Cilostazol Induces eNOS and TM Expression via Activation with Sirtuin 1/Krüppel-like Factor 2 Pathway in Endothelial Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Cilostazol Showed a Negative Correlation with Thrombosis-Related Gene Sets Investigated in GSE19151
2.2. Cilostazol Upregulates KLF2 Expression in Endothelial Cells
2.3. Cilostazol Stimulates eNOS and TM Expression, and, Subsequently, Their Bioavailability
2.4. KLF2 Expression Is Critical for eNOS and TM Induction in Cilostazol-Treated HUVECs
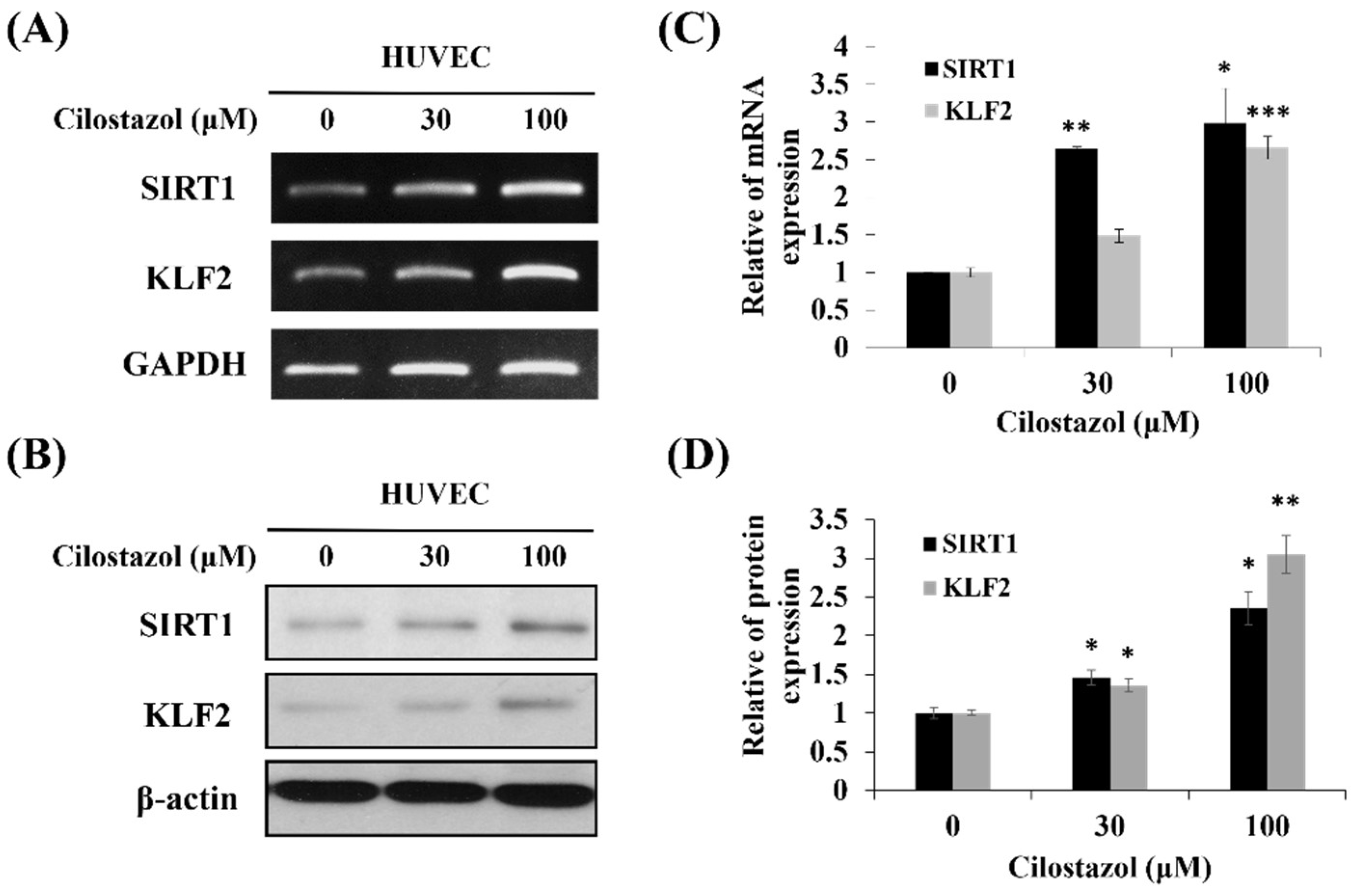
2.5. Cilostazol Induces KLF2 via SIRT1 Activation
3. Discussion
4. Materials and Methods
4.1. Gene Expression Profiling and GSEA
4.2. Cilostazol Preparation and Storage
4.3. Reverse Transcription Polymerase Chain Reaction (RT-PCR) and qRT-PCR
4.4. Western Blot Analysis
4.5. Functional Assay of TM
4.6. Detection of NO by Flow Cytometry
4.7. Genetic Knockdown Using the Lentivirus shRNA System
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Anderson, K.P.; Kern, C.B.; Crable, S.C.; Lingrel, J.B. Isolation of a gene encoding a functional zinc finger protein homologous to erythroid Kruppel-like factor: Identification of a new multigene family. Mol. Cell. Biol. 1995, 15, 5957–5965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, M.A.; Wert, S.E.; Lingrel, J.B. Lung Kruppel-like factor, a zinc finger transcription factor, is essential for normal lung development. J. Biol. Chem. 1999, 274, 21180–21185. [Google Scholar] [CrossRef] [Green Version]
- Nayak, L.; Lin, Z.; Jain, M.K. “Go with the flow”: How Kruppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxid. Redox Signal. 2011, 15, 1449–1461. [Google Scholar] [CrossRef] [Green Version]
- Atkins, G.B.; Jain, M.K. Role of Kruppel-like transcription factors in endothelial biology. Circ. Res. 2007, 100, 1686–1695. [Google Scholar] [CrossRef]
- Dekker, R.J.; Boon, R.A.; Rondaij, M.G.; Kragt, A.; Volger, O.L.; Elderkamp, Y.W.; Meijers, J.C.; Voorberg, J.; Pannekoek, H.; Horrevoets, A.J. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood 2006, 107, 4354–4363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Kumar, A.; SenBanerjee, S.; Staniszewski, K.; Parmar, K.; Vaughan, D.E.; Gimbrone, M.A., Jr.; Balasubramanian, V.; Garcia-Cardena, G.; Jain, M.K. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ. Res. 2005, 96, e48–e57. [Google Scholar] [CrossRef] [Green Version]
- SenBanerjee, S.; Lin, Z.; Atkins, G.B.; Greif, D.M.; Rao, R.M.; Kumar, A.; Feinberg, M.W.; Chen, Z.; Simon, D.I.; Luscinskas, F.W.; et al. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J. Exp. Med. 2004, 199, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Gracia-Sancho, J.; Villarreal, G., Jr.; Zhang, Y.; Garcia-Cardena, G. Activation of SIRT1 by resveratrol induces KLF2 expression conferring an endothelial vasoprotective phenotype. Cardiovasc. Res 2010, 85, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Parmar, K.M.; Nambudiri, V.; Dai, G.; Larman, H.B.; Gimbrone, M.A., Jr.; Garcia-Cardena, G. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. J. Biol. Chem. 2005, 280, 26714–26719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, S. Cilostazol: Potential mechanism of action for antithrombotic effects accompanied by a low rate of bleeding. Atherosclerosis Suppl. 2005, 6, 3–11. [Google Scholar] [CrossRef]
- Ryu, K.H.; Han, H.Y.; Lee, S.Y.; Jeon, S.D.; Im, G.J.; Lee, B.Y.; Kim, K.; Lim, K.M.; Chung, J.H. Ginkgo biloba extract enhances antiplatelet and antithrombotic effects of cilostazol without prolongation of bleeding time. Thromb. Res. 2009, 124, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Pattanaik, S.; Malhotra, S.; Sharma, Y.P.; Ahluwalia, J.; Bhalla, A.; Pandhi, P. Effect of cilostazol on platelet aggregation in patients with non-ST elevation acute coronary syndrome. Int. J. Clin. Pharmacol. Ther. 2010, 48, 93–102. [Google Scholar] [CrossRef]
- Kim, K.Y.; Shin, H.K.; Choi, J.M.; Hong, K.W. Inhibition of lipopolysaccharide-induced apoptosis by cilostazol in human umbilical vein endothelial cells. J. Pharmacol. Exp. Ther. 2002, 300, 709–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Lee, J.H.; Park, S.Y.; Hong, K.W.; Kim, C.D.; Kim, K.Y.; Lee, W.S. Protection from apoptotic cell death by cilostazol, phosphodiesterase type III inhibitor, via cAMP-dependent protein kinase activation. Pharmacol. Res. 2006, 54, 261–267. [Google Scholar] [PubMed]
- Nishio, Y.; Kashiwagi, A.; Takahara, N.; Hidaka, H.; Kikkawa, R. Cilostazol, a cAMP phosphodiesterase inhibitor, attenuates the production of monocyte chemoattractant protein-1 in response to tumor necrosis factor-alpha in vascular endothelial cells. Horm. Metab. Res. 1997, 29, 491–495. [Google Scholar] [PubMed]
- Otsuki, M.; Saito, H.; Xu, X.; Sumitani, S.; Kouhara, H.; Kurabayashi, M.; Kasayama, S. Cilostazol represses vascular cell adhesion molecule-1 gene transcription via inhibiting NF-kappaB binding to its recognition sequence. Atherosclerosis 2001, 158, 121–128. [Google Scholar] [CrossRef]
- Lee, T.M.; Su, S.F.; Tsai, C.H.; Lee, Y.T.; Wang, S.S. Differential effects of cilostazol and pentoxifylline on vascular endothelial growth factor in patients with intermittent claudication. Clin. Sci. 2001, 101, 305–311. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, B.; Xia, L. Effect of low wall shear stress on the morphology of endothelial cells and its evaluation indicators. Comput. Methods Programs Biomed. 2021, 208, 106082. [Google Scholar] [CrossRef]
- Huddleson, J.P.; Srinivasan, S.; Ahmad, N.; Lingrel, J.B. Fluid shear stress induces endothelial KLF2 gene expression through a defined promoter region. Biol. Chem. 2004, 385, 723–729. [Google Scholar] [CrossRef]
- Boon, R.A.; Leyen, T.A.; Fontijn, R.D.; Fledderus, J.O.; Baggen, J.M.; Volger, O.L.; van Nieuw Amerongen, G.P.; Horrevoets, A.J. KLF2-induced actin shear fibers control both alignment to flow and JNK signaling in vascular endothelium. Blood 2010, 115, 2533–2542. [Google Scholar] [CrossRef] [Green Version]
- van Thienen, J.V.; Fledderus, J.O.; Dekker, R.J.; Rohlena, J.; van Ijzendoorn, G.A.; Kootstra, N.A.; Pannekoek, H.; Horrevoets, A.J. Shear stress sustains atheroprotective endothelial KLF2 expression more potently than statins through mRNA stabilization. Cardiovasc. Res 2006, 72, 231–240. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Wang, Z.; Chen, H.Z.; Zhou, S.; Zheng, W.; Liu, G.; Wei, Y.S.; Cai, H.; Liu, D.P.; Liang, C.C. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc. Res. 2008, 80, 191–199. [Google Scholar] [CrossRef]
- Young, A.; Wu, W.; Sun, W.; Benjamin Larman, H.; Wang, N.; Li, Y.S.; Shyy, J.Y.; Chien, S.; Garcia-Cardena, G. Flow activation of AMP-activated protein kinase in vascular endothelium leads to Kruppel-like factor 2 expression. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1902–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimmeler, S.; Assmus, B.; Hermann, C.; Haendeler, J.; Zeiher, A.M. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: Involvement in suppression of apoptosis. Circ. Res. 1998, 83, 334–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, T.H.; Tseng, S.Y.; Li, Y.H.; Liu, P.Y.; Cho, C.L.; Shi, G.Y.; Wu, H.L.; Chen, J.H. A novel vasculo-angiogenic effect of cilostazol mediated by cross-talk between multiple signalling pathways including the ERK/p38 MAPK signalling transduction cascade. Clin. Sci. 2012, 123, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Uchida, K.; Nakanishi, N.; Hattori, Y. Cilostazol activates AMP-activated protein kinase and restores endothelial function in diabetes. Am. J. Hypertens. 2008, 21, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Geng, P.; Zhu, J.; Li, J.; Zhang, L.; Chen, W.; Zhang, D.; Lu, Y.; Xu, X. KLF2 regulates eNOS uncoupling via Nrf2/HO-1 in endothelial cells under hypoxia and reoxygenation. Chem. Biol. Interact. 2019, 305, 105–111. [Google Scholar] [CrossRef]
- Urban, P.; De Benedetti, E. Thrombosis: The last frontier of coronary stenting? Lancet 2007, 369, 619–621. [Google Scholar] [CrossRef]
- Han, Y.; Li, Y.; Wang, S.; Jing, Q.; Wang, Z.; Wang, D.; Shu, Q.; Tang, X. Cilostazol in addition to aspirin and clopidogrel improves long-term outcomes after percutaneous coronary intervention in patients with acute coronary syndromes: A randomized, controlled study. Am. Heart J. 2009, 157, 733–739. [Google Scholar] [CrossRef]
- Cleanthis, M.; Bhattacharya, V.; Smout, J.; Ashour, H.; Stansby, G. Combined aspirin and cilostazol treatment is associated with reduced platelet aggregation and prevention of exercise-induced platelet activation. Eur. J. Vasc. Endovasc. Surg. 2009, 37, 604–610. [Google Scholar] [CrossRef] [Green Version]
- Geng, D.F.; Liu, M.; Jin, D.M.; Wu, W.; Deng, J.; Wang, J.F. Cilostazol-based triple antiplatelet therapy compared to dual antiplatelet therapy in patients with coronary stent implantation: A meta-analysis of 5821 patients. Cardiology 2012, 122, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Jeon, D.S.; Yoo, K.D.; Park, C.S.; Shin, D.I.; Her, S.H.; Park, H.J.; Choi, Y.S.; Kim, D.B.; Lee, C.M.; Park, C.S.; et al. The effect of cilostazol on stent thrombosis after drug-eluting stent implantation. Korean Circ. J. 2010, 40, 10–15. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, M.C.; Liang, M.; Fu, J. Sirt1 protects against thrombomodulin down-regulation and lung coagulation after particulate matter exposure. Blood 2012, 119, 2422–2429. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Eto, M.; Kano, M.R.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. Cilostazol inhibits oxidative stress-induced premature senescence via upregulation of Sirt1 in human endothelial cells. Arter. Thromb. Vasc. Biol. 2008, 28, 1634–1639. [Google Scholar] [CrossRef] [Green Version]
- Parmar, K.M.; Larman, H.B.; Dai, G.; Zhang, Y.; Wang, E.T.; Moorthy, S.N.; Kratz, J.R.; Lin, Z.; Jain, M.K.; Gimbrone, M.A., Jr.; et al. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J. Clin. Investig. 2006, 116, 49–58. [Google Scholar] [CrossRef]
- Blanes, M.G.; Oubaha, M.; Rautureau, Y.; Gratton, J.P. Phosphorylation of tyrosine 801 of vascular endothelial growth factor receptor-2 is necessary for Akt-dependent endothelial nitric-oxide synthase activation and nitric oxide release from endothelial cells. J. Biol. Chem. 2007, 282, 10660–10669. [Google Scholar] [CrossRef] [Green Version]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.S.; Weigelt, J.A. Disaster management. Lessons learned. Surg. Clin. N. Am. 1991, 71, 257–266. [Google Scholar] [CrossRef]
- Tsai, T.N.; Lin, W.S.; Wu, C.H.; Lin, W.Y.; Chu, K.M.; Cheng, C.C.; Hsu, C.H.; Tsai, W.C.; Cheng, S.M.; Yang, S.P. Activation of Kruppel-Like Factor 2 with Ginkgo Biloba Extract Induces eNOS Expression and Increases NO Production in Cultured Human Umbilical Endothelial Cells. Acta Cardiol. Sin. 2014, 30, 215–222. [Google Scholar] [PubMed]
- Lewis, D.A.; Stashenko, G.J.; Akay, O.M.; Price, L.I.; Owzar, K.; Ginsburg, G.S.; Chi, J.T.; Ortel, T.L. Whole blood gene expression analyses in patients with single versus recurrent venous thromboembolism. Thromb. Res. 2011, 128, 536–540. [Google Scholar] [CrossRef] [Green Version]
- Yoo, M.; Shin, J.; Kim, J.; Ryall, K.A.; Lee, K.; Lee, S.; Jeon, M.; Kang, J.; Tan, A.C. DSigDB: Drug signatures database for gene set analysis. Bioinformatics 2015, 31, 3069–3071. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Gene Ontology, C. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Kohler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Chiu, Y.L.; Tsai, W.C.; Wu, C.H.; Wu, C.H.; Cheng, C.C.; Lin, W.S.; Tsai, T.N.; Wu, L.S. Ginkgo biloba Induces Thrombomodulin Expression and Tissue-Type Plasminogen Activator Secretion via the Activation of Kruppel-Like Factor 2 within Endothelial Cells. Am. J. Chin. Med. 2020, 48, 357–372. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-H.; Chiu, Y.-L.; Hsieh, C.-Y.; Tsung, G.-S.; Wu, L.-S.; Cheng, C.-C.; Tsai, T.-N. Cilostazol Induces eNOS and TM Expression via Activation with Sirtuin 1/Krüppel-like Factor 2 Pathway in Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 10287. https://doi.org/10.3390/ijms221910287
Wu C-H, Chiu Y-L, Hsieh C-Y, Tsung G-S, Wu L-S, Cheng C-C, Tsai T-N. Cilostazol Induces eNOS and TM Expression via Activation with Sirtuin 1/Krüppel-like Factor 2 Pathway in Endothelial Cells. International Journal of Molecular Sciences. 2021; 22(19):10287. https://doi.org/10.3390/ijms221910287
Chicago/Turabian StyleWu, Chih-Hsien, Yi-Lin Chiu, Chung-Yueh Hsieh, Guo-Shiang Tsung, Lian-Shan Wu, Cheng-Chung Cheng, and Tsung-Neng Tsai. 2021. "Cilostazol Induces eNOS and TM Expression via Activation with Sirtuin 1/Krüppel-like Factor 2 Pathway in Endothelial Cells" International Journal of Molecular Sciences 22, no. 19: 10287. https://doi.org/10.3390/ijms221910287