
Role of K+ and Ca2+-Permeable Channels in Osteoblast Functions



Abstract
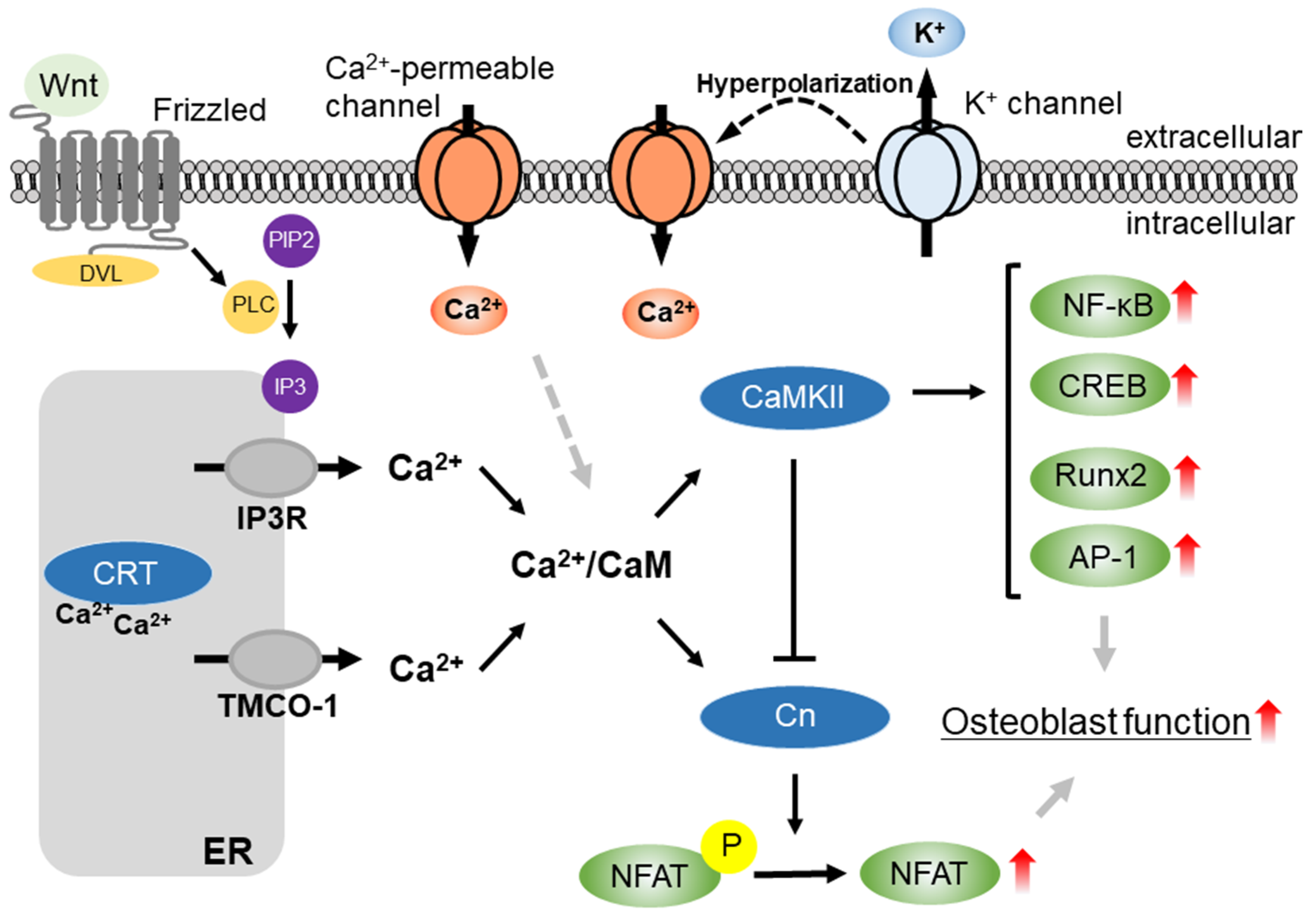
:1. Introduction
2. Regulatory Mechanism of Ca2+ Signaling by Membrane Potential in Non-Excitable Cell
3. K+ Channel Superfamilies in Osteoblast Lineages
3.1. Voltage-Gated K+ (KV) Channel Superfamily
3.2. Inward-Rectifier K+ (Kir) Channel Superfamily
3.3. Ca2+-Activated K+ (KCa) Channel Superfamily
3.4. Two-Pore Domain K+ (K2P) Channel Superfamily
4. Ca2+-Permeable Channel Superfamilies in Osteoblast Lineages
4.1. Orai/STIM
4.2. TRP Channel Superfamilies
4.2.1. TRPM Channels
4.2.2. TRPV Channels
4.2.3. TRPP Channels
4.3. Piezo Channels
5. Conclusions/Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berendsen, A.D.; Olsen, B.R. Bone development. Bone 2015, 80, 14–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salhotra, A.; Shah, H.N.; Levi, B.; Longaker, M.T. Mechanisms of bone development and repair. Nat. Rev. Mol. Cell Biol. 2020, 21, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chen, G.; Li, Y.P. TGF-b and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef] [PubMed]
- Horbelt, D.; Denkis, A.; Knaus, P. A portrait of Transforming Growth Factor b superfamily signalling: Background matters. Int. J. Biochem. Cell Biol. 2012, 44, 469–474. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-b Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Marie, P.J. Targeting integrins to promote bone formation and repair. Nat. Rev. Endocrinol. 2013, 9, 288–295. [Google Scholar] [CrossRef]
- Damsky, C.H. Extracellular matrix-integrin interactions in osteoblast function and tissue remodeling. Bone 1999, 25, 95–96. [Google Scholar] [CrossRef]
- Yuh, D.Y.; Maekawa, T.; Li, X.; Kajikawa, T.; Bdeir, K.; Chavakis, T.; Hajishengallis, G. The secreted protein DEL-1 activates a b3 integrin-FAK-ERK1/2-RUNX2 pathway and promotes osteogenic differentiation and bone regeneration. J. Biol. Chem. 2020, 295, 7261–7273. [Google Scholar] [CrossRef] [Green Version]
- Komori, T. Regulation of Proliferation, Differentiation and Functions of Osteoblasts by Runx2. Int. J. Mol. Sci. 2019, 20, 1694. [Google Scholar] [CrossRef] [Green Version]
- Zayzafoon, M.; Fulzele, K.; McDonald, J.M. Calmodulin and calmodulin-dependent kinase IIalpha regulate osteoblast differentiation by controlling c-fos expression. J. Biol. Chem. 2005, 280, 7049–7059. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Blair, H.C.; Peng, Y.; Zaidi, N.; Adebanjo, O.A.; Wu, X.B.; Wu, X.Y.; Iqbal, J.; Epstein, S.; Abe, E.; et al. Calcineurin regulates bone formation by the osteoblast. Proc. Natl. Acad. Sci. USA 2005, 102, 17130–17135. [Google Scholar] [CrossRef] [Green Version]
- Winslow, M.M.; Pan, M.; Starbuck, M.; Gallo, E.M.; Deng, L.; Karsenty, G.; Crabtree, G.R. Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev. Cell 2006, 10, 771–782. [Google Scholar] [CrossRef] [Green Version]
- Pilquil, C.; Alvandi, Z.; Opas, M. Calreticulin regulates a switch between osteoblast and chondrocyte lineages derived from murine embryonic stem cells. J. Biol. Chem. 2020, 295, 6861–6875. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, C.; Li, Y.; Zheng, Q.; Xu, Y.; Liu, B.; Sun, W.; Li, Y.; Ji, S.; Liu, M.; et al. TMCO1-mediated Ca2+ leak underlies osteoblast functions via CaMKII signaling. Nat. Commun. 2019, 10, 1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houschyar, K.S.; Tapking, C.; Borrelli, M.R.; Popp, D.; Duscher, D.; Maan, Z.N.; Chelliah, M.P.; Li, J.; Harati, K.; Wallner, C.; et al. Wnt Pathway in Bone Repair and Regeneration—What Do We Know So Far. Front. Cell Dev. Biol. 2019, 6, 170. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, E.; Ebe, Y.; Kanaya, S.; Tsuchiya, M.; Nakamura, T.; Tamura, M.; Shimauchi, H. Wnt5a signaling is a substantial constituent in bone morphogenetic protein-2-mediated osteoblastogenesis. Biochem. Biophys. Res. Commun. 2012, 422, 627–632. [Google Scholar] [CrossRef]
- Nakakura, S.; Matsui, M.; Sato, A.; Ishii, M.; Endo, K.; Muragishi, S.; Murase, M.; Kito, H.; Niguma, H.; Kurokawa, N.; et al. Pathophysiological significance of the two-pore domain K+ channel K2P5.1 in splenic CD4+CD25− T cell subset from a chemically-induced murine inflammatory bowel disease model. Front. Physiol. 2015, 6, 299. [Google Scholar] [CrossRef] [Green Version]
- Kito, H.; Yamamura, H.; Suzuki, Y.; Ohya, S.; Asai, K.; Imaizumi, Y. Membrane hyperpolarization induced by endoplasmic reticulum stress facilitates Ca2+ influx to regulate cell cycle progression in brain capillary endothelial cells. J. Pharmacol. Sci. 2014, 125, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohya, S.; Kanatsuka, S.; Hatano, N.; Kito, H.; Matsui, A.; Fujimoto, M.; Matsuba, S.; Niwa, S.; Zhan, P.; Suzuki, T.; et al. Downregulation of the Ca2+-activated K+ channel KCa3.1 by histone deacetylase inhibition in human breast cancer cells. Pharmacol. Res. Perspect. 2016, 4, e00228. [Google Scholar] [CrossRef] [PubMed]
- Sundelacruz, S.; Levin, M.; Kaplan, D.L. Membrane potential controls adipogenic and osteogenic differentiation of mesenchymal stem cells. PLoS ONE 2008, 3, e3737. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Ohba, T.; Sawada, E.; Suzuki, Y.; Yamamura, H.; Ohya, S.; Tsuda, H.; Imaizumi, Y. Enhancement of Ca2+ influx and ciliary beating by membrane hyperpolarization due to ATP-sensitive K+ channel opening in mouse airway epithelial cells. J. Pharmacol. Exp. Ther. 2013, 347, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, D.; Kito, H.; Yamamoto, S.; Ohya, S.; Yamamura, H.; Asai, K.; Imaizumi, Y. Contribution of Kir2 potassium channels to ATP-induced cell death in brain capillary endothelial cells and reconstructed HEK293 cell model. Am. J. Physiol. Cell Physiol. 2011, 300, C75–C86. [Google Scholar] [CrossRef] [PubMed]
- Laprell, L.; Schulze, C.; Brehme, M.L.; Oertner, T.G. The role of microglia membrane potential in chemotaxis. J. Neuroinflamm. 2021, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Wulff, H.; Skolnik, E.Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015, 33, 291–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, C.; Baez-Nieto, D.; Valencia, I.; Oyarzun, I.; Rojas, P.; Naranjo, D.; Latorre, R. K+ channels: Function-structural overview. Compr. Physiol. 2012, 2, 2087–2149. [Google Scholar]
- Yang, J.E.; Song, M.S.; Shen, Y.; Ryu, P.D.; Lee, S.Y. The Role of KV7.3 in Regulating Osteoblast Maturation and Mineralization. Int. J. Mol. Sci. 2016, 17, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Zhong, D.; Fu, X.; Liu, Q.; Kang, L.; Ding, Z. Silencing of Ether a go-go 1 by shRNA inhibits osteosarcoma growth and cell cycle progression. Int. J. Mol. Sci. 2014, 15, 5570–5581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Zhong, D.; Gao, Q.; Zhai, W.; Ding, Z.; Wu, J. MicroRNA-34a inhibits human osteosarcoma proliferation by downregulating ether a go-go 1 expression. Int. J. Med. Sci. 2013, 10, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zheng, S.; Dong, X.; Xiao, J. 17β-Estradiol inhibits outward voltage-gated K⁺ currents in human osteoblast-like MG63 cells. J. Membr. Biol. 2013, 246, 39–45. [Google Scholar] [CrossRef]
- Belus, M.T.; Rogers, M.A.; Elzubeir, A.; Josey, M.; Rose, S.; Andreeva, V.; Yelick, P.C.; Bates, E.A. Kir2.1 is important for efficient BMP signaling in mammalian face development. Dev. Biol. 2018, 444 (Suppl. 1), S297–S307. [Google Scholar] [CrossRef]
- Sacco, S.; Giuliano, S.; Sacconi, S.; Desnuelle, C.; Barhanin, J.; Amri, E.Z.; Bendahhou, S. The inward rectifier potassium channel Kir2.1 is required for osteoblastogenesis. Hum. Mol. Genet. 2015, 24, 471–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehlmann, A.; Bork, S.; Saffrich, R.; Veh, R.W.; Wagner, W.; Derst, C. KATP channels in mesenchymal stromal stem cells: Strong up-regulation of Kir6.2 subunits upon osteogenic differentiation. Tissue Cell 2011, 43, 331–336. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Li, X.; Wang, F.; Xu, X.; Wang, C. Exogenous H2S prevents high glucose-induced damage to osteoblasts through regulation of KATP channels. Biochimie 2017, 137, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Hirukawa, K.; Muraki, K.; Ohya, S.; Imaizumi, Y.; Togari, A. Electrophysiological properties of a novel Ca2+-activated K+ channel expressed in human osteoblasts. Calcif. Tissue Int. 2008, 83, 222–229. [Google Scholar] [CrossRef]
- Hei, H.; Gao, J.; Dong, J.; Tao, J.; Tian, L.; Pan, W.; Wang, H.; Zhang, X. BK Knockout by TALEN-Mediated Gene Targeting in Osteoblasts: KCNMA1 Determines the Proliferation and Differentiation of Osteoblasts. Mol. Cells 2016, 39, 530–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Guo, Q.; Hei, H.; Tao, J.; Zhou, Y.; Dong, J.; Xin, H.; Cai, H.; Gao, J.; Yu, K.; et al. BK ablation attenuates osteoblast bone formation via integrin pathway. Cell Death Dis. 2019, 10, 738. [Google Scholar] [CrossRef] [PubMed]
- Ning, F.L.; Tao, J.; Li, D.D.; Tian, L.L.; Wang, M.L.; Reilly, S.; Liu, C.; Cai, H.; Xin, H.; Zhang, X.M. Activating BK channels ameliorates vascular smooth muscle calcification through Akt signaling. Acta Pharmacol. Sin. 2021, 1–10. [Google Scholar] [CrossRef]
- Freise, C.; Querfeld, U. Inhibition of vascular calcification by block of intermediate conductance calcium-activated potassium channels with TRAM-34. Pharmacol. Res. 2014, 85, 6–14. [Google Scholar] [CrossRef]
- Kito, H.; Morihiro, H.; Sakakibara, Y.; Endo, K.; Kajikuri, J.; Suzuki, T.; Ohya, S. Downregulation of the Ca2+-activated K+ channel KCa3.1 in mouse preosteoblast cells treated with vitamin D receptor agonist. Am. J. Physiol. Cell Physiol. 2020, 319, C345–C358. [Google Scholar] [CrossRef]
- Hughes, S.; Magnay, J.; Foreman, M.; Publicover, S.J.; Dobson, J.P.; El Haj, A.J. Expression of the mechanosensitive 2PK+ channel TREK-1 in human osteoblasts. J. Cell. Physiol. 2006, 206, 738–748. [Google Scholar] [CrossRef]
- Li, X.; Dong, X.; Zheng, S.; Xiao, J. Expression and localization of TASK-1, -2 and -3 channels in MG63 human osteoblast-like cells. Oncol. Lett. 2013, 5, 865–869. [Google Scholar] [CrossRef]
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; McKinnon, D.; Pardo, L.A.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stuhmer, W.; et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 2005, 57, 473–508. [Google Scholar] [CrossRef] [PubMed]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozekin, Y.H.; Isner, T.; Bates, E.A. Ion Channel Contributions to Morphological Development: Insights from the Role of Kir2.1 in Bone Development. Front. Mol. Neurosci. 2020, 13, 99. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.S.; Uzel, S.G.; Akagi, J.; Wlodkowic, D.; Andreeva, V.; Yelick, P.C.; Devitt-Lee, A.; Pare, J.F.; Levin, M. Bioelectric signalling via potassium channels: A mechanism for craniofacial dysmorphogenesis in KCNJ2-associated Andersen-Tawil Syndrome. J. Physiol. 2016, 594, 3245–3270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahal, G.R.; Rawson, J.; Gassaway, B.; Kwok, B.; Tong, Y.; Ptacek, L.J.; Bates, E. An inwardly rectifying K+ channel is required for patterning. Development 2012, 139, 3653–3664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaritsky, J.J.; Eckman, D.M.; Wellman, G.C.; Nelson, M.T.; Schwarz, T.L. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K+ current in K+-mediated vasodilation. Circ. Res. 2000, 87, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Pini, J.; Giuliano, S.; Matonti, J.; Gannoun, L.; Simkin, D.; Rouleau, M.; Bendahhou, S. Osteogenic and Chondrogenic Master Genes Expression Is Dependent on the Kir2.1 Potassium Channel Through the Bone Morphogenetic Protein Pathway. J. Bone Miner. Res. 2018, 33, 1826–1841. [Google Scholar] [CrossRef]
- Wei, A.D.; Gutman, G.A.; Aldrich, R.; Chandy, K.G.; Grissmer, S.; Wulff, H. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol. Rev. 2005, 57, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Berkefeld, H.; Fakler, B.; Schulte, U. Ca2+-activated K+ channels: From protein complexes to function. Physiol. Rev. 2010, 90, 1437–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolton, T.B.; Imaizumi, Y. Spontaneous transient outward currents in smooth muscle cells. Cell Calcium 1996, 20, 141–152. [Google Scholar] [CrossRef]
- Yamazaki, D.; Aoyama, M.; Ohya, S.; Muraki, K.; Asai, K.; Imaizumi, Y. Novel functions of small conductance Ca2+-activated K+ channel in enhanced cell proliferation by ATP in brain endothelial cells. J. Biol. Chem. 2006, 281, 38430–38439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, M.; Kajikuri, J.; Kito, H.; Endo, K.; Hasegawa, Y.; Murate, S.; Ohya, S. Inhibition of Interleukin 10 Transcription through the SMAD2/3 Signaling Pathway by Ca2+-Activated K+ Channel KCa3.1 Activation in Human T-Cell Lymphoma HuT-78 Cells. Mol. Pharmacol. 2019, 95, 294–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sausbier, U.; Dullin, C.; Missbach-Guentner, J.; Kabagema, C.; Flockerzie, K.; Kuscher, G.M.; Stuehmer, W.; Neuhuber, W.; Ruth, P.; Alves, F.; et al. Osteopenia due to enhanced cathepsin K release by BK channel ablation in osteoclasts. PLoS ONE 2011, 6, e21168. [Google Scholar]
- Bi, D.; Toyama, K.; Lemaitre, V.; Takai, J.; Fan, F.; Jenkins, D.P.; Wulff, H.; Gutterman, D.D.; Park, F.; Miura, H. The intermediate conductance calcium-activated potassium channel KCa3.1 regulates vascular smooth muscle cell proliferation via controlling calcium-dependent signaling. J. Biol. Chem. 2013, 288, 15843–15853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Arnaud, R. The direct role of vitamin D on bone homeostasis. Arch. Biochem. Biophys. 2008, 473, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Yoshizawa, T.; Fukuda, T.; Shirode-Fukuda, Y.; Yu, T.; Sekine, K.; Sato, T.; Kawano, H.; Aihara, K.; Nakamichi, Y.; et al. Vitamin D receptor in osteoblasts is a negative regulator of bone mass control. Endocrinology 2013, 154, 1008–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sooy, K.; Sabbagh, Y.; Demay, M.B. Osteoblasts lacking the vitamin D receptor display enhanced osteogenic potential in vitro. J. Cell. Biochem. 2005, 94, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Feliciangeli, S.; Chatelain, F.C.; Bichet, D.; Lesage, F. The family of K2P channels: Salient structural and functional properties. J. Physiol. 2015, 593, 2587–2603. [Google Scholar] [CrossRef] [Green Version]
- Enyedi, P.; Czirjak, G. Molecular background of leak K+ currents: Two-pore domain potassium channels. Physiol. Rev. 2010, 90, 559–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djillani, A.; Mazella, J.; Heurteaux, C.; Borsotto, M. Role of TREK-1 in Health and Disease, Focus on the Central Nervous System. Front. Pharmacol. 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, K.; Kurokawa, N.; Kito, H.; Nakakura, S.; Fujii, M.; Ohya, S. Molecular identification of the dominant-negative, splicing isoform of the two-pore domain K+ channel K2P5.1 in lymphoid cells and enhancement of its expression by splicing inhibition. Biochem. Pharmacol. 2015, 98, 440–452. [Google Scholar] [CrossRef]
- Cid, L.P.; Roa-Rojas, H.A.; Niemeyer, M.I.; Gonzalez, W.; Araki, M.; Araki, K.; Sepulveda, F.V. TASK-2: A K2P K+ channel with complex regulation and diverse physiological functions. Front. Physiol. 2013, 4, 198. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.Y.; Foley, J.; Numaga-Tomita, T.; Petranka, J.G.; Bird, G.S.; Putney, J.W., Jr. Deletion of Orai1 alters expression of multiple genes during osteoclast and osteoblast maturation. Cell Calcium 2012, 52, 488–500. [Google Scholar] [CrossRef] [Green Version]
- Robinson, L.J.; Mancarella, S.; Songsawad, D.; Tourkova, I.L.; Barnett, J.B.; Gill, D.L.; Soboloff, J.; Blair, H.C. Gene disruption of the calcium channel Orai1 results in inhibition of osteoclast and osteoblast differentiation and impairs skeletal development. Lab. Investig. 2012, 92, 1071–1083. [Google Scholar] [CrossRef]
- Choi, H.; Srikanth, S.; Atti, E.; Pirih, F.Q.; Nervina, J.M.; Gwack, Y.; Tetradis, S. Deletion of Orai1 leads to bone loss aggravated with aging and impairs function of osteoblast lineage cells. Bone Rep. 2018, 8, 147–155. [Google Scholar] [CrossRef]
- Liu, S.; Takahashi, M.; Kiyoi, T.; Toyama, K.; Mogi, M. Genetic Manipulation of Calcium Release-Activated Calcium Channel 1 Modulates the Multipotency of Human Cartilage-Derived Mesenchymal Stem Cells. J. Immunol. Res. 2019, 2019, 7510214. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, Y.; Song, M.; Srikanth, S.; Kim, S.; Kang, M.K.; Gwack, Y.; Park, N.H.; Kim, R.H.; Shin, K.H. Orai1 mediates osteogenic differentiation via BMP signaling pathway in bone marrow mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2016, 473, 1309–1314. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Yan, J.; Umbach, A.T.; Fakhri, H.; Fajol, A.; Schmidt, S.; Salker, M.S.; Chen, H.; Alexander, D.; Spichtig, D.; et al. NFκB-sensitive Orai1 expression in the regulation of FGF23 release. J. Mol. Med. 2016, 94, 557–566. [Google Scholar] [CrossRef]
- Feger, M.; Hase, P.; Zhang, B.; Hirche, F.; Glosse, P.; Lang, F.; Föller, M. The production of fibroblast growth factor 23 is controlled by TGF-β2. Sci. Rep. 2017, 7, 4982. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Ramachandran, A.; Zhang, Y.; Koshy, R.; George, A. The ER Ca2+ sensor STIM1 can activate osteoblast and odontoblast differentiation in mineralized tissues. Connect. Tissue Res. 2018, 59, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Roy, B.; Das, T.; Mishra, D.; Maiti, T.K.; Chakraborty, S. Oscillatory shear stress induced calcium flickers in osteoblast cells. Integr. Biol. 2014, 6, 289–299. [Google Scholar] [CrossRef]
- Xiao, E.; Yang, H.Q.; Gan, Y.H.; Duan, D.H.; He, L.H.; Guo, Y.; Wang, S.Q.; Zhang, Y. Brief reports: TRPM7 Senses mechanical stimulation inducing osteogenesis in human bone marrow mesenchymal stem cells. Stem Cells 2015, 33, 615–621. [Google Scholar] [CrossRef]
- Liu, Y.S.; Liu, Y.A.; Huang, C.J.; Yen, M.H.; Tseng, C.T.; Chien, S.; Lee, O.K. Mechanosensitive TRPM7 mediates shear stress and modulates osteogenic differentiation of mesenchymal stromal cells through Osterix pathway. Sci. Rep. 2015, 5, 16522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohde, M.; Ziebart, J.; Kirschstein, T.; Sellmann, T.; Porath, K.; Kühl, F.; Delenda, B.; Bahls, C.; van Rienen, U.; Bader, R.; et al. Human Osteoblast Migration in DC Electrical Fields Depends on Store Operated Ca2+-Release and Is Correlated to Upregulation of Stretch-Activated TRPM7 Channels. Front. Bioeng. Biotechnol. 2019, 7, 422. [Google Scholar] [CrossRef] [Green Version]
- Son, A.; Kang, N.; Kang, J.Y.; Kim, K.W.; Yang, Y.M.; Shin, D.M. TRPM3/TRPV4 regulates Ca2+-mediated RANKL/NFATc1 expression in osteoblasts. J. Mol. Endocrinol. 2018, 61, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.H.; Liu, M.; He, Y.; Xiao, E.; Zhao, L.; Zhang, T.; Yang, H.Q.; Zhang, Y. TRPV1 deletion impaired fracture healing and inhibited osteoclast and osteoblast differentiation. Sci. Rep. 2017, 7, 42385. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Kawasaki, M.; Tsukamoto, M.; Menuki, K.; Suzuki, H.; Matsuura, T.; Baba, K.; Motojima, Y.; Fujitani, T.; Ohnishi, H.; et al. Transient receptor potential vanilloid 1 and 4 double knockout leads to increased bone mass in mice. Bone Rep. 2020, 12, 100268. [Google Scholar] [CrossRef]
- Han, X.; Kato, H.; Sato, H.; Hirofuji, Y.; Fukumoto, S.; Masuda, K. Accelerated osteoblastic differentiation in patient-derived dental pulp stem cells carrying a gain-of-function mutation of TRPV4 associated with metatropic dysplasia. Biochem. Biophys. Res. Commun. 2020, 523, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Pozo, A.; Regnier, M.; Lizotte, J.; Martineau, C.; Scorza, T.; Moreau, R. Cyclic adenosine monophosphate-dependent activation of transient receptor potential vanilloid 4 (TRPV4) channels in osteoblast-like MG-63 cells. Cell. Signal. 2020, 66, 109486. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Baudry, J.; Cao, L.; Huang, J.; Chen, H.; Yates, C.R.; Li, W.; Dong, B.; Waters, C.M.; Smith, J.C.; et al. Polycystin-1 interacts with TAZ to stimulate osteoblastogenesis and inhibit adipogenesis. J. Clin. Investig. 2018, 128, 157–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrick, D.; Mistry, K.; Wu, J.; Gresko, N.; Baggs, J.E.; Hogenesch, J.B.; Sun, Z.; Caplan, M.J. Polycystin-1 regulates bone development through an interaction with the transcriptional coactivator TAZ. Hum. Mol. Genet. 2019, 28, 16–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, N.; Zhou, H.; Xiao, Z. Downregulation of PKD1 by shRNA results in defective osteogenic differentiation via cAMP/PKA pathway in human MG-63 cells. J. Cell. Biochem. 2012, 113, 967–976. [Google Scholar] [CrossRef] [Green Version]
- Mesner, L.D.; Ray, B.; Hsu, Y.H.; Manichaikul, A.; Lum, E.; Bryda, E.C.; Rich, S.S.; Rosen, C.J.; Criqui, M.H.; Allison, M.; et al. Bicc1 is a genetic determinant of osteoblastogenesis and bone mineral density. J. Clin. Investig. 2014, 124, 2736–2749. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Cao, L.; Liang, Y.; Huang, J.; Stern, A.R.; Dallas, M.; Johnson, M.; Quarles, L.D. Osteoblast-specific deletion of Pkd2 leads to low-turnover osteopenia and reduced bone marrow adiposity. PLoS ONE 2014, 9, e114198. [Google Scholar] [CrossRef]
- Sugimoto, A.; Miyazaki, A.; Kawarabayashi, K.; Shono, M.; Akazawa, Y.; Hasegawa, T.; Ueda-Yamaguchi, K.; Kitamura, T.; Yoshizaki, K.; Fukumoto, S.; et al. Piezo type mechanosensitive ion channel component 1 functions as a regulator of the cell fate determination of mesenchymal stem cells. Sci. Rep. 2017, 7, 17696. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, M.; Suzuki, H.; Hatano, N.; Nakano, S.; Muraki, Y.; Miyazawa, K.; Goto, S.; Muraki, K. PIEZO1 and TRPV4, which Are Distinct Mechano-Sensors in the Osteoblastic MC3T3-E1 Cells, Modify Cell-Proliferation. Int. J. Mol. Sci. 2019, 20, 4960. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Liu, L.; Lv, L.; Hu, S.; Tariq, A.; Wang, W.; Dang, X. Fluid shear stress induces Runx-2 expression via upregulation of PIEZO1 in MC3T3-E1 cells. Cell Biol. Int. 2020, 44, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Chi, S.; Li, Y.; Ling, S.; Tan, Y.; Xu, Y.; Jiang, F.; Li, J.; Liu, C.; Zhong, G.; et al. The mechanosensitive Piezo1 channel is required for bone formation. eLife 2019, 8, e47454. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Han, L.; Nookaew, I.; Mannen, E.; Silva, M.J.; Almeida, M.; Xiong, J. Stimulation of Piezo1 by mechanical signals promotes bone anabolism. eLife 2019, 8, e49631. [Google Scholar] [CrossRef]
- Sasaki, F.; Hayashi, M.; Mouri, Y.; Nakamura, S.; Adachi, T.; Nakashima, T. Mechanotransduction via the Piezo1-Akt pathway underlies Sost suppression in osteocytes. Biochem. Biophys. Res. Commun. 2020, 521, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, G.; Fischer, V.; Liedert, A.; von Kroge, S.; Haffner-Luntzer, M.; Brylka, L.; Pawlus, E.; Schweizer, M.; Yorgan, T.; Baranowsky, A.; et al. Piezo1 Inactivation in Chondrocytes Impairs Trabecular Bone Formation. J. Bone Miner. Res. 2021, 36, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Gao, B.; Fan, Y.; Liu, Y.; Feng, S.; Cong, Q.; Zhang, X.; Zhou, Y.; Yadav, P.S.; Lin, J.; et al. Piezo1/2 mediate mechanotransduction essential for bone formation through concerted activation of NFAT-YAP1-ss-catenin. eLife 2020, 9, e52779. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Zuo, C.; Huang, Y.; Bajis, R.; Sahih, M.; Li, Y.P.; Dai, K.; Zhang, X. Osteoblastogenesis regulation signals in bone remodeling. Osteoporos. Int. 2012, 23, 1653–1663. [Google Scholar] [CrossRef]
- Salazar, V.S.; Gamer, L.W.; Rosen, V. BMP signalling in skeletal development, disease and repair. Nat. Rev. Endocrinol. 2016, 12, 203–221. [Google Scholar] [CrossRef]
- Kuro, O.M.; Moe, O.W. FGF23-alphaKlotho as a paradigm for a kidney-bone network. Bone 2017, 100, 4–18. [Google Scholar] [CrossRef]
- Raya, A.I.; Rios, R.; Pineda, C.; Rodriguez-Ortiz, M.E.; Diez, E.; Almaden, Y.; Munoz-Castaneda, J.R.; Rodriguez, M.; Aguilera-Tejero, E.; Lopez, I. Energy-dense diets increase FGF23, lead to phosphorus retention and promote vascular calcifications in rats. Sci. Rep. 2016, 6, 36881. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.R.; Holt, S.G.; Hewitson, T.D. alphaKlotho-FGF23 interactions and their role in kidney disease: A molecular insight. Cell. Mol. Life Sci. 2019, 76, 4705–4724. [Google Scholar] [CrossRef]
- Glosse, P.; Feger, M.; Mutig, K.; Chen, H.; Hirche, F.; Hasan, A.A.; Gaballa, M.M.S.; Hocher, B.; Lang, F.; Föller, M. AMP-activated kinase is a regulator of fibroblast growth factor 23 production. Kidney Int. 2018, 94, 491–501. [Google Scholar] [CrossRef]
- Li, S.; Yang, B.; Du, Y.; Lin, Y.; Liu, J.; Huang, S.; Zhang, A.; Jia, Z.; Zhang, Y. Targeting PPARα for the Treatment and Understanding of Cardiovascular Diseases. Cell Physiol. Biochem. 2018, 51, 2760–2775. [Google Scholar] [CrossRef]
- Syversen, U.; Stunes, A.K.; Gustafsson, B.I.; Obrant, K.J.; Nordsletten, L.; Berge, R.; Thommesen, L.; Reseland, J.E. Different skeletal effects of the peroxisome proliferator activated receptor (PPAR)α agonist fenofibrate and the PPARg agonist pioglitazone. BMC Endocr. Disord. 2009, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Ewendt, F.; Hirche, F.; Feger, M.; Föller, M. Peroxisome proliferator-activated receptor α (PPARα)-dependent regulation of fibroblast growth factor 23 (FGF23). Pflug. Arch. Eur. J. Physiol. 2020, 472, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Rojas, R.; Laporte, J.; Bohm, J. STIM1/ORAI1 Loss-of-Function and Gain-of-Function Mutations Inversely Impact on SOCE and Calcium Homeostasis and Cause Multi-Systemic Mirror Diseases. Front. Physiol. 2020, 11, 604941. [Google Scholar] [CrossRef] [PubMed]
- Misceo, D.; Holmgren, A.; Louch, W.E.; Holme, P.A.; Mizobuchi, M.; Morales, R.J.; De Paula, A.M.; Stray-Pedersen, A.; Lyle, R.; Dalhus, B.; et al. A dominant STIM1 mutation causes Stormorken syndrome. Hum. Mutat. 2014, 35, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Gamage, T.H.; Lengle, E.; Gunnes, G.; Pullisaar, H.; Holmgren, A.; Reseland, J.E.; Merckoll, E.; Corti, S.; Mizobuchi, M.; Morales, R.J.; et al. STIM1 R304W in mice causes subgingival hair growth and an increased fraction of trabecular bone. Cell Calcium 2020, 85, 102110. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, Q.; Feng, Z.; Zheng, L. STIM1 controls calcineurin/Akt/mTOR/NFATC2-mediated osteoclastogenesis induced by RANKL/M-CSF. Exp. Ther. Med. 2020, 20, 736–747. [Google Scholar] [CrossRef]
- Li, H. TRP Channel Classification. Adv. Exp. Med. Biol. 2017, 976, 1–8. [Google Scholar]
- Lieben, L.; Carmeliet, G. The Involvement of TRP Channels in Bone Homeostasis. Front. Endocrinol. 2012, 3, 99. [Google Scholar] [CrossRef] [Green Version]
- Wittkowske, C.; Reilly, G.C.; Lacroix, D.; Perrault, C.M. In Vitro Bone Cell Models: Impact of Fluid Shear Stress on Bone Formation. Front. Bioeng. Biotechnol. 2016, 4, 87. [Google Scholar] [CrossRef] [Green Version]
- Cadossi, R.; Massari, L.; Racine-Avila, J.; Aaron, R.K. Pulsed Electromagnetic Field Stimulation of Bone Healing and Joint Preservation: Cellular Mechanisms of Skeletal Response. J. Am. Acad. Orthop. Surg. Glob. Res. Rev. 2020, 4, e1900155. [Google Scholar] [CrossRef] [PubMed]
- Hiemer, B.; Ziebart, J.; Jonitz-Heincke, A.; Grunert, P.C.; Su, Y.; Hansmann, D.; Bader, R. Magnetically induced electrostimulation of human osteoblasts results in enhanced cell viability and osteogenic differentiation. Int. J. Mol. Med. 2016, 38, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wu, H.; Yang, Y.; Song, M. Effects of electromagnetic fields on osteoporosis: A systematic literature review. Electromagn. Biol. Med. 2016, 35, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Baek, K.; Baek, J.H.; Kim, H.R. The cooperation of CREB and NFAT is required for PTHrP-induced RANKL expression in mouse osteoblastic cells. J. Cell. Physiol. 2015, 230, 667–679. [Google Scholar] [CrossRef]
- Nilius, B.; Voets, T. The puzzle of TRPV4 channelopathies. EMBO Rep. 2013, 14, 152–163. [Google Scholar] [CrossRef]
- Nonaka, K.; Han, X.; Kato, H.; Sato, H.; Yamaza, H.; Hirofuji, Y.; Masuda, K. Novel gain-of-function mutation of TRPV4 associated with accelerated chondrogenic differentiation of dental pulp stem cells derived from a patient with metatropic dysplasia. Biochem. Biophys. Rep. 2019, 19, 100648. [Google Scholar] [CrossRef]
- Jilka, R.L. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone 2007, 40, 1434–1446. [Google Scholar] [CrossRef] [Green Version]
- Jilka, R.L.; Weinstein, R.S.; Bellido, T.; Roberson, P.; Parfitt, A.M.; Manolagas, S.C. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Investig. 1999, 104, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Pajevic, P.D.; Selig, M.; Barry, K.J.; Yang, J.Y.; Shin, C.S.; Baek, W.Y.; Kim, J.E.; Kronenberg, H.M. Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. J. Bone Miner. Res. 2012, 27, 2075–2084. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.E.; Harris, P.C.; Pirson, Y. Autosomal dominant polycystic kidney disease. Lancet 2007, 369, 1287–1301. [Google Scholar] [CrossRef]
- Wang, Z.; Ng, C.; Liu, X.; Wang, Y.; Li, B.; Kashyap, P.; Chaudhry, H.A.; Castro, A.; Kalontar, E.M.; Ilyayev, L.; et al. The ion channel function of polycystin-1 in the polycystin-1/polycystin-2 complex. EMBO Rep. 2019, 20, e48336. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.A.; Boletta, A. Role of the polycystins as mechanosensors of extracellular stiffness. Am. J. Physiol. Ren. Physiol. 2021, 320, F693–F705. [Google Scholar] [CrossRef] [PubMed]
- Dalagiorgou, G.; Piperi, C.; Adamopoulos, C.; Georgopoulou, U.; Gargalionis, A.N.; Spyropoulou, A.; Zoi, I.; Nokhbehsaim, M.; Damanaki, A.; Deschner, J.; et al. Mechanosensor polycystin-1 potentiates differentiation of human osteoblastic cells by upregulating Runx2 expression via induction of JAK2/STAT3 signaling axis. Cell. Mol. Life Sci. 2017, 74, 921–936. [Google Scholar] [CrossRef]
- Gudipaty, S.A.; Lindblom, J.; Loftus, P.D.; Redd, M.J.; Edes, K.; Davey, C.F.; Krishnegowda, V.; Rosenblatt, J. Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature 2017, 543, 118–121. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Luo, J.; Yang, P.; Du, J.; Kim, B.S.; Hu, H. Piezo2 channel-Merkel cell signaling modulates the conversion of touch to itch. Science 2018, 360, 530–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Hou, B.; Tumova, S.; Muraki, K.; Bruns, A.; Ludlow, M.J.; Sedo, A.; Hyman, A.J.; McKeown, L.; Young, R.S.; et al. Piezo1 integration of vascular architecture with physiological force. Nature 2014, 515, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Gnanasambandam, R.; Bae, C.; Gottlieb, P.A.; Sachs, F. Ionic Selectivity and Permeation Properties of Human PIEZO1 Channels. PLoS ONE 2015, 10, e0125503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marouli, E.; Graff, M.; Medina-Gomez, C.; Lo, K.S.; Wood, A.R.; Kjaer, T.R.; Fine, R.S.; Lu, Y.; Schurmann, C.; Highland, H.M.; et al. Rare and low-frequency coding variants alter human adult height. Nature 2017, 542, 186–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.A.; Kemp, J.P.; Youlten, S.E.; Laurent, L.; Logan, J.G.; Chai, R.C.; Vulpescu, N.A.; Forgetta, V.; Kleinman, A.; Mohanty, S.T.; et al. An atlas of genetic influences on osteoporosis in humans and mice. Nat. Genet. 2019, 51, 258–266. [Google Scholar] [CrossRef]
- Wadhwa, S.; Godwin, S.L.; Peterson, D.R.; Epstein, M.A.; Raisz, L.G.; Pilbeam, C.C. Fluid flow induction of cyclo-oxygenase 2 gene expression in osteoblasts is dependent on an extracellular signal-regulated kinase signaling pathway. J. Bone Miner. Res. 2002, 17, 266–274. [Google Scholar] [CrossRef]
- Zhao, N.; Nociti, F.H., Jr.; Duan, P.; Prideaux, M.; Zhao, H.; Foster, B.L.; Somerman, M.J.; Bonewald, L.F. Isolation and Functional Analysis of an Immortalized Murine Cementocyte Cell Line, IDG-CM6. J. Bone Miner. Res. 2016, 31, 430–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Leddy, H.A.; Chen, Y.; Lee, S.H.; Zelenski, N.A.; McNulty, A.L.; Wu, J.; Beicker, K.N.; Coles, J.; Zauscher, S.; et al. Synergy between Piezo1 and Piezo2 channels confers high-strain mechanosensitivity to articular cartilage. Proc. Natl. Acad. Sci. USA 2014, 111, E5114–E5122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; You, X.; Lotinun, S.; Zhang, L.; Wu, N.; Zou, W. Mechanical sensing protein PIEZO1 regulates bone homeostasis via osteoblast-osteoclast crosstalk. Nat. Commun. 2020, 11, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alejandro, P.; Constantinescu, F. A Review of Osteoporosis in the Older Adult: An Update. Rheum. Dis. Clin. 2018, 44, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Landowski, C.P.; Hediger, M.A. Mechanisms and regulation of epithelial Ca2+ absorption in health and disease. Annu. Rev. Physiol. 2008, 70, 257–271. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Member | Cell Types | Function | Reference |
|---|---|---|---|---|
| voltage-gated K+ channel | Kv7.3 | human mesenchymal stem cells, MG-63, and Saos-2 cells | osteoblast differentiation and mineralization | [27] |
| Kv10.1 | MG-63 cells | proliferation/ cell cycle progression | [28] | |
| MG-63, Saos-2, and hFOB 1.19 cells | prolifeation | [29] | ||
| Kv2.1 | MG-63 cells | functional expression | [30] | |
| inward rectifier K+ channel | Kir2.1 | myoblasts form Andersen-Tawil syndrome (ATS) patients | Mineralization | [31] |
| induced pluripotent stem cells form ATS patients | osteoblast differentiation and mineralization | [32] | ||
| Kir6.2 | human mesenchamal stromal cells | osteogenic diferentiation | [33] | |
| Kir6/SUR1 | rat calvarial osteoblasts | proliferation, apoptosis, and mineralization | [34] | |
| Ca2+-activated K+ channel | KCa1.1 | SaM-1, MG-63, SaOS-2, and HOS cells | functional expression | [35] |
| ROS17/2.8 and MC3T3-E1 cells | osteoblast proliferation and differentiation | [36] | ||
| mouse bone marrow mesenchymal stem cells, ROS17/2.8, and MC3T3-E1 cells | bone loss and defects in osteoblast formation | [37] | ||
| rat/mouse vascular smooth muscle cells | vascular calcification (-) | [38] | ||
| KCa3.1 | rat/mouse vascular smooth muscle cells | vascular calcification (+) | [39] | |
| MC3T3-E1 cells | prolifeataion | [40] | ||
| two-pore K+ channel | K2P2.1 | human osteoblasts and MG-63 cells | functional expression | [41] |
| K2P3.1, K2P5.1, K2P9.1 | MG-63 cells | prolifeartion | [42] |
| IUPHAR | HUGO | Synonyms |
|---|---|---|
| TRPC | ||
| TRPC1 TRPC2 TRPC3 TRPC4 TRPC5 TRPC6 TRPC7 | TRPC1 pseudogene TRPC3 TRPC4 TRPC5 TRPC6 TRPC7 | TRP1 TRP3 TRP4 TRP5 TRP6 TRP7 |
| TRPV | ||
| TRPV1 TRPV2 TRPV3 TRPV4 TRPV5 TRPV6 | TRPV1 TRPV2 TRPV3 TRPV4 TRPV5 TRPV6 | VR1 VRL/VRL1 VR3 VR2/TRP12/OTRPC4 ECAC1/CAT2/OTRPC3 ECAC2/CAT1/CATL |
| TRPM | ||
| TRPM1 TRPM2 TRPM3 TRPM4 TRPM5 TRPM6 TRPM7 TRPM8 | TRPM1 TRPM2 TRPM3 TRPM4 TRPM5 TRPM6 TRPM7 TRPM8 | LTRPC1/MLSN1 LTRPC2/TRPC7 LTRPC3/ MLSN3 LTRPC4 LTRPC5/MTR1 CHAK2/HOMG1 LTRPC7/CHAK1 LTRPC6/TRPP8 |
| TRPA | ||
| TRPA1 | TRPA1 | ANK/TM1 |
| TRPN | ||
| TRPN1 | trpn1 (fish) | nompC |
| TRPML | ||
| TRPML1 TRPML2 TRPML3 | MCOLN1 MCOLN2 MCOLN3 | mucolipin1 mucolipin2 mucolipin3 |
| TRPP | ||
| TRPP2 TRPP3 TRPP5 | PKD2 PKD2L1 PKD2L2 | polycystin 2 polycystin 2L1 polycystin 2L2 |
| Orai | ||
| Orai1 Orai2 Orai3 | ORAI1 ORAI2 ORAI3 | CRACM1/TMEM142A TMEM142B TMEM142C |
| Piezo | ||
| Piezo1 Piezo2 | PIEZO1 PIEZO2 |
| Member | Cell Types | Function | Reference |
|---|---|---|---|
| Orai1 | mouse bone marrow mesenchymal stromal cells and MC3T3-E1 cells | osteoblast differentiation and mineralization | [65] |
| human osteoprogenitor cells (CC-2538) | osteoblast differentiation and mineralization | [66] | |
| mouse calvaria osteoblasts and mesenchymal progenitors | osteoblast differentiation and mineralization | [67] | |
| human cartilage derived mesenchamal stem cells | osteoblast differentiation | [68] | |
| mouse bone marrow mesenchymal stromal cells | osteoblast differentiation and mineralization | [69] | |
| UMR106 cells | FGF23 expression | [70,71] | |
| STIM1 | MC3T3-E1 cells | osteoblast differentiation and mineralization | [72] |
| Member | Cell Types | Function | Reference |
|---|---|---|---|
| TRPM7 | MG-63 cells human mesenchymal stem cells human osteoblasts human osteoblasts | Ca2+ flicker activity osteoblast differentiation osteoblast differentiation migration | [73] [74,75] [75] [76] |
| TRPM3, TRPV4 | mouse calvarial osteoblasts | RANKL and NFATc1 expression | [77] |
| TRPV1 | mouse bone marrow stromal cells | osteoblast differentiation and mineralization | [78] |
| TRPV1, TRPV4 | mouse bone marrow cells | osteoblast differentiation | [79] |
| TRPV4 | mesenchymal stem cells from metatropic dysplasia patients MG-63 cells | osteoblast differentiation osteoblast proliferation and differentiation | [80] [81] |
| TRPP1 | human osteoblastic cells MG-63 cells | osteoblast differentiation osteoblast differentiation and mineralization | [82,83] [84] |
| TRPP1, TRPP2 | mouse calvarial osteoblasts | osteoblast differentiation and mineralization | [85] |
| TRPP2 | mouse calvarial osteoblasts | osteoblast differentiation and mineralization | [86] |
| Member | Cell Types | Function | Reference |
|---|---|---|---|
| Piezo1 | human mesenchymal stem cells, UE7T-13 cells, and SDP11 cell | osteoblast differentiation | [87] |
| MC3T3-E1 cells | proliferation | [88] | |
| MC3T3-E1 cells | Runx2 expression | [89] | |
| MC3T3-E1 cells and mouse calvarial osteoblasts | osteoblast differentiation | [90] | |
| mouse bone marrow stromal cells | osteoblast differentiation | [91] | |
| MLO-Y4 cells | mechanotransduction | [91] | |
| IDG-SW3 cells | Sost expression | [92] | |
| mouse calvarial osteoblasts | osteoblast differentiation | [93] | |
| Piezo1, Piezo2 | mouse bone marrow stromal cells | osteoblast differentiation | [94] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kito, H.; Ohya, S. Role of K+ and Ca2+-Permeable Channels in Osteoblast Functions. Int. J. Mol. Sci. 2021, 22, 10459. https://doi.org/10.3390/ijms221910459
Kito H, Ohya S. Role of K+ and Ca2+-Permeable Channels in Osteoblast Functions. International Journal of Molecular Sciences. 2021; 22(19):10459. https://doi.org/10.3390/ijms221910459
Chicago/Turabian StyleKito, Hiroaki, and Susumu Ohya. 2021. "Role of K+ and Ca2+-Permeable Channels in Osteoblast Functions" International Journal of Molecular Sciences 22, no. 19: 10459. https://doi.org/10.3390/ijms221910459
APA StyleKito, H., & Ohya, S. (2021). Role of K+ and Ca2+-Permeable Channels in Osteoblast Functions. International Journal of Molecular Sciences, 22(19), 10459. https://doi.org/10.3390/ijms221910459