neurons from ischemic injury. Antioxid. Redox Signal. 2015, 22, 135–148. [Google Scholar] [CrossRef] [Green Version]
- Stetler, R.A.; Gao, Y.; Leak, R.K.; Weng, Z.; Shi, Y.; Zhang, L.; Pu, H.; Zhang, F.; Hu, X.; Hassan, S.; et al. APE1/Ref-1 facilitates recovery of gray and white matter and neurological function after mild stroke injury. Proc. Natl. Acad. Sci. USA 2016, 113, E3558–E3567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, M.R.; Jiang, Y.; Guo, C.; Reed, A.; Meng, H.; Vasko, M.R. Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PLoS ONE 2014, 9, e106485. [Google Scholar] [CrossRef]
- Mijiti, M.; Caston, R.; Gampala, S.; Fishel, M.L.; Fehrenbacher, J.C.; Kelley, M.R. APE1/Ref-1—One target with multiple indications: Emerging aspects and new directions. J. Cell. Signal. 2021, 2, 151–161. [Google Scholar]
- Sahakian, L.; Filippone, R.T.; Stavely, R.; Robinson, A.M.; Yan, X.S.; Abalo, R.; Eri, R.; Bornstein, J.C.; Kelley, M.R.; Nurgali, K. Inhibition of APE1/Ref-1 redox signaling alleviates intestinal dysfunction and damage to myenteric neurons in a mouse model of spontaneous chronic colitis. Inflamm. Bowel Dis. 2021, 27, 388–406. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A. Ocular neovascularization. J. Mol. Med. 2013, 91, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, I.; Lutty, G. Understanding age-related macular degeneration (AMD): Relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Aspects Med. 2012, 33, 295–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.; Cruickshanks, K.J.; Myers, C.E.; Sivakumaran, T.A.; Iyengar, S.K.; Meuer, S.M.; Schubert, C.R.; Gangnon, R.E.; Klein, B.E. The relationship of atherosclerosis to the 10-year cumulative incidence of age-related macular degeneration: The Beaver Dam studies. Ophthalmology 2013, 120, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (AMD): Associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. 2016, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. Primers 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.; Lutty, G.A. Bruch’s membrane and the choroid in age-related macular degeneration. Adv. Exp. Med. Biol. 2021, 1256, 89–119. [Google Scholar]
- Spaide, R.F.; Jaffe, G.J.; Sarraf, D.; Freund, K.B.; Sadda, S.R.; Staurenghi, G.; Waheed, N.K.; Chakravarthy, U.; Rosenfeld, P.J.; Holz, F.G.; et al. Consensus nomenclature for reporting neovascular age-related macular degeneration data: Consensus on neovascular age-related macular degeneration nomenclature study group. Ophthalmology 2020, 127, 616–636. [Google Scholar] [CrossRef]
- Seddon, J.M. Macular degeneration epidemiology: Nature-nurture, lifestyle factors, genetic risk, and gene-environment interactions—The Weisenfeld Award Lecture. Invest. Ophthalmol. Vis. Sci. 2017, 58, 6513–6528. [Google Scholar] [CrossRef]
- Ung, C.; Lains, I.; Miller, J.W.; Kim, I.K. Current management of age-related macular degeneration. Adv. Exp. Med. Biol. 2021, 1256, 295–314. [Google Scholar] [PubMed]
- Klein, B.E. Overview of epidemiologic studies of diabetic retinopathy. Ophthalmic. Epidemiol. 2007, 14, 179–183. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peet, D.J.; Kittipassorn, T.; Wood, J.P.; Chidlow, G.; Casson, R.J. HIF signalling: The eyes have it. Exp. Cell Res. 2017, 356, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Hendrick, A.M.; Gibson, M.V.; Kulshreshtha, A. Diabetic retinopathy. Prim. Care 2015, 42, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Capitao, M.; Soares, R. Angiogenesis and inflammation crosstalk in diabetic retinopathy. J. Cell Biochem. 2016, 117, 2443–2453. [Google Scholar] [CrossRef]
- Simo, R.; Sundstrom, J.M.; Antonetti, D.A. Ocular anti-VEGF therapy for diabetic retinopathy: The role of VEGF in the pathogenesis of diabetic retinopathy. Diabetes Care 2014, 37, 893–899. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, Q.D.; Shah, S.M.; Khwaja, A.A.; Channa, R.; Hatef, E.; Do, D.V.; Boyer, D.; Heier, J.S.; Abraham, P.; Thach, A.B.; et al. Two-year outcomes of the ranibizumab for edema of the macula in diabetes (READ-2) study. Ophthalmology 2010, 117, 2146–2151. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, M.; Kaines, A.; Hamilton, R.D.; Fraser-Bell, S.; Rajendram, R.; Quhill, F.; Boos, C.J.; Xing, W.; Egan, C.; Peto, T.; et al. A prospective randomized trial of intravitreal bevacizumab or laser therapy in the management of diabetic macular edema (BOLT study) 12-month data: Report 2. Ophthalmology 2010, 117, 1078–1086. [Google Scholar] [CrossRef]
- Wallsh, J.O.; Gallemore, R.P. Anti-VEGF-resistant retinal diseases: A review of the latest treatment options. Cells 2021, 10, 1049. [Google Scholar] [CrossRef]
- Ludwig, C.A.; Chen, T.A.; Hernandez-Boussard, T.; Moshfeghi, A.A.; Moshfeghi, D.M. The epidemiology of retinopathy of prematurity in the United States. Ophthalmic. Surg. Lasers Imaging Retin. 2017, 48, 553–562. [Google Scholar] [CrossRef]
- Good, W.V.; Hardy, R.J.; Dobson, V.; Palmer, E.A.; Phelps, D.L.; Quintos, M.; Tung, B. Early Treatment for Retinopathy of Prematurity Cooperative Group, The incidence and course of retinopathy of prematurity: Findings from the Early Treatment for Retinopathy of Prematurity Study. Pediatrics 2005, 116, 15–23. [Google Scholar]
- Gupta, V.P.; Dhaliwal, U.; Sharma, R.; Gupta, P.; Rohatgi, J. Retinopathy of prematurity—Risk factors. Indian J. Pediatr. 2004, 71, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.E. Pathogenesis of retinopathy of prematurity. Semin. Neonatol. 2003, 8, 469–473. [Google Scholar] [CrossRef]
- Chen, J.; Stahl, A.; Hellstrom, A.; Smith, L.E. Current update on retinopathy of prematurity: Screening and treatment. Curr. Opin. Pediatr. 2011, 23, 173–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.K.; Dean, T.W.; Hartnett, M.E.; Kong, L.; Smith, L.E.; Hubbard, G.B.; McGregor, M.L.; Jordan, C.O.; Mantagos, I.S.; Bell, E.F.; et al. A dosing study of bevacizumab for retinopathy of prematurity: Late recurrences and additional treatments. Ophthalmology 2018, 125, 1961–1966. [Google Scholar] [CrossRef]
- Wallace, D.K.; Kraker, R.T.; Freedman, S.F.; Crouch, E.R.; Bhatt, A.R.; Hartnett, M.E.; Yang, M.B.; Rogers, D.L.; Hutchinson, A.K.; VanderVeen, D.K.; et al. Short-term outcomes after very low-dose intravitreous bevacizumab for retinopathy of prematurity. JAMA Ophthalmol. 2020, 138, 698–701. [Google Scholar] [CrossRef] [Green Version]
- Cornel, S.; Adriana, I.D.; Mihaela, T.C.; Speranta, S.; Algerino, S.; Mehdi, B.; Jalaladin, H.R. Anti-vascular endothelial growth factor indications in ocular disease. Rom. J. Ophthalmol. 2015, 59, 235–242. [Google Scholar]
- Ammar, M.J.; Hsu, J.; Chiang, A.; Ho, A.C.; Regillo, C.D. Age-related macular degeneration therapy: A review. Curr. Opin. Ophthalmol. 2020, 31, 215–221. [Google Scholar] [CrossRef]
- Bloch, S.B.; Larsen, M.; Munch, I.C. Incidence of legal blindness from age-related macular degeneration in Denmark: Year 2000 to 2010. Am. J. Ophthalmol. 2012, 153, 209–213. [Google Scholar] [CrossRef]
- Avery, R.L.; Pearlman, J.; Pieramici, D.J.; Rabena, M.D.; Castellarin, A.A.; Nasir, M.A.; Giust, M.J.; Wendel, R.; Patel, A. Intravitreal bevacizumab (Avastin) in the treatment of proliferative diabetic retinopathy. Ophthalmology 2006, 113, 1695–1705. [Google Scholar] [CrossRef]
- Ricci, F.; Bandello, F.; Navarra, P.; Staurenghi, G.; Stumpp, M.; Zarbin, M. Neovascular age-related macular degeneration: Therapeutic management and new-upcoming approaches. Int. J. Mol. Sci. 2020, 21, 8242. [Google Scholar] [CrossRef]
- Stewart, M.W. The expanding role of vascular endothelial growth factor inhibitors in ophthalmology. Mayo Clin. Proc. 2012, 87, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Schlenker, M.B.; Thiruchelvam, D.; Redelmeier, D.A. Intravitreal anti-vascular endothelial growth factor treatment and the risk of thromboembolism. Am. J. Ophthalmol. 2015, 160, 569–580. [Google Scholar] [CrossRef] [PubMed]
- VanderVeen, D.K.; Cataltepe, S.U. Anti-vascular endothelial growth factor intravitreal therapy for retinopathy of prematurity. Semin. Perinatol. 2019, 43, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Day, S.; Acquah, K.; Mruthyunjaya, P.; Grossman, D.S.; Lee, P.P.; Sloan, F.A. Ocular complications after anti-vascular endothelial growth factor therapy in Medicare patients with age-related macular degeneration. Am. J. Ophthalmol. 2011, 152, 266–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, N.; Jacobs, B.; Shetty, T.; Dimaras, H.; Hajrasouliha, A.R.; Jusufbegovic, D.; Corson, T.W. Patient preferences in retinal drug delivery. Sci. Rep. 2021, 11, 18996. [Google Scholar]
- Sulaiman, R.S.; Basavarajappa, H.D.; Corson, T.W. Natural product inhibitors of ocular angiogenesis. Exp. Eye Res. 2014, 129, 161–171. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, G.; Duplan, E.; Boyer, N.; Vigne, P.; Frelin, C. Hypoxia up-regulates prolyl hydroxylase activity: A feedback mechanism that limits HIF-1 responses during reoxygenation. J. Biol. Chem. 2003, 278, 38183–38187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campochiaro, P.A. Molecular pathogenesis of retinal and choroidal vascular diseases. Prog. Retin. Eye Res. 2015, 49, 67–81. [Google Scholar] [CrossRef] [Green Version]
- Kelly, B.D.; Hackett, S.F.; Hirota, K.; Oshima, Y.; Cai, Z.; Berg-Dixon, S.; Rowan, A.; Yan, Z.; Campochiaro, P.A.; Semenza, G.L. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ. Res. 2003, 93, 1074–1081. [Google Scholar] [CrossRef] [Green Version]
- Campochiaro, P.A.; Aiello, L.P.; Rosenfeld, P.J. Anti-vascular endothelial growth factor agents in the treatment of retinal disease: From bench to bedside. Ophthalmology 2016, 123, S78–S88. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, R.S.; Merrigan, S.; Quigley, J.; Qi, X.; Lee, B.; Boulton, M.E.; Kennedy, B.; Seo, S.Y.; Corson, T.W. A novel small molecule ameliorates ocular neovascularisation and synergises with anti-VEGF therapy. Sci. Rep. 2016, 6, 25509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basavarajappa, H.D.; Lee, B.; Lee, H.; Sulaiman, R.S.; An, H.; Magana, C.; Shadmand, M.; Vayl, A.; Rajashekhar, G.; Kim, E.Y.; et al. Synthesis and biological evaluation of novel homoisoflavonoids for retinal neovascularization. J. Med. Chem. 2015, 58, 5015–5027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulaiman, R.S.; Quigley, J.; Qi, X.; O’Hare, M.N.; Grant, M.B.; Boulton, M.E.; Corson, T.W. A simple optical coherence tomography quantification method for choroidal neovascularization. J. Ocul. Pharmacol. Ther. 2015, 31, 447–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Ma, J.X. Canonical Wnt signaling in diabetic retinopathy. Vis. Res. 2017, 139, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Rubsam, A.; Parikh, S.; Fort, P.E. Role of inflammation in diabetic retinopathy. Int. J. Mol. Sci. 2018, 19, 942. [Google Scholar] [CrossRef] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, T.D. The Rel/NF-kappaB signal transduction pathway: Introduction. Oncogene 1999, 18, 6842–6844. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene 2006, 25, 6706–6716. [Google Scholar] [CrossRef] [Green Version]
- Park, M.S.; Choi, S.; Lee, Y.R.; Joo, H.K.; Kang, G.; Kim, C.S.; Kim, S.J.; Lee, S.D.; Jeon, B.H. Secreted APE1/Ref-1 inhibits TNF-alpha-stimulated endothelial inflammation via thiol-disulfide exchange in TNF receptor. Sci. Rep. 2016, 6, 23015. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [Green Version]
- Batliwala, S.; Xavier, C.; Liu, Y.; Wu, H.; Pang, I.H. Involvement of Nrf2 in ocular diseases. Oxid. Med. Cell Longev. 2017, 2017, 1703810. [Google Scholar] [CrossRef]
- Dong, L.; Lin, T.; Li, W.; Hong, Y.; Ren, X.; Ke, Y.; Zhang, X.; Li, X. Antioxidative effects of polypyrimidine tract-binding protein-associated splicing factor against pathological retinal angiogenesis through promotion of mitochondrial function. J. Mol. Med. 2021, 99, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Xu, X.; Bi, H.; Zhu, Q.; Wu, J.; Xia, X.; Qiushi, R.; Ho, P.C. Expression modification of uncoupling proteins and MnSOD in retinal endothelial cells and pericytes induced by high glucose: The role of reactive oxygen species in diabetic retinopathy. Exp. Eye Res. 2006, 83, 807–816. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Chan, P.S. Oxidative stress and diabetic retinopathy. Exp. Diabetes Res. 2007, 2007, 43603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, A.D.; Harris-Hayes, M.; Schootman, M. Epidemiology of diabetes and diabetes-related complications. Phys. Ther. 2008, 88, 1254–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.W.; Byzova, T.V. Oxidative stress in angiogenesis and vascular disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.S.; Mahapatra, C.; Park, J.S.; Dashnyam, K.; Kim, J.W.; Ahn, J.C.; Chung, P.S.; Yoon, D.S.; Mandakhbayar, N.; Singh, R.K.; et al. Revascularization and limb salvage following critical limb ischemia by nanoceria-induced Ref-1/APE1-dependent angiogenesis. Biomaterials 2020, 242, 119919. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Kim, K.C.; Kang, K.A.; Zhang, R.; Piao, M.J.; Kim, G.Y.; Kang, M.Y.; Lee, S.J.; Lee, N.H.; Surh, Y.J.; Hyun, J.W. Up-regulation of Nrf2-mediated heme oxygenase-1 expression by eckol, a phlorotannin compound, through activation of Erk and PI3K/Akt. Int. J. Biochem. Cell Biol. 2010, 42, 297–305. [Google Scholar] [CrossRef]
- Wei, Y.; Gong, J.; Yoshida, T.; Eberhart, C.G.; Xu, Z.; Kombairaju, P.; Sporn, M.B.; Handa, J.T.; Duh, E.J. Nrf2 has a protective role against neuronal and capillary degeneration in retinal ischemia-reperfusion injury. Free Radic. Biol. Med. 2011, 51, 216–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Wei, Y.; Gong, J.; Cho, H.; Park, J.K.; Sung, E.R.; Huang, H.; Wu, L.; Eberhart, C.; Handa, J.T.; et al. NRF2 plays a protective role in diabetic retinopathy in mice. Diabetologia 2014, 57, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Lenox, A.R.; Bhootada, Y.; Gorbatyuk, O.; Fullard, R.; Gorbatyuk, M. Unfolded protein response is activated in aged retinas. Neurosci. Lett. 2015, 609, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Li, J.; Zha, D.; Zhang, L.; Gao, P.; Yao, T.; Wu, X. Chlorogenic acid prevents diabetic nephropathy by inhibiting oxidative stress and inflammation through modulation of the Nrf2/HO-1 and NF-kB pathways. Int. Immunopharmacol. 2018, 54, 245–253. [Google Scholar] [CrossRef]
- Blasiak, J.; Piechota, M.; Pawlowska, E.; Szatkowska, M.; Sikora, E.; Kaarniranta, K. Cellular senescence in age-related macular degeneration: Can autophagy and DNA damage response play a role? Oxid. Med. Cell Longev. 2017, 2017, 5293258. [Google Scholar] [CrossRef]
- Silva, L.L.; Lambert-Cheatham, N.; Stratford, R.E.; Quinney, S.K.; Corson, T.W.; Kelley, M.R. Oral APX3330 treatment reduces L-CNV lesions in a preclinical mouse model and confirms Phase 2 DR/DME clinical dose with sufficient distribution to human retina using PBPK modeling. Invest. Ophthalmol. Vis. Sci. 2021, 62, 1073. [Google Scholar]
- Fishel, M.L.; Cheng, H.; Shahda, S.; Kelley, M.R. APX3330 Drug Development for Clinical Trials Targeting APE1/Ref-1 in Pancreatic Cancer [abstract]. In Proceedings of the AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, Boston, MA, USA, 5–9 November 2015. [Google Scholar]
- Oshima, Y.; Oshima, S.; Nambu, H.; Kachi, S.; Hackett, S.F.; Melia, M.; Kaleko, M.; Connelly, S.; Esumi, N.; Zack, D.J.; et al. Increased expression of VEGF in retinal pigmented epithelial cells is not sufficient to cause choroidal neovascularization. J. Cell Physiol. 2004, 201, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Xanthoudakis, S.; Smeyne, R.J.; Wallace, J.D.; Curran, T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 8919–8923. [Google Scholar] [CrossRef] [Green Version]
- Kelley, M.R.; Fishel, M.L.; (Indiana University School of Medicine, Indianapolis, IN, USA). Personal Communication, 2021.



{kind=link}
{kind=link}
{kind=link}
| Intervention | System | Findings | Reference |
|---|---|---|---|
| In vitro | |||
| APX3330 | RVECs | Dose-dependently suppressed proliferation, migration and tube formation | [30] |
| APX3330 | RVECs | Reduced CI | [30] |
| APX3330 | ARPE-19 | Prevented apoptosis and reduced the upsurge of MCP-1 following induction of pathological stress with oxLDL | [28] |
| APX3330 | ARPE-19 | Reduced accumulation of intracellular ROS, secretion of VEGF and effectively blocked the upsurge of NF-κB in response to induced pathological stress by oxLDL | [28] |
| APX3330 | ARPE-19 | Protected the cells from a stress-induced senescence-like phenotype | [28] |
| APX3330 | ARPE-19 | Decreased the transcription activities of Nrf2/Nrf1, p53, NF-κB, HIF-1, CBF/NF-Y, YY1, MTF1 and HSF-1 | [28] |
| APX3330 | Rf/6a | Reduced p65 expression and NF-κB transcriptional activity | [55] |
| APX3330 | Rf/6a | Dose-dependently downregulated the production of MCP-1 | [55] |
| APX3330 | Rf/6a | Reduced STAT3 and NF-κB DNA binding activity | [55] |
| APX3330 | Rf/6a | Dose-dependently suppressed angiogenesis (proliferation, migration and tube formation) | [55] |
| APX3330 | Rf/6a | Did not induce apoptosis | [55] |
| APX3330 and Bevacizumab (antibody) | Rf/6a | Additive decline in migration, tube formation and proliferation | [55] |
| APX2009APX2014 | HRECs; Rf/6a | Dose-dependently decreased choroidal sprouting, proliferation, tube formation and endothelial cell migration | [44] |
| APX2009APX2014 | HRECs | Dose-dependently reduced translocation of the p65 subunit of NF-κB into the nucleus and decreased downstream mRNA targets of NF-κB including VCAM1, CCL20 and VEGFA | [44] |
| APX2009APX2014 | HRECs | Did not induce apoptosis in a TUNEL assay and blocked cells from entering the S phase | [44] |
| In vivo | |||
| APX3330 | Vldlr−/− mice | Single IVT injection of 20 μM decreased neovascularization | [30] |
| APX3330 | L-CNV mice | Single IVT injection for final intraocular concentration of 20 μM suppressed L-CNV lesion area | [28,55] |
| APX3330 | L-CNV mice | IP injection twice a day at 50 mg/kg for 5 days on and 2 days off for two weeks reduced L-CNV volume by 25% | [44] |
| APX2009 | L-CNV mice | IP injection (25 mg/kg twice daily for two weeks) decreased L-CNV volume without causing systemic toxicity | [44] |
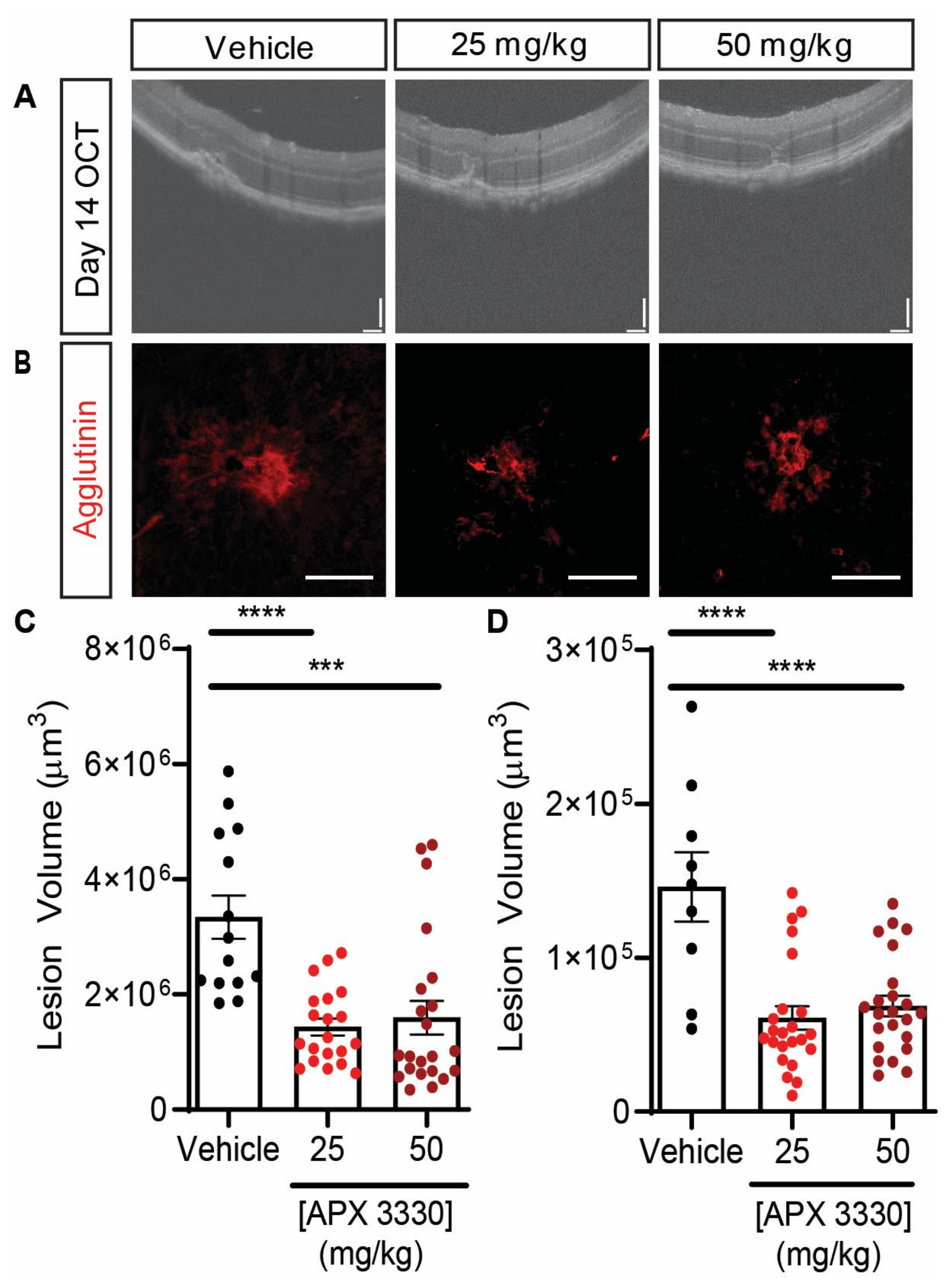
| APX3330 | L-CNV mice | Gavage administration of either 25 mg/kg or 50 mg/kg gavage of APX3330 twice daily for 14 days resulted in decrease of lesion size by >50% | Figure 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hartman, G.D.; Lambert-Cheatham, N.A.; Kelley, M.R.; Corson, T.W. Inhibition of APE1/Ref-1 for Neovascular Eye Diseases: From Biology to Therapy. Int. J. Mol. Sci. 2021, 22, 10279. https://doi.org/10.3390/ijms221910279
Hartman GD, Lambert-Cheatham NA, Kelley MR, Corson TW. Inhibition of APE1/Ref-1 for Neovascular Eye Diseases: From Biology to Therapy. International Journal of Molecular Sciences. 2021; 22(19):10279. https://doi.org/10.3390/ijms221910279
Chicago/Turabian StyleHartman, Gabriella D., Nathan A. Lambert-Cheatham, Mark R. Kelley, and Timothy W. Corson. 2021. "Inhibition of APE1/Ref-1 for Neovascular Eye Diseases: From Biology to Therapy" International Journal of Molecular Sciences 22, no. 19: 10279. https://doi.org/10.3390/ijms221910279
APA StyleHartman, G. D., Lambert-Cheatham, N. A., Kelley, M. R., & Corson, T. W. (2021). Inhibition of APE1/Ref-1 for Neovascular Eye Diseases: From Biology to Therapy. International Journal of Molecular Sciences, 22(19), 10279. https://doi.org/10.3390/ijms221910279