
TNAP: A New Multitask Enzyme in Energy Metabolism


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. TNAP’s Pathophysiological Functions in the Liver, Bile and Intestinal Lumen
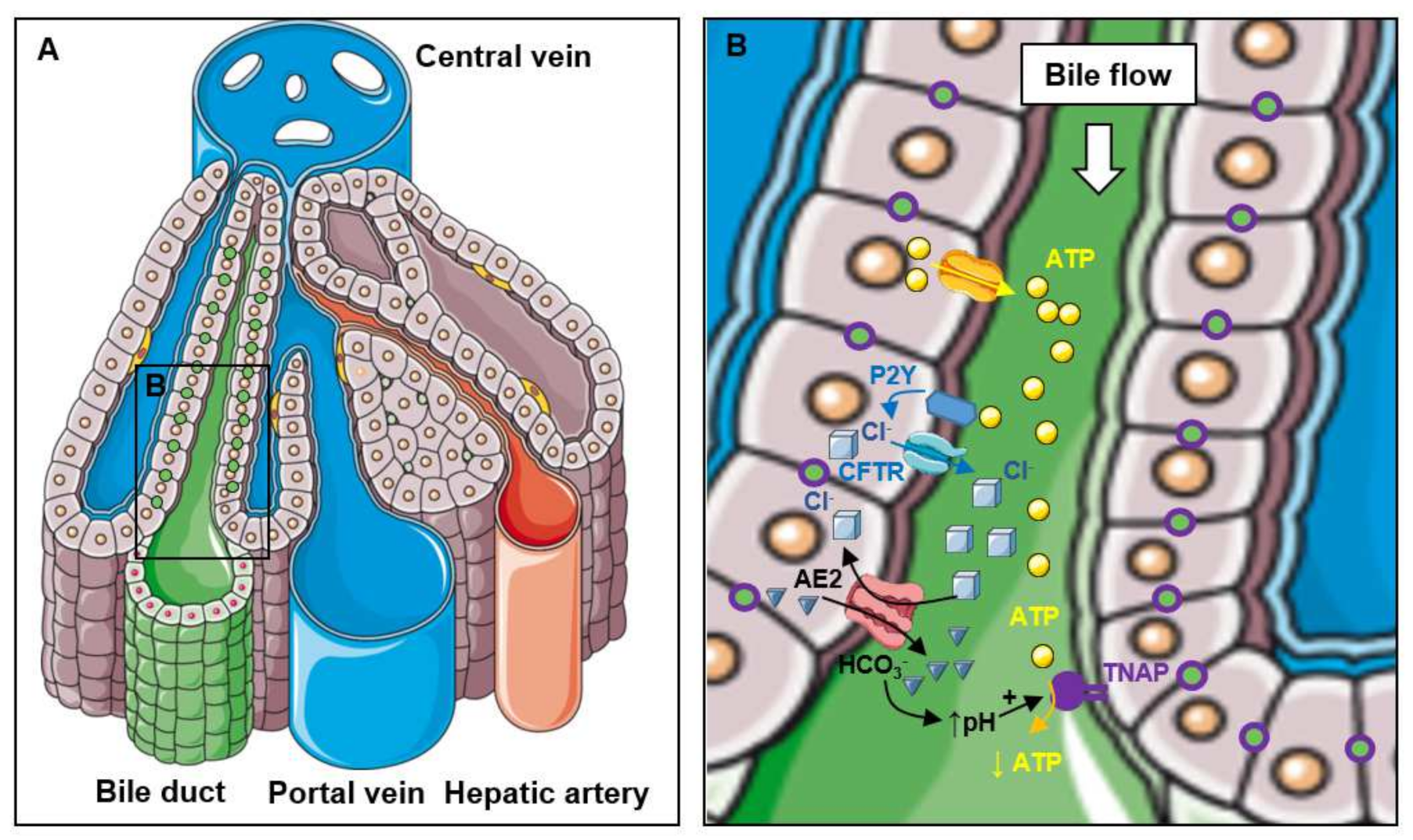
2.1. Localization of TNAP in the Liver
2.2. Function of TNAP in Bile Excretion
2.3. Function of TNAP in the Bile and the Intestinal Lumen
2.4. Functions of TNAP in Liver Metabolism
3. Pathophysiological TNAP’s Function in Adipocytes
3.1. Function of TNAP in Adipocyte Differentiation
3.2. Function of TNAP in Thermogenesis
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buchet, R.; Millán, J.L.; Magne, D. Multisystemic functions of alkaline phosphatases. Methods Mol. Biol. 2013, 1053, 27–51. [Google Scholar] [PubMed]
- Magnusson, P.; Degerblad, M.; Sääf, M.; Larsson, L.; Thorén, M. Different responses of bone alkaline phosphatase isoforms during recombinant insulin-like growth factor-I (IGF-I) and during growth hormone therapy in adults with growth hormone deficiency. J. Bone Miner. Res. 1997, 12, 210–220. [Google Scholar] [CrossRef]
- Poupon, R. Liver alkaline phosphatase: A missing link between choleresis and biliary inflammation. Hepatology 2015, 61, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Curhan, G.; Pfeffer, M.; Sacks, F.; Thadhani, R.; Melamed, M.L.; Wiebe, N.; Muntner, P. Relation between alkaline phosphatase, serum phosphate, and all-cause or cardiovascular mortality. Circulation 2009, 120, 1784–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramowitz, M.; Muntner, P.; Coco, M.; Southern, W.; Lotwin, I.; Hostetter, T.H.; Melamed, M.L. Serum alkaline phosphatase and phosphate and risk of mortality and hospitalization. Clin. J. Am. Soc. Nephrol. 2010, 5, 1064–1071. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, V.R.; Baird, B.C.; Wei, G.; Greene, T.; Raphael, K.; Beddhu, S. Associations of serum alkaline phosphatase with metabolic syndrome and mortality. Am. J. Med. 2011, 124, 566.e1–566.e7. [Google Scholar] [CrossRef] [Green Version]
- Filipowicz, R.; Greene, T.; Wei, G.; Cheung, A.K.; Raphael, K.L.; Baird, B.C.; Beddhu, S. Associations of serum skeletal alkaline phosphatase with elevated C-reactive protein and mortality. Clin. J. Am. Soc. Nephrol. 2013, 8, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Wannamethee, S.G.; Sattar, N.; Papcosta, O.; Lennon, L.; Whincup, P.H. Alkaline phosphatase, serum phosphate, and incident cardiovascular disease and total mortality in older men. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1070–1076. [Google Scholar] [CrossRef] [Green Version]
- Lammers, W.J.; van Buuren, H.R.; Hirschfield, G.M.; Janssen, H.L.; Invernizzi, P.; Mason, A.L.; Ponsoien, C.Y.; Floreani, A.; Corpechot, C.; Mayo, M.J.; et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: An international follow-up study. Gastroenterology 2014, 147, 1338–1349. [Google Scholar] [CrossRef] [Green Version]
- Panh, L.; Ruidavets, J.B.; Rousseau, H.; Petermann, A.; Bongard, V.; Bérard, E.; Taraszkiewicz, D.; Lairez, O.; Galinier, M.; Carrié, D.; et al. Association between serum alkaline phosphatase and coronary artery calcification in a sample of primary cardiovascular prevention patients. Atherosclerosis 2017, 260, 81–86. [Google Scholar] [CrossRef]
- Goettsch, C.; Strzelecka-Kiliszek, A.; Bessueille, L.; Quillard, T.; Mechtouff, L.; Pikula, S.; Canet-Soulas, E.; Millan, J.L.; Fonta, C.; Magne, D. TNAP as a therapeutic target for cardiovascular calcification—A discussion of its pleiotropic functions in the body. Cardiovasc. Res. 2020, 10, cvaa299. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, K.; Amizuka, N.; Oda, K.; Ikehara, Y.; Ozawa, H. Immunolocalization of tissue non-specific alkaline phosphatase in mice. Histochem. Cell Biol. 1997, 107, 183–191. [Google Scholar] [CrossRef]
- Halling Linder, C.; Englund, U.H.; Narisawa, S.; Millán, J.L.; Magnusson, P. Isozyme profile and tissue-origin of alkaline phosphatases in mouse serum. Bone 2013, 53, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gámez-Belmonte, R.; Tena-Garitaonaindia, M.; Hernández-Chirlaque, C.; Córdova, S.; Ceacero-Heras, D.; de Medina, F.S.; Martínez-Augustin, O. Deficiency in Tissue Non-Specific Alkaline Phosphatase Leads to Steatohepatitis in Mice Fed a High Fat Diet Similar to That Produced by a Methionine and Choline Deficient Diet. Int. J. Mol. Sci. 2020, 22, 51. [Google Scholar] [CrossRef]
- Whyte, M.P. Hypophosphatasia—Aetiology, nosology, pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2016, 12, 233–246. [Google Scholar] [CrossRef]
- Graser, S.; Liedtke, D.; Jakob, F. TNAP as a New Player in Chronic Inflammatory Conditions and Metabolism. Int. J. Mol. Sci. 2021, 22, 919. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; Greenberg, C.R.; Salman, N.J.; Bober, M.B.; McAlister, W.H.; Wenkert, D.; Van Sickle, B.J.; Simmons, J.H.; Edgar, T.S.; Bauer, M.L.; et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N. Engl. J. Med. 2012, 366, 904–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, N.; Takashima, Y.; Makita, T. Redistribution and fate of colchicine-induced alkaline phosphatase in rat hepatocytes: Possible formation of autophagosomes whose membrane is derived from excess plasma membrane. Histochem. Cell Biol. 1995, 104, 257–265. [Google Scholar] [CrossRef]
- Chida, K.; Taguchi, M. Localization of alkaline phosphatase and proteins related to intercellular junctions in primary cultures of fetal rat hepatocytes. Anat. Embryol. 2005, 210, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar]
- Alvaro, D.; Benedetti, A.; Marucci, L.; Delle Monache, M.; Monterubbianesi, R.; Di Cosimo, E.; Perego, L.; Macarri, G.; Glaser, S.; Le Sage, G.; et al. The function of alkaline phosphatase in the liver: Regulation of intrahepatic biliary epithelium secretory activities in the rat. Hepatology 2000, 32, 174–184. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, M.M.; Righetti, A. Induction of rat liver alkaline phosphatase: The mechanism of the serum elevation in bile duct obstruction. J. Clin. Investig. 1970, 49, 508–516. [Google Scholar] [CrossRef]
- Hatoff, D.E.; Hardison, W.G. Induced synthesis of alkaline phosphatase by bile acids in rat liver cell culture. Gastroenterology 1979, 77, 1062–1067. [Google Scholar] [CrossRef]
- Khan, K.N.; Tsutsumi, T.; Nakata, K.; Nakao, K.; Kato, Y.; Nagataki, S. Regulation of alkaline phosphatase gene expression in human hepatoma cells by bile acids. J. Gastroenterol. Hepatol. 1998, 13, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Hohenester, S.; de Buy Wenniger, L.J.; Kremer, A.E.; Jansen, P.L.; Elferink, R.P. The biliary HCO(3)(-) umbrella: A unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology 2010, 52, 1489–1496. [Google Scholar] [CrossRef]
- Banales, J.M.; Huebert, R.C.; Karlsen, T.; Strazzabosco, M.; LaRusso, N.F.; Gores, G.J. Cholangiocyte pathobiology. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 269–281. [Google Scholar] [CrossRef]
- Woo, K.; Dutta, A.K.; Patel, V.; Kresge, C.; Feranchak, A.P. Fluid flow induces mechanosensitive ATP release, calcium signalling and Cl- transport in biliary epithelial cells through a PKCzeta-dependent pathway. J. Physiol. 2008, 586, 2779–2798. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, D.; Cho, W.K.; Mennone, A.; Boyer, J.L. Effect of secretion on intracellular pH regulation in isolated rat bile duct epithelial cells. J. Clin. Investig. 1993, 92, 1314–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizumori, M.; Ham, M.; Guth, P.H.; Engel, E.; Kaunitz, J.D.; Akiba, Y. Intestinal alkaline phosphatase regulates protective surface microclimate pH in rat duodenum. J. Physiol. 2009, 587 Pt 14, 3651–3663. [Google Scholar] [CrossRef] [PubMed]
- Hatoff, D.E.; Hardison, W.G. Bile acid-dependent secretion of alkaline phosphatase in rat bile. Hepatology 1982, 2, 433–439. [Google Scholar] [CrossRef]
- Blom, E.; Ali, M.M.; Mortensen, B.; Huseby, N.E. Elimination of alkaline phosphatases from circulation by the galactose receptor. Different isoforms are cleared at various rates. Clin. Chim. Acta 1998, 270, 125–137. [Google Scholar] [CrossRef]
- Nosjean, O.; Koyama, I.; Goseki, M.; Roux, B.; Komoda, T. Human tissue non-specific alkaline phosphatases: Sugar-moiety-induced enzymic and antigenic modulations and genetic aspects. Biochem. J. 1997, 321 Pt 2, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Halling Linder, C.; Narisawa, S.; Millán, J.L.; Magnusson, P. Glycosylation differences contribute to distinct catalytic properties among bone alkaline phosphatase isoforms. Bone 2009, 45, 987–993. [Google Scholar] [CrossRef] [Green Version]
- Kaliannan, K.; Hamarneh, S.R.; Economopoulos, K.P.; Nasrin Alam, S.; Moaven, O.; Patel, P.; Malo, N.S.; Ray, M.; Abtahi, S.M.; Muhammad, M.; et al. Intestinal alkaline phosphatase prevents metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 7003–7008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühn, F.; Adiliaghdam, F.; Cavallaro, P.M.; Hamarneh, S.R.; Tsurumi, A.; Hoda, R.S.; Munoz, A.R.; Dhole, Y.; Ramirez, J.M.; Liu, E.; et al. Intestinal alkaline phosphatase targets the gut barrier to prevent aging. JCI Insight 2020, 5, e134049. [Google Scholar] [CrossRef] [PubMed]
- Herieka, M.; Erridge, C. High-fat meal induced postprandial inflammation. Mol. Nutr. Food Res. 2014, 58, 136–146. [Google Scholar] [CrossRef]
- Schromm, A.B.; Brandenburg, K.; Loppnow, H.; Zähringer, U.; Rietschel, E.T.; Carroll, S.F.; Koch, M.H.; Kusumoto, S.; Seydel, U. The charge of endotoxin molecules influences their conformation and IL-6-inducing capacity. J. Immunol. 1998, 161, 5464–5471. [Google Scholar]
- Poelstra, K.; Bakker, W.W.; Klok, P.A.; Hardonk, M.J.; Meijer, D.K. A physiologic function for alkaline phosphatase: Endotoxin detoxification. Lab. Investig. 1997, 76, 319–327. [Google Scholar]
- Poelstra, K.; Bakker, W.W.; Klok, P.A.; Kamps, J.A.; Hardonk, M.J.; Meijer, D.K. Dephosphorylation of endotoxin by alkaline phosphatase in vivo. Am. J. Pathol. 1997, 151, 1163–1169. [Google Scholar]
- Yang, W.H.; Heithoff, D.M.; Aziz, P.V.; Sperandio, M.; Nizet, V.; Mahan, M.J.; Marth, J.D. Recurrent infection progressively disables host protection against intestinal inflammation. Science 2017, 358, eaao5610. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.W.; Kim, J.H.; Lee, C.; Im, J.P.; Kim, J.S. Intestinal alkaline phosphatase ameliorates experimental colitis via toll-like receptor 4-dependent pathway. Eur. J. Pharmacol. 2018, 820, 156–166. [Google Scholar] [CrossRef]
- Liu, Y.; Cavallaro, P.M.; Kim, B.M.; Liu, T.; Wang, H.; Kühn, F.; Adiliaghdam, S.; Liu, E.; Vasan, R.; Samarbafzadeh, E.; et al. A role for intestinal alkaline phosphatase in preventing liver fibrosis. Theranostics 2021, 11, 14–26. [Google Scholar] [CrossRef]
- Ghosh, S.S.; He, H.; Wang, J.; Korzun, W.; Yannie, P.J.; Ghosh, S. Intestine-specific expression of human chimeric intestinal alkaline phosphatase attenuates Western diet-induced barrier dysfunction and glucose intolerance. Physiol. Rep. 2018, 6, e13790. [Google Scholar] [CrossRef] [Green Version]
- Lynes, M.; Narisawa, S.; Millán, J.L.; Widmaier, E.P. Interactions between CD36 and global intestinal alkaline phosphatase in mouse small intestine and effects of high-fat diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1738–R1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
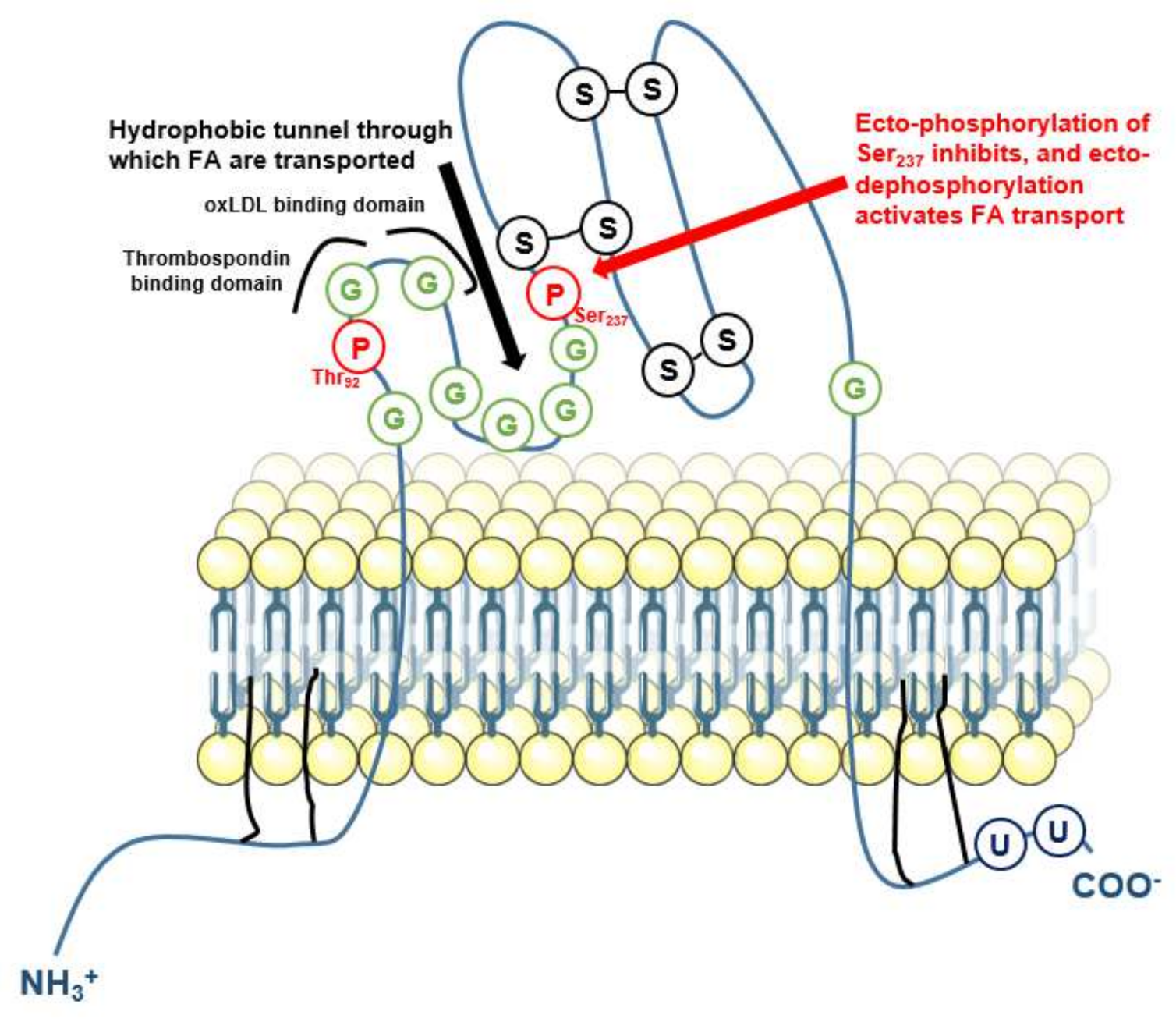
- Lynes, M.D.; Widmaier, E.P. Involvement of CD36 and intestinal alkaline phosphatases in fatty acid transport in enterocytes, and the response to a high-fat diet. Life Sci. 2011, 88, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Luiken, J.J.; Chanda, D.; Nabben, M.; Neumann, D.; Glatz, J.F. Post-translational modifications of CD36 (SR-B2): Implications for regulation of myocellular fatty acid uptake. Biochim. Biophys. Acta 2016, 1862, 2253–2258. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.C.; Luiken, J.J.F.P. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guthmann, F.; Maehl, P.; Preiss, J.; Kolleck, I.; Rüstow, B. Ectoprotein kinase-mediated phosphorylation of FAT/CD36 regulates palmitate uptake by human platelets. Cell. Mol. Life Sci. 2002, 59, 1999–2003. [Google Scholar] [CrossRef]
- Nakano, T.; Inoue, I.; Koyama, I.; Kanazawa, K.; Nakamura, K.; Narisawa, S.; Tanaka, K.; Akita, M.; Masuyama, T.; Seo, M.; et al. Disruption of the murine intestinal alkaline phosphatase gene Akp3 impairs lipid transcytosis and induces visceral fat accumulation and hepatic steatosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1439–G1449. [Google Scholar] [CrossRef] [Green Version]
- Asch, A.S.; Liu, I.; Briccetti, F.M.; Barnwell, J.W.; Kwakye-Berko, F.; Dokun, A.; Goldberger, J.; Pernambuco, M. Analysis of CD36 binding domains: Ligand specificity controlled by dephosphorylation of an ectodomain. Science 1993, 262, 1436–1440. [Google Scholar] [CrossRef]
- Kuehn, F.; Adiliaghdam, F.; Hamarneh, S.R.; Vasan, R.; Liu, E.; Liu, Y.; Ramirez, J.M.; Hoda, R.S.; Munoz, A.R.; Ko, F.C.; et al. Loss of Intestinal Alkaline Phosphatase Leads to Distinct Chronic Changes in Bone Phenotype. J. Surg. Res. 2018, 232, 325–331. [Google Scholar] [CrossRef]
- Kiffer-Moreira, T.; Sheen, C.R.; Gasque, K.C.; Bolean, M.; Ciancaglini, P.; van Elsas, A.; Hoylaerts, M.F.; Millan, J.L. Catalytic signature of a heat-stable, chimeric human alkaline phosphatase with therapeutic potential. PLoS ONE 2014, 9, e89374. [Google Scholar]
- Verpooten, G.F.; Nouwen, E.J.; Hoylaerts, M.F.; Hendrix, P.G.; de Broe, M.E. Segment-specific localization of intestinal-type alkaline phosphatase in human kidney. Kidney Int. 1989, 36, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Posadas, R.; González, R.; Ballester, I.; Martínez-Moya, P.; Romero-Calvo, I.; Suárez, M.D.; Zarzuelo, A.; Martinez-Augustin, O.; Sanchez de Medina, F. Tissue-nonspecific alkaline phosphatase is activated in enterocytes by oxidative stress via changes in glycosylation. Inflamm. Bowel Dis. 2011, 17, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Young, G.P.; Friedman, S.; Yedlin, S.T.; Allers, D.H. Effect of fat feeding on intestinal alkaline phosphatase activity in tissue and serum. Am. J. Physiol. 1981, 241, G461–G468. [Google Scholar] [CrossRef] [PubMed]
- Hansen, G.H.; Niels-Christiansen, L.L.; Immerdal, L.; Nystrøm, B.T.; Danielsen, E.M. Intestinal alkaline phosphatase: Selective endocytosis from the enterocyte brush border during fat absorption. Am. J. Physiol. Gastrointest. Liver. Physiol. 2007, 293, G1325–G1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, M.; Harajiri, S.; Tabata, S.; Yukimasa, N.; Muramoto, Y.; Komoda, T. [Alkaline phosphatase activity in blood group B or O secretors is fluctuated by the dinner intake of previous night]. Rinsho Byori 2013, 61, 307–312. [Google Scholar]
- Ruiter, D.J.; van der Meulen, J.; Brouwer, A.; Hummel, M.J.; Mauw, B.J.; van der Ploeg, J.C.; Wisse, E. Uptake by liver cells of endotoxin following its intravenous injection. Lab. Investig. 1981, 45, 38–45. [Google Scholar]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Pang, J.; Xu, W.; Zhang, X.; Wong, G.L.; Chan, A.W.; Chan, H.Y.; Tse, C.H.; Shu, S.S.; Choi, P.C.; Chan, H.L.; et al. Significant positive association of endotoxemia with histological severity in 237 patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2017, 46, 175–182. [Google Scholar] [CrossRef]
- Jin, C.J.; Baumann, A.; Brandt, A.; Engstler, A.J.; Nier, A.; Hege, M.; Schmeer, C.; Kehm, R.; Hohn, A.; Grune, T.; et al. Aging-related liver degeneration is associated with increased bacterial endotoxin and lipopolysaccharide binding protein levels. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G736–G747. [Google Scholar] [CrossRef] [PubMed]
- Nier, A.; Huber, Y.; Labenz, C.; Michel, M.; Bergheim, I.; Schattenberg, J.M. Adipokines and Endotoxemia Correlate with Hepatic Steatosis in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2020, 12, 699. [Google Scholar] [CrossRef] [Green Version]
- Tuin, A.; Huizinga-Van der Vlag, A.; van Loenen-Weemaes, A.M.; Meijer, D.K.; Poelstra, K. On the role and fate of LPS-dephosphorylating activity in the rat liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G377–G385. [Google Scholar] [CrossRef]
- Schippers, M.; Post, E.; Eichhorn, I.; Langeland, J.; Beljaars, L.; Malo, M.S.; Hodin, R.A.; Millan, J.L.; Popov, Y.; Schuppan, D.; et al. Phosphate Groups in the Lipid A Moiety Determine the Effects of LPS on Hepatic Stellate Cells: A Role for LPS-Dephosphorylating Activity in Liver Fibrosis. Cells 2020, 9, 2708. [Google Scholar] [CrossRef]
- Pettengill, M.; Matute, J.D.; Tresenriter, M.; Hibbert, J.; Burgner, D.; Richmond, P.; Millan, J.L.; Ozonoff, A.; Strunk, T.; Currie, A.; et al. Human alkaline phosphatase dephosphorylates microbial products and is elevated in preterm neonates with a history of late-onset sepsis. PLoS ONE 2017, 12, e0175936. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.F.; Henriques, M.C.; Oliveira, M.C.; Menezes-Garcia, Z.; Marques, P.E.; Souza, D.a.G.; Menezes, G.B.; Teixeira, M.M.; Ferreira, A.V. Acute intake of a high-fructose diet alters the balance of adipokine concentrations and induces neutrophil influx in the liver. J. Nutr. Biochem. 2014, 25, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebler, J.; Galle, P.R.; Weber, M.M. The gut-liver-axis: Endotoxemia, inflammation, insulin resistance and NASH. J. Hepatol. 2008, 48, 1032–1034. [Google Scholar] [CrossRef]
- Imajo, K.; Fujita, K.; Yoneda, M.; Nozaki, Y.; Ogawa, Y.; Shinohara, Y.; Kato, S.; Mawatari, H.; Shibata, W.; Kitani, H.; et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012, 16, 44–54. [Google Scholar]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thévenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Chirambo, G.M.; van Niekerk, C.; Crowther, N.J. The role of alkaline phosphatase in intracellular lipid accumulation in the human hepatocarcinoma cell line, HepG2. Exp. Mol. Pathol. 2017, 102, 224–229. [Google Scholar] [CrossRef]
- Su, R.C.; Lad, A.; Breidenbach, J.D.; Blomquist, T.M.; Gunning, W.T.; Dube, P.; Kleinhenz, A.L.; Malhotra, D.; Haller, S.T.; Kennedy, D.J. Hyperglycemia induces key genetic and phenotypic changes in human liver epithelial HepG2 cells which parallel the Leprdb/J mouse model of non-alcoholic fatty liver disease (NAFLD). PLoS ONE 2019, 14, e0225604. [Google Scholar] [CrossRef] [Green Version]
- Chirambo, G.; van Niekerk, C.; Crowther, N.J. Specific knock-down of tissue non-specific alkaline phosphatase mRNA levels inhibits intracellular lipid accumulation in 3T3-L1 and HepG2 cells. Int. J. Exp. Pathol. 2017, 98, 260–268. [Google Scholar] [CrossRef]
- Coburn, C.T.; Knapp, F.F.; Febbraio, M.; Beets, A.L.; Silverstein, R.L.; Abumrad, N.A. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J. Biol. Chem. 2000, 275, 32523–32529. [Google Scholar] [CrossRef] [Green Version]
- Hajri, T.; Han, X.X.; Bonen, A.; Abumrad, N.A. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J. Clin. Investig. 2002, 109, 1381–1389. [Google Scholar] [CrossRef]
- Goudriaan, J.R.; Dahlmans, V.E.; Teusink, B.; Ouwens, D.M.; Febbraio, M.; Maassen, J.A.; Romijn, J.A.; Havekes, L.M.; Voshol, P.J. CD36 deficiency increases insulin sensitivity in muscle, but induces insulin resistance in the liver in mice. J. Lipid Res. 2003, 44, 2270–2277. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Vance, D.E. Phosphatidylcholine and choline homeostasis. J. Lipid Res. 2008, 49, 1187–1194. [Google Scholar] [CrossRef] [Green Version]
- Corbin, K.D.; Zeisel, S.H. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr. Opin. Gastroenterol. 2012, 28, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, S.H.; Chan, M.L.; Marathe, G.K.; Parveen, F.; Chen, C.H.; Ke, L.Y. An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases. Int. J. Mol. Sci. 2019, 20, 1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakagami, H.; Aoki, J.; Natori, Y.; Nishikawa, K.; Kakehi, Y.; Arai, H. Biochemical and molecular characterization of a novel choline-specific glycerophosphodiester phosphodiesterase belonging to the nucleotide pyrophosphatase/phosphodiesterase family. J. Biol. Chem. 2005, 280, 23084–23093. [Google Scholar] [PubMed] [Green Version]
- Stefan, C.; Jansen, S.; Bollen, M. NPP-type ectophosphodiesterases: Unity in diversity. Trends Biochem. Sci. 2005, 30, 542–550. [Google Scholar] [CrossRef]
- Morita, J.; Kano, K.; Kato, K.; Takita, H.; Sakagami, H.; Yamamoto, Y.; Mihara, E.; Ueda, H.; Sato, T.; Tokuyama, H.; et al. Structure and biological function of ENPP6, a choline-specific glycerophosphodiester-phosphodiesterase. Sci. Rep. 2016, 6, 20995. [Google Scholar] [CrossRef] [Green Version]
- Hedtke, V.; Bakovic, M. Choline transport for phospholipid synthesis: An emerging role of choline transporter-like protein 1. Exp. Biol. Med. 2019, 244, 655–662. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.; Grapentine, S.; Ichhpuniani, J.; Bakovic, M. Choline transporter-like proteins 1 and 2 are newly identified plasma membrane and mitochondrial ethanolamine transporters. J. Biol. Chem. 2021, 296, 100604. [Google Scholar] [CrossRef] [PubMed]
- Noga, A.A.; Zhao, Y.; Vance, D.E. An unexpected requirement for phosphatidylethanolamine N-methyltransferase in the secretion of very low density lipoproteins. J. Biol. Chem. 2002, 277, 42358–42365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, R.L.; Zhao, Y.; Koonen, D.P.; Sletten, T.; Su, B.; Lingrell, S.; Cao, G.; Peake, D.A.; Kuo, M.S.; Proctor, S.D.; et al. Impaired de novo choline synthesis explains why phosphatidylethanolamine N-methyltransferase-deficient mice are protected from diet-induced obesity. J. Biol. Chem. 2010, 285, 22403–22413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, S.; Kuipers, F.; Havinga, R.; Ando, H.; Vance, D.E.; Jacobs, R.L.; van der Veen, J.N. Impaired Hepatic Phosphatidylcholine Synthesis Leads to Cholestasis in Mice Challenged With a High-Fat Diet. Hepatol. Commun. 2019, 3, 262–276. [Google Scholar] [CrossRef]
- Whyte, M.P. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann. N. Y. Acad. Sci. 2010, 1192, 190–200. [Google Scholar] [CrossRef]
- Coburn, S.P. Vitamin B-6 Metabolism and Interactions with TNAP. Subcell Biochem. 2015, 76, 207–238. [Google Scholar]
- Ikeda, K.; Maretich, P.; Kajimura, S. The Common and Distinct Features of Brown and Beige Adipocytes. Trends Endocrinol. Metab. 2018, 29, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Estève, D.; Galitzky, J.; Bouloumié, A.; Fonta, C.; Buchet, R.; Magne, D. Multiple Functions of MSCA-1/TNAP in Adult Mesenchymal Progenitor/Stromal Cells. Stem Cells Int. 2016, 2016, 1815982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Zhang, L.; Xuan, K.; Hu, C.; Li, L.; Zhang, Y.; Jin, F.; Jin, Y. Alkaline Phosphatase Controls Lineage Switching of Mesenchymal Stem Cells by Regulating the LRP6/GSK3β Complex in Hypophosphatasia. Theranostics 2018, 8, 5575–5592. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, L.; Xuan, K.; Hu, C.; Liu, S.; Liao, L.; Li, B.; Jin, F.; Shi, S.; Jin, Y. Alpl prevents bone ageing sensitivity by specifically regulating senescence and differentiation in mesenchymal stem cells. Bone Res. 2018, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Nam, H.K.; Crouch, S.; Hatch, N.E. Tissue Nonspecific Alkaline Phosphatase Function in Bone and Muscle Progenitor Cells: Control of Mitochondrial Respiration and ATP Production. Int. J. Mol. Sci. 2021, 22, 1140. [Google Scholar] [CrossRef]
- Estève, D.; Boulet, N.; Volat, F.; Zakaroff-Girard, A.; Ledoux, S.; Coupaye, M.; Decaunes, P.; Belles, C.; Gaits-Iacovoni, F.; Iacovoni, J.S.; et al. Human White and Brite Adipogenesis is Supported by MSCA1 and is Impaired by Immune Cells. Stem Cells 2014, 33, 1277–1291. [Google Scholar] [CrossRef]
- Ali, A.T.; Penny, C.B.; Paiker, J.E.; van Niekerk, C.; Smit, A.; Ferris, W.F.; Crowther, N.J. Alkaline phosphatase is involved in the control of adipogenesis in the murine preadipocyte cell line, 3T3-L1. Clin. Chim. Acta 2005, 354, 101–109. [Google Scholar] [CrossRef]
- Ali, A.T.; Penny, C.B.; Paiker, J.E.; Psaras, G.; Ikram, F.; Crowther, N.J. The effect of alkaline phosphatase inhibitors on intracellular lipid accumulation in preadipocytes isolated from human mammary tissue. Ann. Clin. Biochem. 2006, 43 Pt 3, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Mosqueira, C.; Velez-delValle, C.; Kuri-Harcuch, W. Tissue alkaline phosphatase is involved in lipid metabolism and gene expression and secretion of adipokines in adipocytes. Biochim. Biophys. Acta 2015, 1850, 2485–2496. [Google Scholar] [CrossRef] [PubMed]
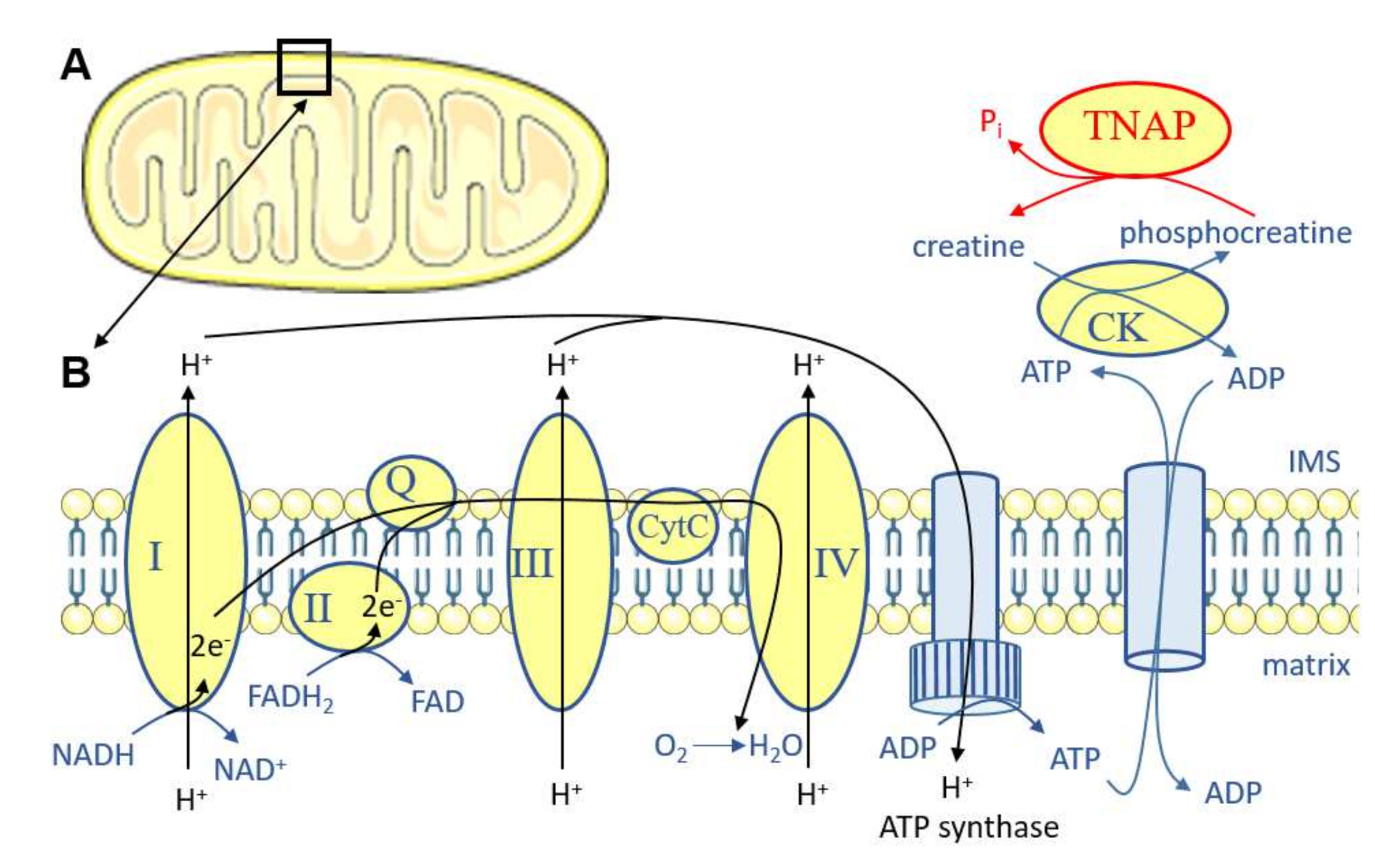
- Sun, Y.; Rahbani, J.F.; Jedrychowski, M.P.; Riley, C.L.; Vidoni, S.; Bogoslavski, D.; Hu, B.; Dumesic, P.A.; Zeng, X.; Wang, A.B.; et al. Mitochondrial TNAP controls thermogenesis by hydrolysis of phosphocreatine. Nature 2021, 593, 580–585. [Google Scholar] [CrossRef]
- Ikeda, K.; Yamada, T. UCP1 Dependent and Independent Thermogenesis in Brown and Beige Adipocytes. Front. Endocrinol. 2020, 11, 498. [Google Scholar] [CrossRef] [PubMed]
- Kazak, L.; Chouchani, E.T.; Jedrychowski, M.P.; Erickson, B.K.; Shinoda, K.; Cohen, P.; Vetrivelan, R.; Lu, G.Z.; Laznik-Bogoslavski, D.; Hasenfuss, S.C.; et al. A creatine-driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell 2015, 163, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahbani, J.F.; Roesler, A.; Hussain, M.F.; Samborska, B.; Dykstra, C.B.; Tsai, L.; Jedrychowski, M.P.; Vergnes, L.; Reue, K.; Spiegelman, B.M.; et al. Creatine kinase B controls futile creatine cycling in thermogenic fat. Nature 2021, 590, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.K.; Pinzón, C.; Huggins, S.; Pryor, J.H.; Falck, A.; Herman, F.; Oldeschulte, J.; Chavez, M.B.; Foster, B.L.; White, S.H.; et al. Genetic engineering a large animal model of human hypophosphatasia in sheep. Sci. Rep. 2018, 8, 16945. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Briolay, A.; Bessueille, L.; Magne, D. TNAP: A New Multitask Enzyme in Energy Metabolism. Int. J. Mol. Sci. 2021, 22, 10470. https://doi.org/10.3390/ijms221910470
Briolay A, Bessueille L, Magne D. TNAP: A New Multitask Enzyme in Energy Metabolism. International Journal of Molecular Sciences. 2021; 22(19):10470. https://doi.org/10.3390/ijms221910470
Chicago/Turabian StyleBriolay, Anne, Laurence Bessueille, and David Magne. 2021. "TNAP: A New Multitask Enzyme in Energy Metabolism" International Journal of Molecular Sciences 22, no. 19: 10470. https://doi.org/10.3390/ijms221910470