
Human CD22-Transgenic, Primary Murine Lymphoma Challenges Immunotherapies in Organ-Specific Tumor Microenvironments

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
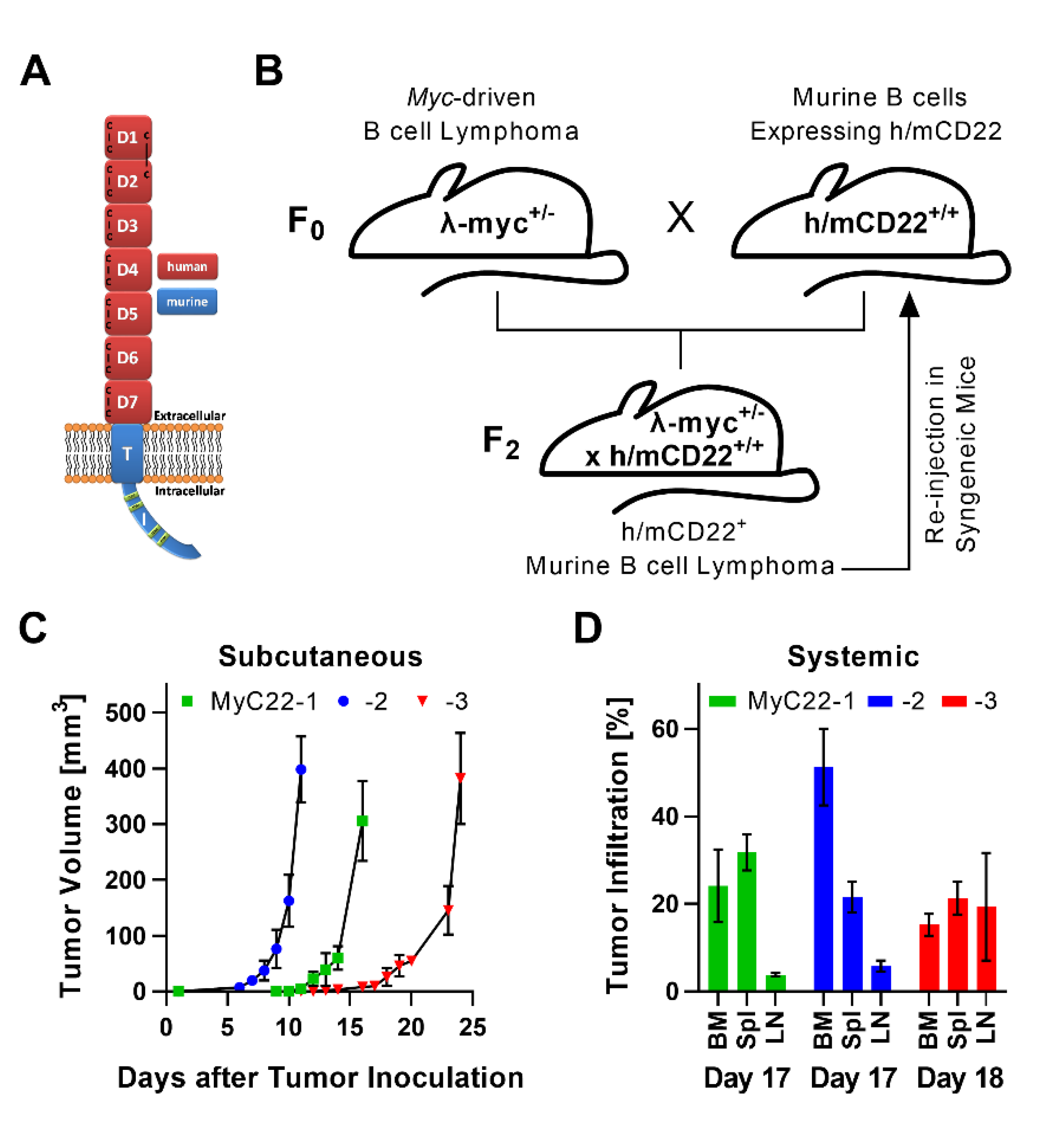
2.1. Development of a Myc-Driven, h/mCD22 Expressing, Primary Lymphoma Model
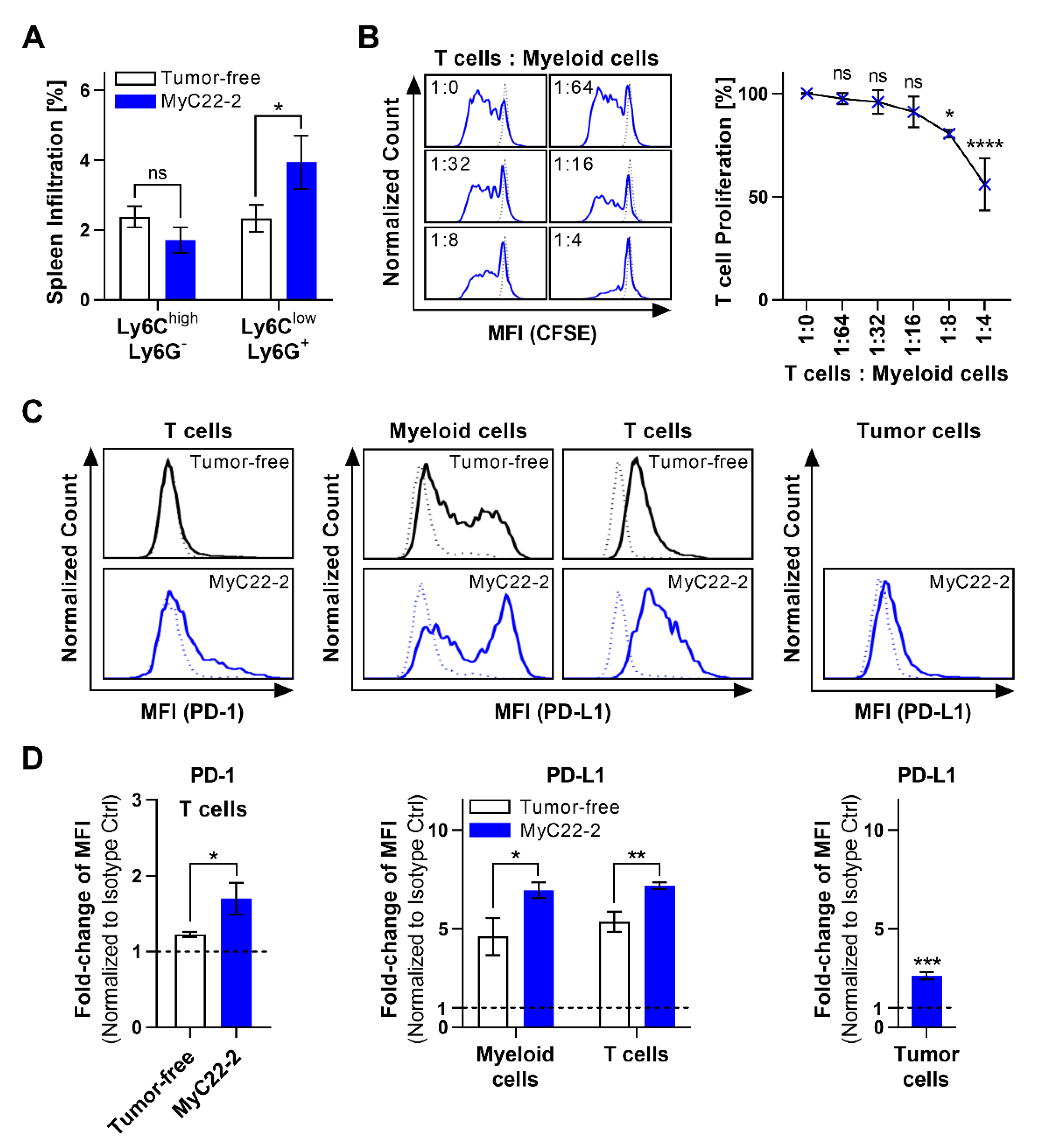
2.2. Myeloid Cells Support Tumor Growth and Contribute to an Immunosuppressive TME
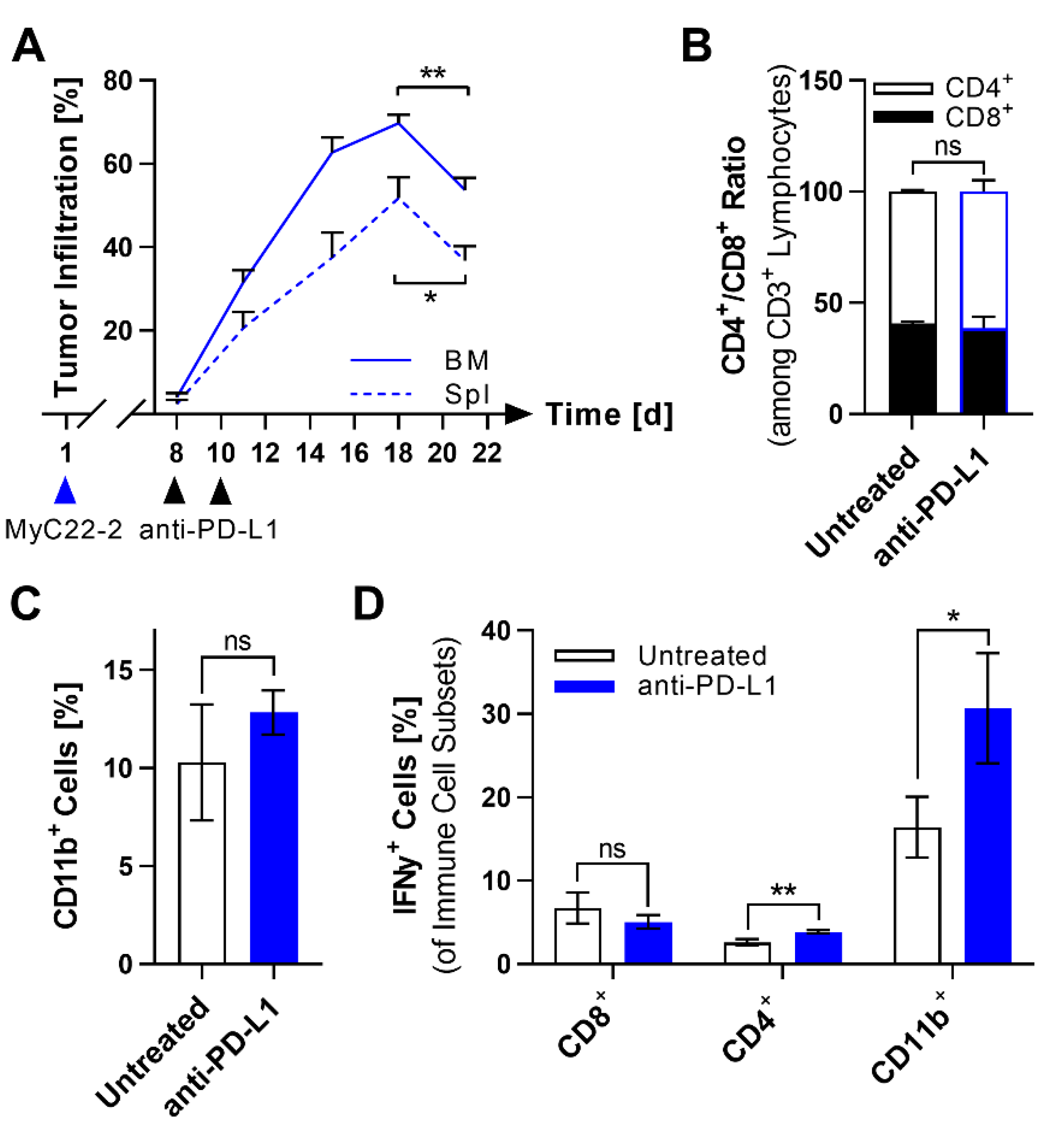
2.3. PD-L1 Checkpoint Blockade Shows Limited Efficacy against MyC22
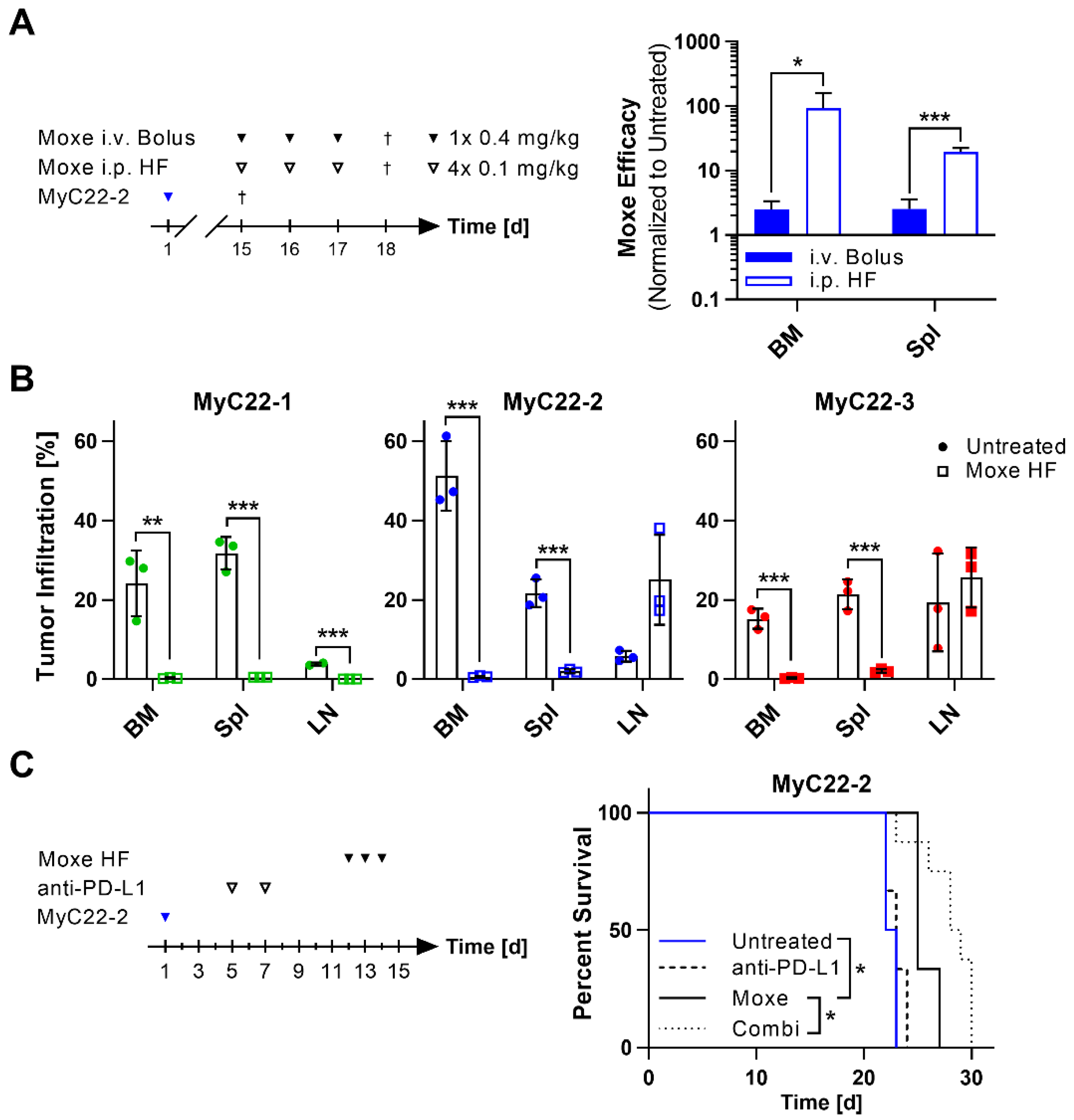
2.4. Efficacy of Moxetumomab Was Clone and Organ Dependent
2.5. Anti-PD-L1 Blockade Enhances Survival of Moxetumomab Treated Mice
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Systemic and Subcutaneous Tumor Growth
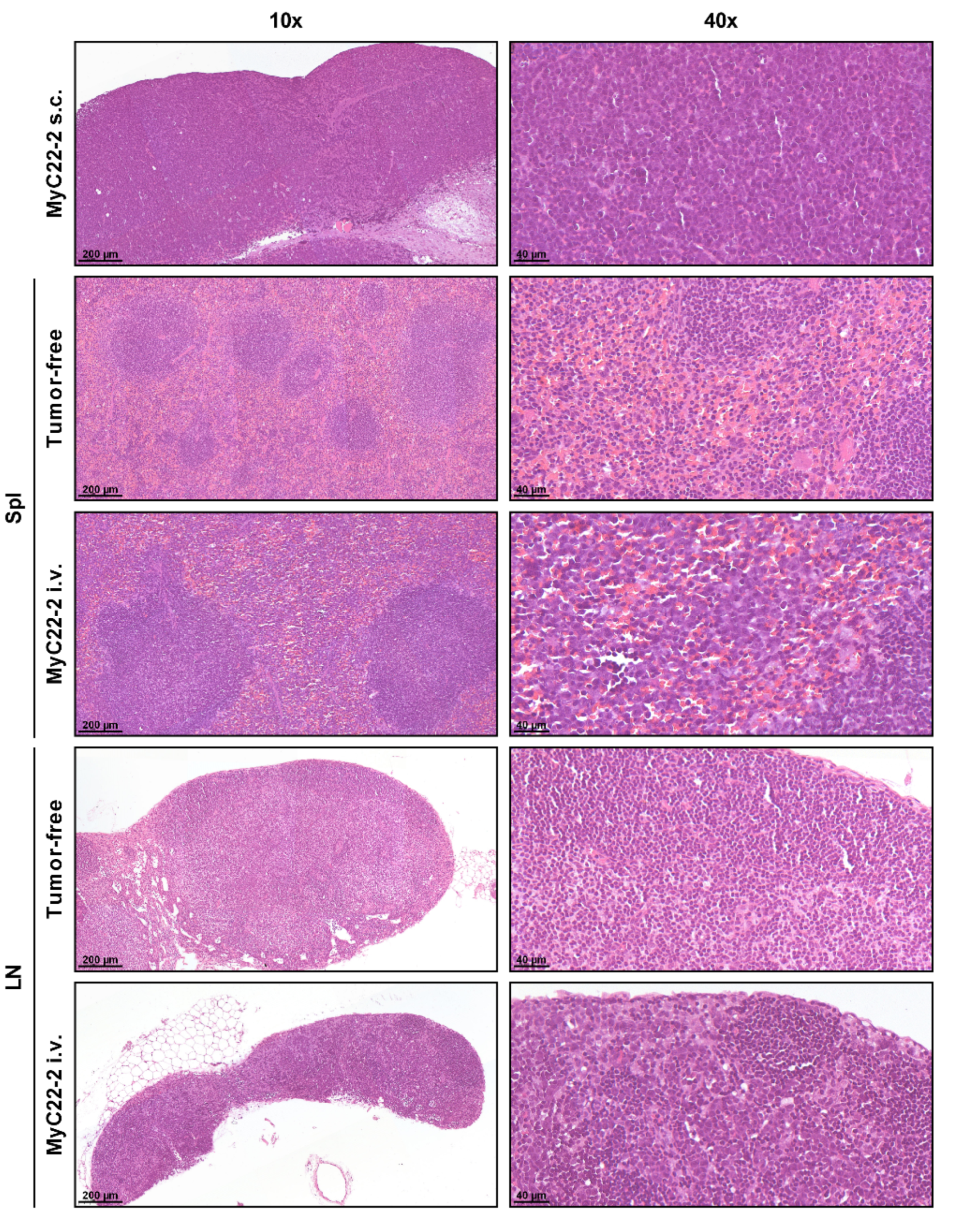
4.3. Histology
4.4. Flow Cytometric Analysis
4.5. T Cell Suppression Assay
4.6. Immune Cell Depletion
4.7. Treatment Studies
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.W.; Gascoyne, R.D. The tumour microenvironment in B cell lymphomas. Nat. Rev. Cancer 2014, 14, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Tarte, K. Role of the microenvironment across histological subtypes of NHL. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 610–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, A.J.; Lau, P.K.H.; Unsworth, A.S.; Loi, S.; Darcy, P.K.; Kershaw, M.H.; Slaney, C.Y. Tissue-Dependent Tumor Microenvironments and Their Impact on Immunotherapy Responses. Front. Immunol. 2018, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef]
- Burkitt, D. A sarcoma involving the jaws in African children. Br. J. Surg. 1958, 46, 218–223. [Google Scholar] [CrossRef]
- Xiu, B.; Lin, Y.; Grote, D.M.; Ziesmer, S.C.; Gustafson, M.P.; Maas, M.L.; Zhang, Z.; Dietz, A.B.; Porrata, L.F.; Novak, A.J.; et al. IL-10 induces the development of immunosuppressive CD14+HLA-DRlow/− monocytes in B-cell non-Hodgkin lymphoma. Blood Cancer J. 2015, 5, e328. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.; Han, J.J.; Stenson, M.; Maurer, M.; Wellik, L.; Hu, G.; Ziesmer, S.; Dogan, A.; Witzig, T.E. Elevated serum IL-10 levels in diffuse large B-cell lymphoma: A mechanism of aberrant JAK2 activation. Blood 2012, 119, 2844–2853. [Google Scholar] [CrossRef]
- Roussel, M.; Irish, J.M.; Menard, C.; Lhomme, F.; Tarte, K.; Fest, T. Regulatory myeloid cells: An underexplored continent in B-cell lymphomas. Cancer Immunol. Immunother. 2017, 66, 1103–1111. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, M.T.; Dolgalev, I.; Evensen, N.A.; Ma, C.; Chambers, T.; Roberts, K.G.; Sreeram, S.; Dai, Y.; Tikhonova, A.N.; Lasry, A.; et al. Extensive Remodeling of the Immune Microenvironment in B Cell Acute Lymphoblastic Leukemia. Cancer Cell 2020, 37, 867–882.e12. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Gustafson, M.P.; Bulur, P.A.; Gastineau, D.A.; Witzig, T.E.; Dietz, A.B. Immunosuppressive CD14+HLA-DR(low)/- monocytes in B-cell non-Hodgkin lymphoma. Blood 2011, 117, 872–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadmor, T.; Fell, R.; Polliack, A.; Attias, D. Absolute monocytosis at diagnosis correlates with survival in diffuse large B-cell lymphoma-possible link with monocytic myeloid-derived suppressor cells. Hematol. Oncol. 2013, 31, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Azzaoui, I.; Uhel, F.; Rossille, D.; Pangault, C.; Dulong, J.; Le Priol, J.; Lamy, T.; Houot, R.; Le Gouill, S.; Cartron, G.; et al. T-cell defect in diffuse large B-cell lymphomas involves expansion of myeloid-derived suppressor cells. Blood 2016, 128, 1081–1092. [Google Scholar] [CrossRef]
- Marini, O.; Spina, C.; Mimiola, E.; Cassaro, A.; Malerba, G.; Todeschini, G.; Perbellini, O.; Scupoli, M.; Carli, G.; Facchinelli, D.; et al. Identification of granulocytic myeloid-derived suppressor cells (G-MDSCs) in the peripheral blood of Hodgkin and non-Hodgkin lymphoma patients. Oncotarget 2016, 7, 27676–27688. [Google Scholar] [CrossRef] [Green Version]
- Saito, R.; Kobayashi, T.; Kashima, S.; Matsumoto, K.; Ogawa, O. Faithful preclinical mouse models for better translation to bedside in the field of immuno-oncology. Int. J. Clin. Oncol. 2020, 25, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Sullivan-Chang, L.; O’Donnell, R.T.; Tuscano, J.M. Targeting CD22 in B-cell malignancies: Current status and clinical outlook. BioDrugs 2013, 27, 293–304. [Google Scholar] [CrossRef]
- Haso, W.; Lee, D.W.; Shah, N.N.; Stetler-Stevenson, M.; Yuan, C.M.; Pastan, I.H.; Dimitrov, D.S.; Morgan, R.A.; FitzGerald, D.J.; Barrett, D.M.; et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 2013, 121, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Barisone, G.A.; Vang, D.P.; Murphy, W.J.; Tuscano, J. Bispecific anti-CD22 T-cell engager efficiently promotes cell-mediated cytotoxicity of lymphoma, leukemia and sarcoma targets. J. Immunol. 2020, 204 (Suppl. 1), 239.27. [Google Scholar]
- Lamb, Y.N. Inotuzumab Ozogamicin: First Global Approval. Drugs 2017, 77, 1603–1610. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Antibody fusion proteins: Anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer Res. 2011, 17, 6398–6405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wöhner, M.; Born, S.; Nitschke, L. Human CD22 cannot fully substitute murine CD22 functions in vivo, as shown in a new knockin mouse model. Eur. J. Immunol. 2012, 42, 3009–3018. [Google Scholar] [CrossRef] [PubMed]
- Brandl, C.; Angermuller, S.; Nitschke, L. Humanized CD22 transgenic mouse model for in vivo analysis of anti-CD22-based immunotherapy. Eur. J. Immunol. 2020, 50, 1834–1837. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Cheng, H.; Watahiki, A.; Ducar, M.D.; Tan, Y.; Chen, L.; Roemer, M.G.; Ouyang, J.; Christie, A.L.; Zhang, L.; et al. Diffuse large B-cell lymphoma patient-derived xenograft models capture the molecular and biological heterogeneity of the disease. Blood 2016, 127, 2203–2213. [Google Scholar] [CrossRef]
- Knittel, G.; Liedgens, P.; Korovkina, D.; Seeger, J.M.; Al-Baldawi, Y.; Al-Maarri, M.; Fritz, C.; Vlantis, K.; Bezhanova, S.; Scheel, A.H.; et al. B-cell-specific conditional expression of Myd88p.L252P leads to the development of diffuse large B-cell lymphoma in mice. Blood 2016, 127, 2732–2741. [Google Scholar] [CrossRef] [Green Version]
- Ott, G.; Rosenwald, A.; Campo, E. Understanding MYC-driven aggressive B-cell lymphomas: Pathogenesis and classification. Blood 2013, 122, 3884–3891. [Google Scholar] [CrossRef] [Green Version]
- Kovalchuk, A.L.; Qi, C.F.; Torrey, T.A.; Taddesse-Heath, L.; Feigenbaum, L.; Park, S.S.; Gerbitz, A.; Klobeck, G.; Hoertnagel, K.; Polack, A.; et al. Burkitt lymphoma in the mouse. J. Exp. Med. 2000, 192, 1183–1190. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Casulo, C.; Santoro, A.; Ando, K.; Le Gouill, S.; Ruan, J.; Radford, J.; Arcaini, L.; Pinto, A.; Bouabdallah, R.; Izutsu, K.; et al. Durvalumab (Anti PD-L1) As Monotherapy or in Combination Therapy for Relapsed/Refractory (r/r) Diffuse Large B-Cell Lymphoma (DLBCL) and Follicular Lymphoma (FL): A Subgroup Analysis from the Phase 1/2 Fusion NHL-001 Global Multicenter Trial. Blood 2019, 134 (Suppl. 1), 5320. [Google Scholar] [CrossRef]
- Muller, F.; Cunningham, T.; Liu, X.F.; Wayne, A.S.; Pastan, I. Wide Variability in the Time Required for Immunotoxins to Kill B Lineage Acute Lymphoblastic Leukemia Cells: Implications for Trial Design. Clin. Cancer Res. 2016, 22, 4913–4922. [Google Scholar] [CrossRef] [Green Version]
- Müller, F.; Stookey, S.; Cunningham, T.; Pastan, I. Paclitaxel synergizes with exposure time adjusted CD22-targeting immunotoxins against B-cell malignancies. Oncotarget 2017, 8, 30644–30655. [Google Scholar] [CrossRef] [Green Version]
- Muller, F.; Cunningham, T.; Beers, R.; Bera, T.K.; Wayne, A.S.; Pastan, I. Domain II of Pseudomonas Exotoxin Is Critical for Efficacy of Bolus Doses in a Xenograft Model of Acute Lymphoblastic Leukemia. Toxins 2018, 10, 210. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Sander, S.; Calado, D.P.; Srinivasan, L.; Kochert, K.; Zhang, B.; Rosolowski, M.; Rodig, S.J.; Holzmann, K.; Stilgenbauer, S.; Siebert, R.; et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell 2012, 22, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Katsuta, E.; Rashid, O.M.; Takabe, K. Clinical relevance of tumor microenvironment: Immune cells, vessels, and mouse models. Human Cell 2020, 33, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Ooi, C.-H.; Birzele, F.; Ruefli-Brasse, A.; Cannarile, M.A.; Reis, B.; Scharf, S.H.; Schubert, D.A.; Hatje, K.; Pelletier, N.; et al. Tissue-Specific Immunoregulation: A Call for Better Understanding of the “Immunostat” in the Context of Cancer. Cancer Discovery 2018, 8, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betsch, A.; Rutgeerts, O.; Fevery, S.; Sprangers, B.; Verhoef, G.; Dierickx, D.; Beckers, M. Myeloid-derived suppressor cells in lymphoma: The good, the bad and the ugly. Blood Rev. 2018, 32, 490–498. [Google Scholar] [CrossRef]
- Audzevich, T.; Bashford-Rogers, R.; Mabbott, N.A.; Frampton, D.; Freeman, T.C.; Potocnik, A.; Kellam, P.; Gilroy, D.W. Pre/pro-B cells generate macrophage populations during homeostasis and inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, E3954–E3963. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Minnema, M.C.; Johnson, P.; Timmerman, J.M.; Armand, P.; Shipp, M.A.; Rodig, S.J.; Ligon, A.H.; Roemer, M.G.M.; Reddy, N.; et al. Nivolumab for Relapsed/Refractory Diffuse Large B-Cell Lymphoma in Patients Ineligible for or Having Failed Autologous Transplantation: A Single-Arm, Phase II Study. J. Clin. Oncol. 2019, 37, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Frigault, M.J.; Armand, P.; Redd, R.A.; Jeter, E.; Merryman, R.W.; Coleman, K.C.; Herrera, A.F.; Dahi, P.; Nieto, Y.; LaCasce, A.S.; et al. PD-1 blockade for diffuse large B-cell lymphoma after autologous stem cell transplantation. Blood Adv. 2020, 4, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Ghafoor, A.; Mian, I.; Rathkey, D.; Thomas, A.; Alewine, C.; Sengupta, M.; Ahlman, M.A.; Zhang, J.; Morrow, B.; et al. Enhanced efficacy of mesothelin-targeted immunotoxin LMB-100 and anti-PD-1 antibody in patients with mesothelioma and mouse tumor models. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Leshem, Y.; O’Brien, J.; Liu, X.; Bera, T.K.; Terabe, M.; Berzofsky, J.A.; Bossenmaier, B.; Niederfellner, G.; Tai, C.H.; Reiter, Y.; et al. Combining Local Immunotoxins Targeting Mesothelin with CTLA-4 Blockade Synergistically Eradicates Murine Cancer by Promoting Anticancer Immunity. Cancer Immunol. Res. 2017, 5, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Biernacki, M.A.; Bleakley, M. Neoantigens in Hematologic Malignancies. Front. Immunol. 2020, 11, 121. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Jitschin, R.; Braun, M.; Buttner, M.; Dettmer-Wilde, K.; Bricks, J.; Berger, J.; Eckart, M.J.; Krause, S.W.; Oefner, P.J.; Le Blanc, K.; et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 2014, 124, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Devaud, C.; Westwood, J.A.; John, L.B.; Flynn, J.K.; Paquet-Fifield, S.; Duong, C.P.M.; Yong, C.S.M.; Pegram, H.J.; Stacker, S.A.; Achen, M.G.; et al. Tissues in Different Anatomical Sites Can Sculpt and Vary the Tumor Microenvironment to Affect Responses to Therapy. Mol. Ther. 2014, 22, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Li, L.; Starr, T.K.; Subramanian, S. Tumor location impacts immune response in mouse models of colon cancer. Oncotarget 2017, 8, 54775–54787. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.; Biburger, M.; Brückner, C.; Ipsen-Escobedo, A.; Gordan, S.; Lehmann, C.; Voehringer, D.; Winkler, T.; Schaft, N.; Dudziak, D.; et al. Tumor location determines tissue-specific recruitment of tumor-associated macrophages and antibody-dependent immunotherapy response. Sci. Immunol. 2017, 2, eaah6413. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Hellmann, M.D.; Hamid, O.; Tsai, K.K.; Loo, K.L.; Gubens, M.A.; Rosenblum, M.; Harview, C.L.; Taube, J.M.; Handley, N.; et al. Liver Metastasis and Treatment Outcome with Anti-PD-1 Monoclonal Antibody in Patients with Melanoma and NSCLC. Cancer Immunol. Res. 2017, 5, 417–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires da Silva, I.; Lo, S.; Quek, C.; Gonzalez, M.; Carlino, M.S.; Long, G.V.; Menzies, A.M. Site-specific response patterns, pseudoprogression, and acquired resistance in patients with melanoma treated with ipilimumab combined with anti-PD-1 therapy. Cancer 2020, 126, 86–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gsottberger, F.; Brandl, C.; Wendland, K.; Petkovic, S.; Emmerich, C.; Erber, R.; Geppert, C.; Hartmann, A.; Mackensen, A.; Nitschke, L.; et al. Human CD22-Transgenic, Primary Murine Lymphoma Challenges Immunotherapies in Organ-Specific Tumor Microenvironments. Int. J. Mol. Sci. 2021, 22, 10433. https://doi.org/10.3390/ijms221910433
Gsottberger F, Brandl C, Wendland K, Petkovic S, Emmerich C, Erber R, Geppert C, Hartmann A, Mackensen A, Nitschke L, et al. Human CD22-Transgenic, Primary Murine Lymphoma Challenges Immunotherapies in Organ-Specific Tumor Microenvironments. International Journal of Molecular Sciences. 2021; 22(19):10433. https://doi.org/10.3390/ijms221910433
Chicago/Turabian StyleGsottberger, Franziska, Carolin Brandl, Kerstin Wendland, Srdjan Petkovic, Charlotte Emmerich, Ramona Erber, Carol Geppert, Arndt Hartmann, Andreas Mackensen, Lars Nitschke, and et al. 2021. "Human CD22-Transgenic, Primary Murine Lymphoma Challenges Immunotherapies in Organ-Specific Tumor Microenvironments" International Journal of Molecular Sciences 22, no. 19: 10433. https://doi.org/10.3390/ijms221910433