
Metformin Affects Olaparib Sensitivity through Induction of Apoptosis in Epithelial Ovarian Cancer Cell Lines


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
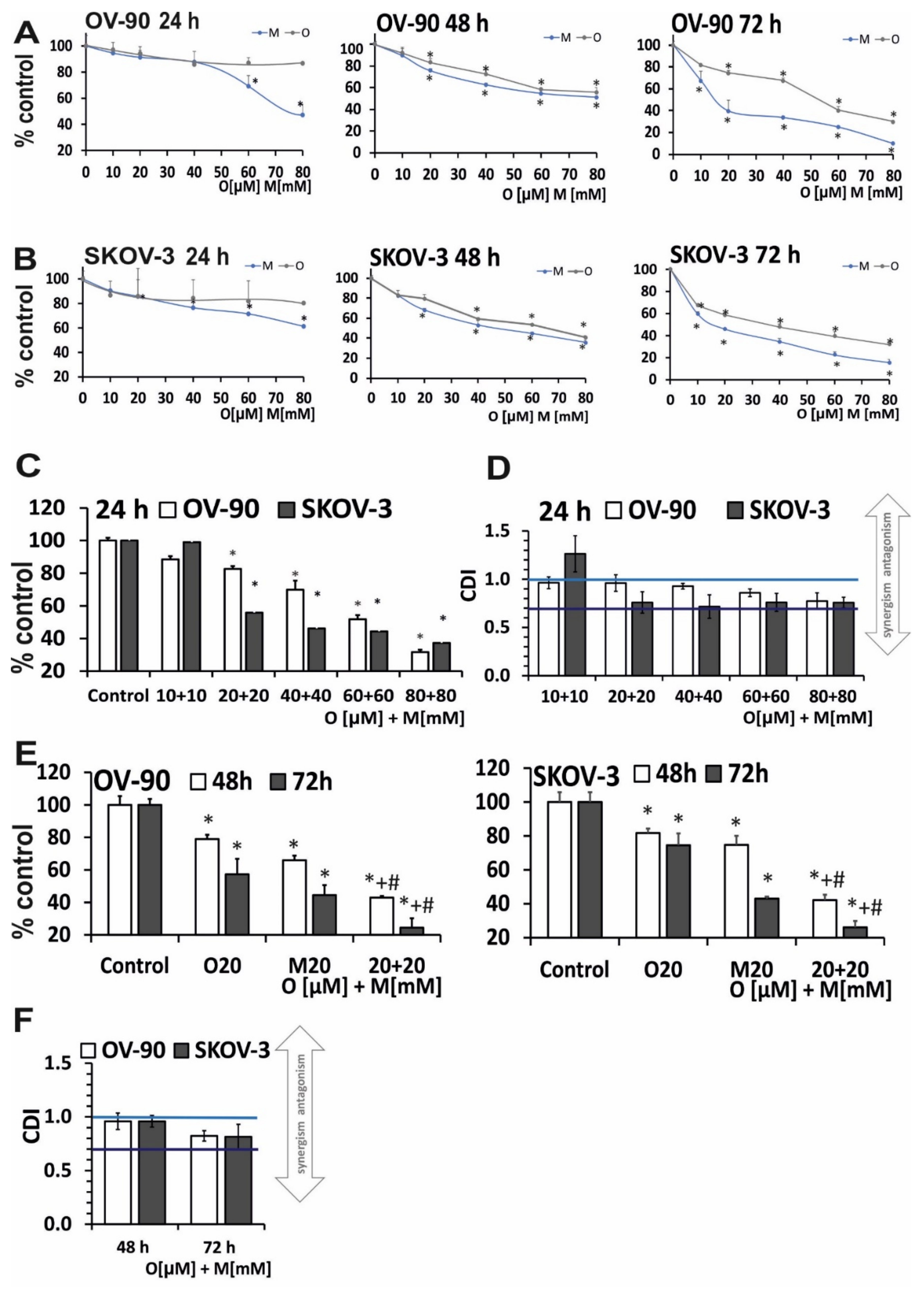
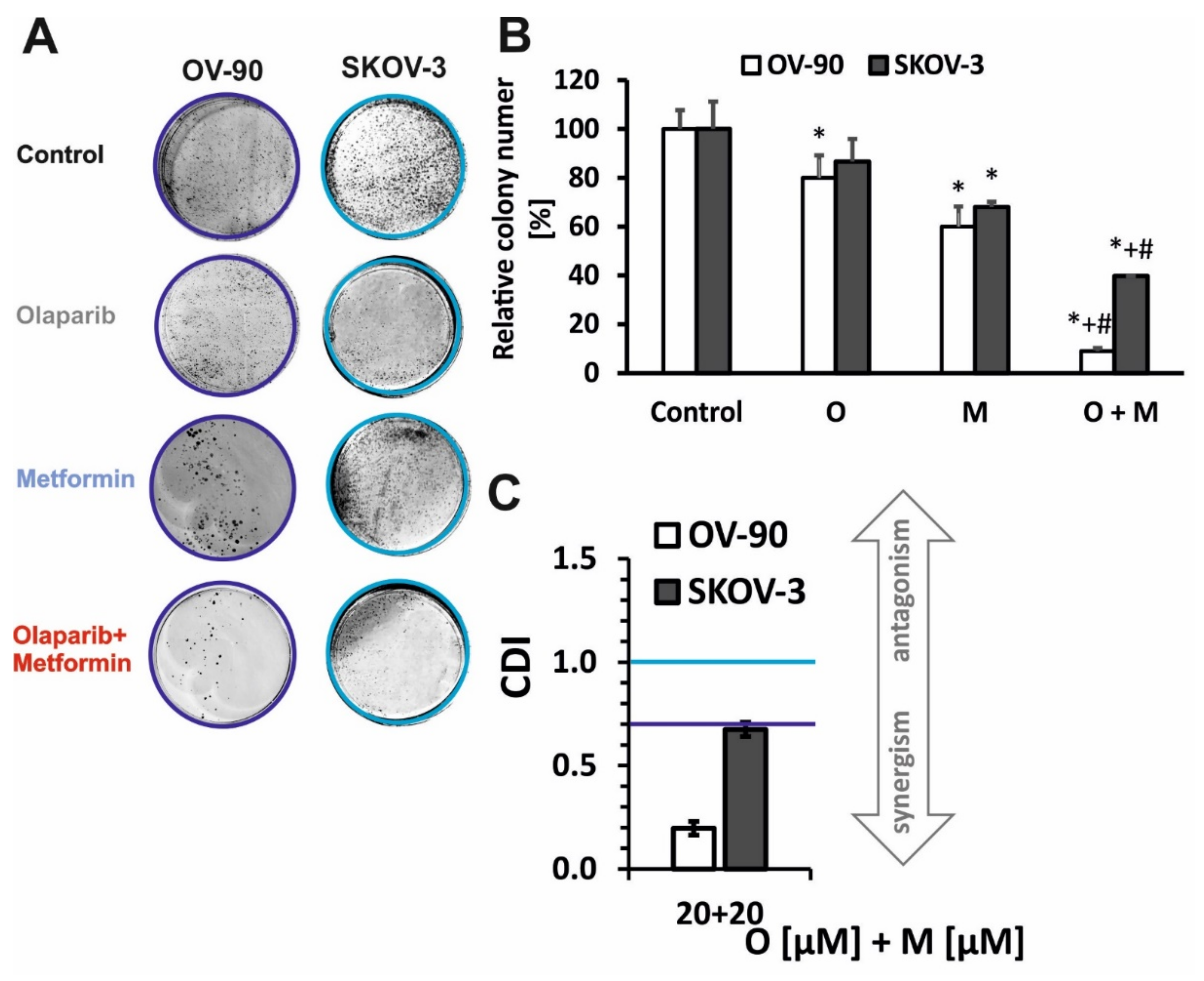
2.1. Olaparib and Metformin Reduce the Viability of Ovarian Cancer Cells
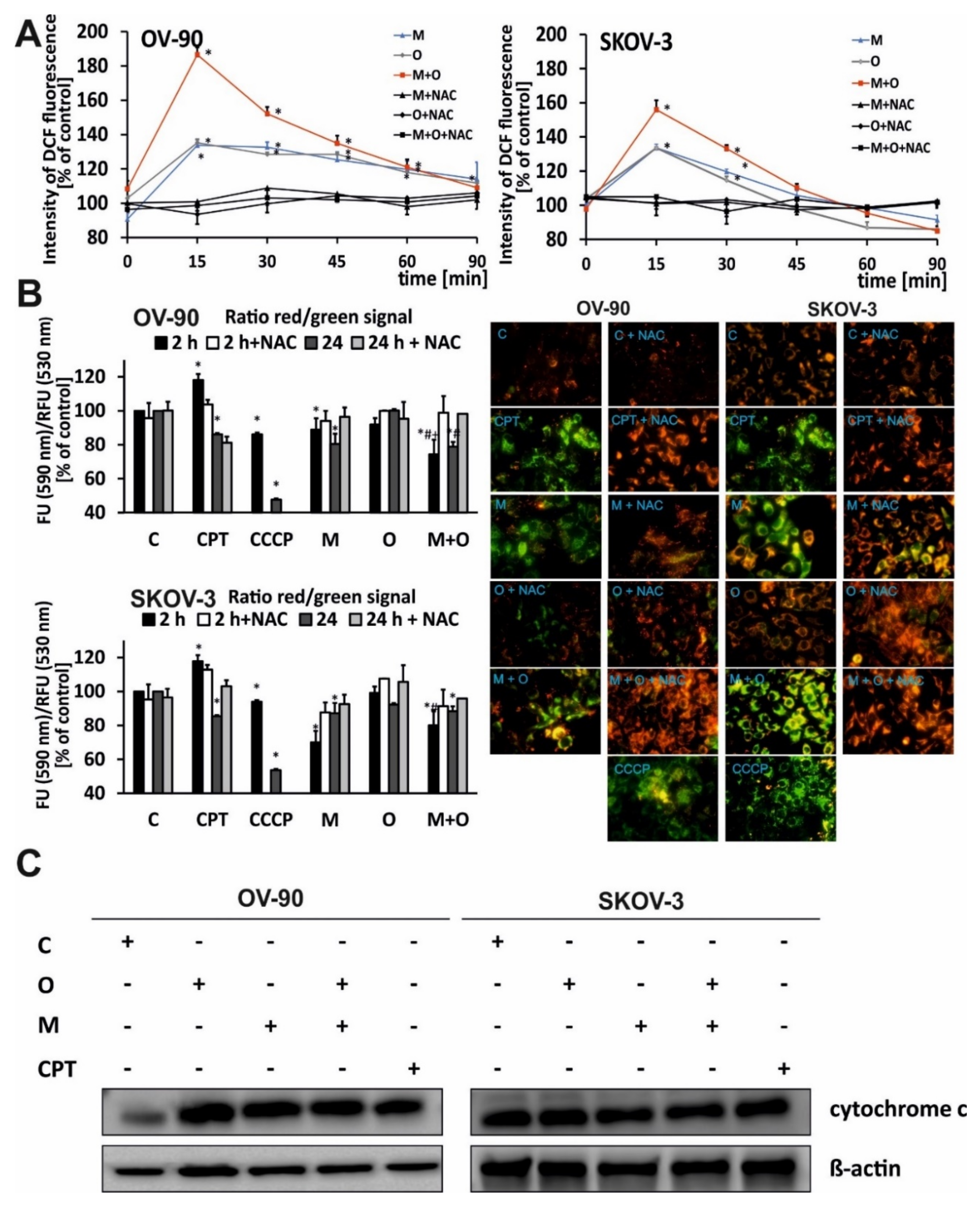
2.2. Treatment with Olaparib and Metformin Leads to Increased Oxidative Stress
2.3. The Combination of Metformin and Olaparib Modulate the Release of Cytochrome c and Causes loss of Mitochondrial Membrane Potential
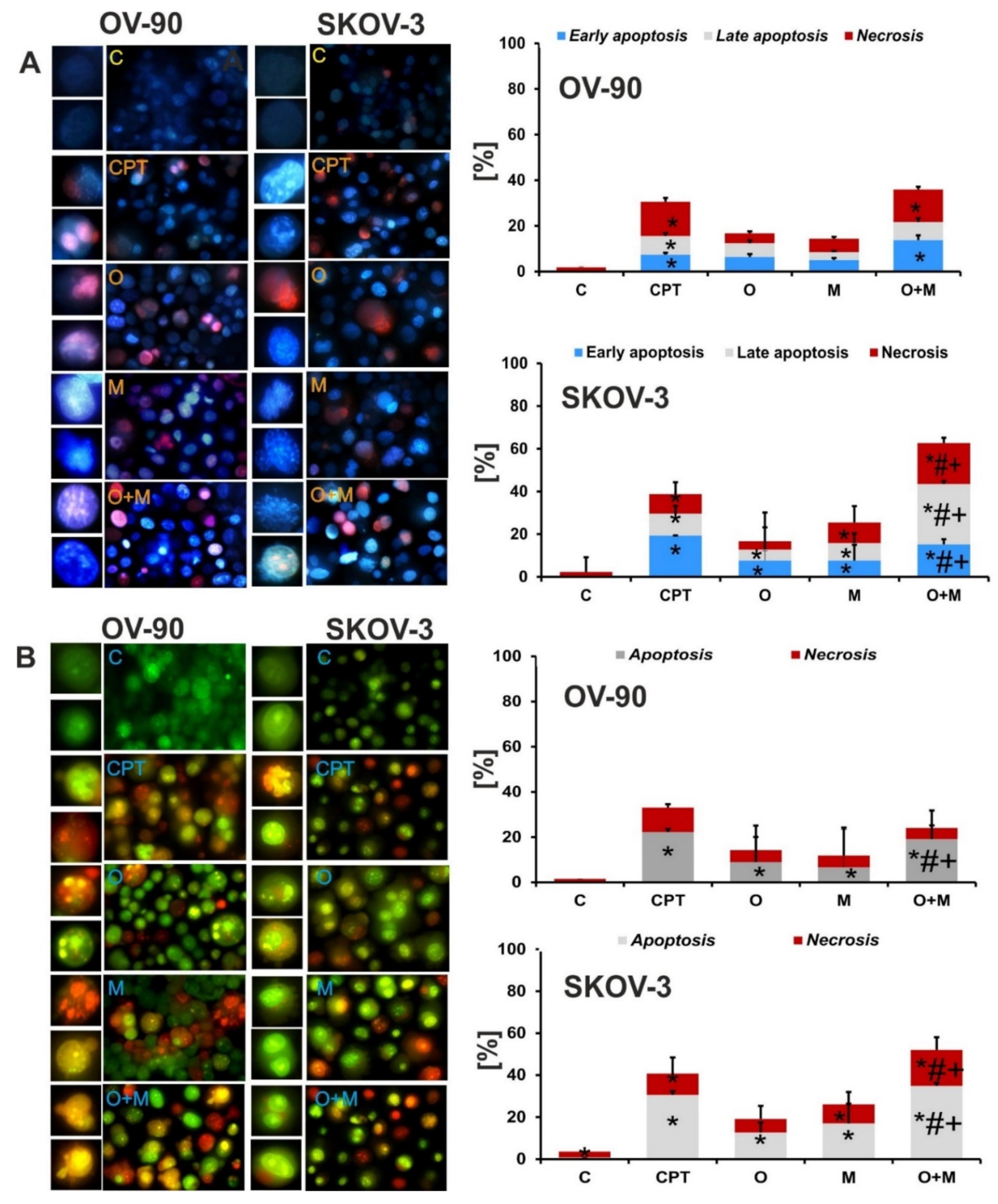
2.4. Metformin Increase the Sensitivity of Ovarian Cancer Cells to Olaparib—Assessment of Apoptosis and Necrosis by Double Staining
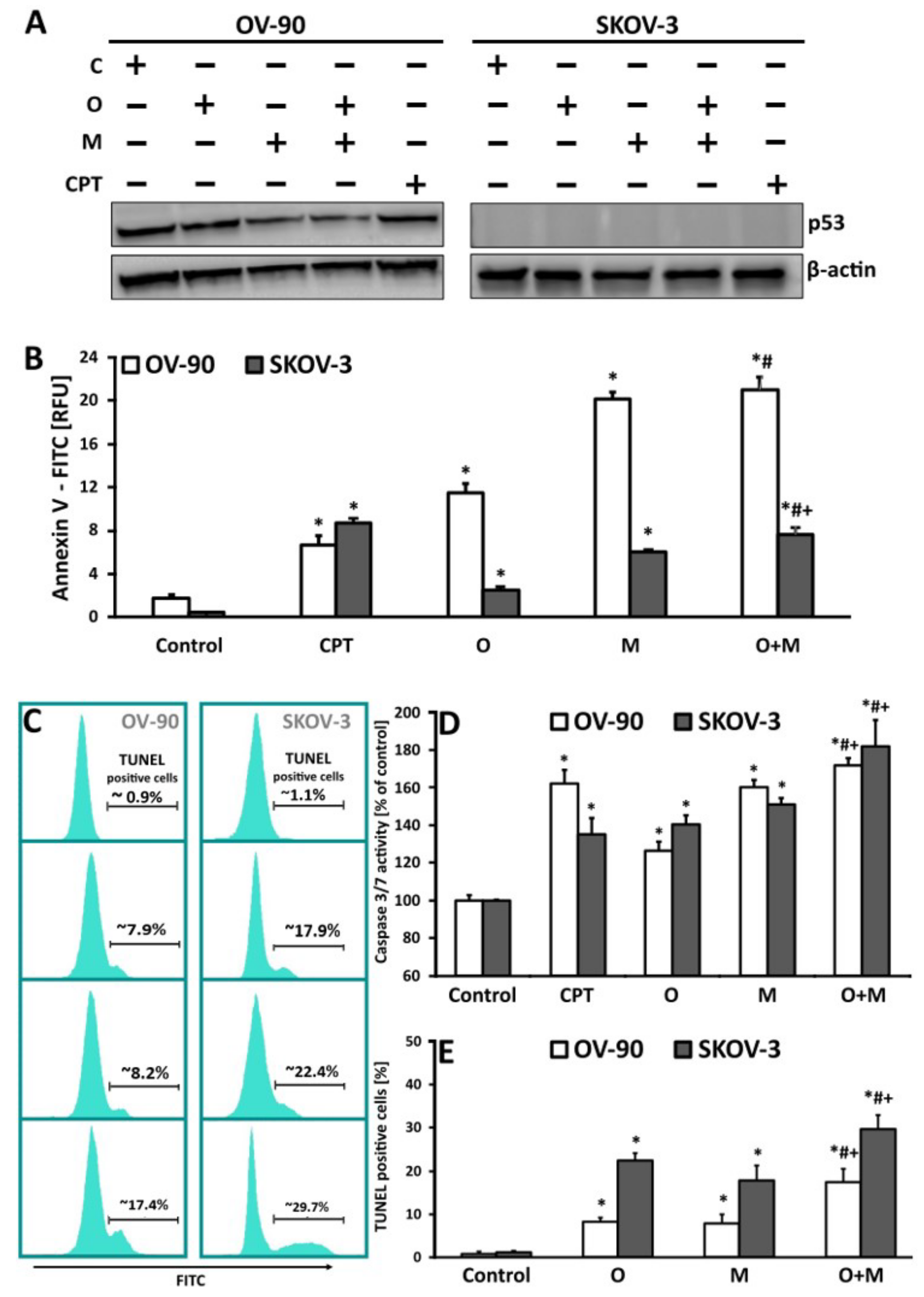
2.5. Metformin in Combination with Olaparib Increases Caspase 3/7 Dependent DNA Damage and Phosphatidylserine Externalization in Ovarian Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Drug Administration
4.3. MTT Assay
4.4. Clonogenic Assay
4.5. Measurement of ROS Production
4.6. Mitochondrial Membrane Potential (ΔΨm)
4.7. Morphological Assessment of Apoptosis and Necrosis—Double staining with Hoechst 3325 and Propidium Iodide and Double Staining with Orange Acridine and Ethidium Bromide
4.8. Caspase 3/7 Assay
4.9. Measurements of DNA Damage during Apoptosis—TUNEL Assay
4.10. Phosphatidylserine Externalization Measurement
4.11. Western Blot Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Momenimovahed, Z.; Tiznobaik, A.; Taheri, S.; Salehiniya, H. Ovarian cancer in the world: Epidemiology and risk factors. Int. J. Womens Health 2019, 11, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Budiana, I.N.G.; Angelina, M.; Pemayun, T.G.A. Ovarian cancer: Pathogenesis and current recommendations for prophylactic surgery. J. Turk. Ger. Gynecol. Assoc. 2019, 20, 47–54. [Google Scholar] [CrossRef]
- McGee, J.; Peart, T.M.; Foley, N.; Bertrand, M.; Prefontaine, M.; Sugimoto, A.; Ettler, H.; Welch, S.; Panabaker, K. Direct Genetics Referral Pathway for High-Grade Serous Ovarian Cancer Patients: The “Opt-Out” Process. J. Oncol. 2019, 2019, 6029097. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Buttarelli, M.; De Donato, M.; Raspaglio, G.; Babini, G.; Ciucci, A.; Martinelli, E.; Baccaro, P.; Pasciuto, T.; Fagotti, A.; Scambia, G.; et al. Clinical Value of lncRNA MEG3 in High-Grade Serous Ovarian Cancer. Cancers 2020, 12, 966. [Google Scholar] [CrossRef]
- van Zyl, B.; Tang, D.; Bowden, N.A. Biomarkers of platinum resistance in ovarian cancer: What can we use to improve treatment. Endocrine.-Relat. Cancer 2018, 25, R303–R318. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Li, Y.; Guo, Z.; Zeng, Y.; Zhang, W.; Wang, H. Metformin: Current clinical applications in nondiabetic patients with cancer. Aging 2020, 12, 3993–4009. [Google Scholar] [CrossRef]
- Cioce, M.; Pulito, C.; Strano, S.; Blandino, G.; Fazio, V.M. Metformin: Metabolic Rewiring Faces Tumor Heterogeneity. Cells 2020, 9, 2439. [Google Scholar] [CrossRef]
- Yin, G.; Liu, Z.; Wang, Y.; Sun, L.; Wang, L.; Yao, B.; Liu, R.; Chen, T.; Niu, Y.; Liu, Q. ZNF503 accelerates aggressiveness of hepatocellular carcinoma cells by down-regulation of GATA3 expression and regulated by microRNA-495. Am. J. Transl. Res. 2019, 11, 3426–3437. [Google Scholar]
- El Shorbagy, S.; abuTaleb, F.; Labib, H.A.; Ebian, H.; Harb, O.A.; Mohammed, M.S.; Rashied, H.A.; Elbana, K.A.; Haggag, R. Prognostic Significance of VEGF and HIF-1 α in Hepatocellular Carcinoma Patients Receiving Sorafenib Versus Metformin Sorafenib Combination. J. Gastrointest. Cancer 2020, 52, 269–279. [Google Scholar] [CrossRef]
- Orecchioni, S.; Reggiani, F.; Talarico, G.; Mancuso, P.; Calleri, A.; Gregato, G.; Labanca, V.; Noonan, D.M.; Dallaglio, K.; Albini, A.; et al. The biguanides metformin and phenformin inhibit angiogenesis, local and metastatic growth of breast cancer by targeting both neoplastic and microenvironment cells. Int. J. Cancer 2015, 136, E534–E544. [Google Scholar] [CrossRef]
- Petrushev, B.; Tomuleasa, C.; Soritau, O.; Aldea, M.; Pop, T.; Susman, S.; Kacso, G.; Berindan, I.; Irimie, A.; Cristea, V. Metformin plus PIAF combination chemotherapy for hepatocellular carcinoma. Exp. Oncol. 2012, 34, 17–24. [Google Scholar] [PubMed]
- Markowska, A.; Leracz-Jacczak, A. Metformin in cancer treatment. Curr. Gynecol. Oncol. 2018, 16, 46–49. [Google Scholar] [CrossRef]
- Baloch, T.; López-Ozuna, V.M.; Wang, Q.; Matanis, E.; Kessous, R.; Kogan, L.; Yasmeen, A.; Gotlieb, W.H. Sequential therapeutic targeting of ovarian Cancer harboring dysfunctional BRCA1. BMC Cancer 2019, 19, 44. [Google Scholar] [CrossRef] [PubMed]
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Klotz, D.M.; Wimberger, P. Overcoming PARP inhibitor resistance in ovarian cancer: What are the most promising strategies? Arch. Gynecol. Obstet. 2020, 302, 1087–1102. [Google Scholar] [CrossRef]
- Pellegrino, B.; Mateo, J.; Serra, V.; Balmaña, J. Controversies in oncology: Are genomic tests quantifying homologous recombination repair deficiency (HRD) useful for treatment decision making? ESMO Open 2019, 4, e000480. [Google Scholar] [CrossRef]
- Ahmad, A.; Lin, Z.P.; Zhu, Y.-L.; Lo, Y.-C.; Moscarelli, J.; Xiong, A.; Korayem, Y.; Huang, P.H.; Giri, S.; LoRusso, P.; et al. Combination of triapine, olaparib, and cediranib suppresses progression of BRCA-wild type and PARP inhibitor-resistant epithelial ovarian cancer. PLoS ONE 2018, 13, e0207399. [Google Scholar] [CrossRef]
- Randall, M.; Burgess, K.; Buckingham, L.; Usha, L. Exceptional Response to Olaparib in a Patient With Recurrent Ovarian Cancer and an Entire BRCA1 Germline Gene Deletion. J. Natl. Compr. Cancer Netw. 2020, 18, 223–228. [Google Scholar] [CrossRef]
- Carneiro, B.A.; Collier, K.A.; Nagy, R.J.; Pamarthy, S.; Sagar, V.; Fairclough, S.; Odegaard, J.; Lanman, R.B.; Costa, R.; Taxter, T.; et al. Acquired Resistance to Poly (ADP-ribose) Polymerase Inhibitor Olaparib in BRCA2-Associated Prostate Cancer Resulting From Biallelic BRCA2 Reversion Mutations Restores Both Germline and Somatic Loss-of-Function Mutations. JCO Precis. Oncol. 2018, 2, PO.17.00176. [Google Scholar] [CrossRef]
- Mengwasser, K.E.; Adeyemi, R.O.; Leng, Y.; Choi, M.Y.; Clairmont, C.; D’Andrea, A.D.; Elledge, S.J. Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol. Cell 2019, 73, 885–899.e6. [Google Scholar] [CrossRef]
- Kaelin, W.G. The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Gentric, G.; Kieffer, Y.; Mieulet, V.; Goundiam, O.; Bonneau, C.; Nemati, F.; Hurbain, I.; Raposo, G.; Popova, T.; Stern, M.H.; et al. PML-Regulated Mitochondrial Metabolism Enhances Chemosensitivity in Human Ovarian Cancers. Cell Metab. 2019, 29, 156–173.e10. [Google Scholar] [CrossRef]
- Wilson, A.J.; Stubbs, M.; Liu, P.; Ruggeri, B.; Khabele, D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol. Oncol. 2018, 149, 575–584. [Google Scholar] [CrossRef]
- Hurley, R.M.; Wahner Hendrickson, A.E.; Visscher, D.W.; Ansell, P.; Harrell, M.I.; Wagner, J.M.; Negron, V.; Goergen, K.M.; Maurer, M.J.; Oberg, A.L.; et al. 53BP1 as a potential predictor of response in PARP inhibitor-treated homologous recombination-deficient ovarian cancer. Gynecol. Oncol. 2019, 153, 127–134. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Sanij, E.; Hannan, K.M.; Xuan, J.; Yan, S.; Ahern, J.E.; Trigos, A.S.; Brajanovski, N.; Son, J.; Chan, K.T.; Kondrashova, O.; et al. CX-5461 activates the DNA damage response and demonstrates therapeutic efficacy in high-grade serous ovarian cancer. Nat. Commun. 2020, 11, 2641. [Google Scholar] [CrossRef]
- Loo, D.T. TUNEL assay. An overview of techniques. Methods Mol. Biol. 2002, 203, 21–30. [Google Scholar] [CrossRef]
- Hjortkjær, M.; Malik Aagaard Jørgensen, M.; Waldstrøm, M.; Ørnskov, D.; Søgaard-Andersen, E.; Jakobsen, A.; Dahl-Steffensen, K. The clinical importance of BRCAness in a population-based cohort of Danish epithelial ovarian cancer. Int. J. Gynecol. Cancer 2019, 29, 166–173. [Google Scholar] [CrossRef]
- Pizzo, S.V.; Dahmani, Z.; Addou-Klouche, L.; Gizard, F.; Dahou, S.; Messaoud, A.; Chahinez Djebri, N.; Benaissti, M.I.; Mostefaoui, M.; Terbeche, H.; et al. Metformin partially reverses the inhibitory effect of co-culture with ER-/PR-/HER2+ breast cancer cells on biomarkers of monocyte antitumor activity. PLoS ONE 2020, 15, e0240982. [Google Scholar] [CrossRef]
- Peuget, S.; Bonacci, T.; Soubeyran, P.; Iovanna, J.; Dusetti, N.J. Oxidative stress-induced p53 activity is enhanced by a redox-sensitive TP53INP1 SUMOylation. Cell Death Differ. 2014, 21, 1107–1118. [Google Scholar] [CrossRef]
- Hou, D.; Liu, Z.; Xu, X.; Liu, Q.; Zhang, X.; Kong, B.; Wei, J.-J.; Gong, Y.; Shao, C. Increased oxidative stress mediates the antitumor effect of PARP inhibition in ovarian cancer. Redox Biol. 2018, 17, 99–111. [Google Scholar] [CrossRef]
- Alhourani, A.H.; Tidwell, T.R.; Bokil, A.A.; Rosland, G.V.; Tronstad, K.J.; Soreide, K.; Hagland, H.R. Metformin treatment response is dependent on glucose growth conditions and metabolic phenotype in colorectal cancer cells. Sci. Rep. 2021, 11, 10487. [Google Scholar] [CrossRef]
- Ma, L.; Wei, J.; Wan, J.; Wang, W.; Wang, L.; Yuan, Y.; Yang, Z.; Liu, X.; Ming, L. Low glucose and metformin-induced apoptosis of human ovarian cancer cells is connected to ASK1 via mitochondrial and endoplasmic reticulum stress-associated pathways. J. Exp. Clin. Cancer Res. 2019, 38, 77. [Google Scholar] [CrossRef]
- Guo, E.; Ishii, Y.; Mueller, J.; Srivatsan, A.; Gahman, T.; Putnam, C.D.; Wang, J.Y.J.; Kolodner, R.D. FEN1 endonuclease as a therapeutic target for human cancers with defects in homologous recombination. Proc. Natl. Acad. Sci. USA 2020, 117, 19415–19424. [Google Scholar] [CrossRef]
- Beaufort, C.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van, I.W.F.; Heine, A.A.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed]
- Silwal-Pandit, L.; Langerod, A.; Borresen-Dale, A.L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026252. [Google Scholar] [CrossRef]
- Angeli, D.; Salvi, S.; Tedaldi, G. Genetic Predisposition to Breast and Ovarian Cancers: How Many and Which Genes to Test? Int. J. Mol. Sci. 2020, 21, 1128. [Google Scholar] [CrossRef]
- Cantor, S.B.; Guillemette, S. Hereditary breast cancer and the BRCA1-associated FANCJ/BACH1/BRIP1. Future Oncol. 2011, 7, 253–261. [Google Scholar] [CrossRef]
- Niraj, J.; Farkkila, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 Inhibition Increases Mitochondrial Metabolism through SIRT1 Activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Verhagen, C.V.M.; de Haan, R.; Hageman, F.; Oostendorp, T.P.D.; Carli, A.L.E.; O’Connor, M.J.; Jonkers, J.; Verheij, M.; van den Brekel, M.W.; Vens, C. Extent of radiosensitization by the PARP inhibitor olaparib depends on its dose, the radiation dose and the integrity of the homologous recombination pathway of tumor cells. Radiother. Oncol. 2015, 116, 358–365. [Google Scholar] [CrossRef]
- Giovannini, S.; Weller, M.-C.; Repmann, S.; Moch, H.; Jiricny, J. Synthetic lethality between BRCA1 deficiency and poly(ADP-ribose) polymerase inhibition is modulated by processing of endogenous oxidative DNA damage. Nucleic Acids Res. 2019, 47, 9132–9143. [Google Scholar] [CrossRef]
- Daugan, M.; Wojcicki, A.D.; d’Hayer, B.; Boudy, V. Metformin: An anti-diabetic drug to fight cancer. Pharmacol. Res. 2016, 113, 675–685. [Google Scholar] [CrossRef]
- Chan, D.K.; Miskimins, W. Metformin and phenethyl isothiocyanate combined treatment in vitro is cytotoxic to ovarian cancer cultures. J. Ovarian Res. 2012, 5, 19. [Google Scholar] [CrossRef]
- Ohnishi, S.; Mizutani, H.; Kawanishi, S. The enhancement of oxidative DNA damage by anti-diabetic metformin, buformin, and phenformin, via nitrogen-centered radicals. Free Radic. Res. 2016, 50, 929–937. [Google Scholar] [CrossRef]
- Lahiguera, Á.; Hyroššová, P.; Figueras, A.; Garzón, D.; Moreno, R.; Soto-Cerrato, V.; McNeish, I.; Serra, V.; Lazaro, C.; Barretina, P.; et al. Tumors defective in homologous recombination rely on oxidative metabolism: Relevance to treatments with PARP inhibitors. EMBO Mol. Med. 2020, 12, e11217. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef]
- Goh, A.M.; Coffill, C.R.; Lane, D.P. The role of mutant p53 in human cancer. J. Pathol. 2011, 223, 116–126. [Google Scholar] [CrossRef]
- Gu, B.; Zhu, W.-G. Surf the Post-translational Modification Network of p53 Regulation. Int. J. Biol. Sci. 2012, 8, 672–684. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Rosen, D.G.; Yang, G.; Liu, G.; Mercado-Uribe, I.; Chang, B.; Xiao, X.S.; Zheng, J.; Xue, F.X.; Liu, J. Ovarian cancer: Pathology, biology, and disease models. Front. Biosci. 2009, 14, 2089–2102. [Google Scholar] [CrossRef]
- Moll, U.M.; Wolff, S.; Speidel, D.; Deppert, W. Transcription-independent pro-apoptotic functions of p53. Current Opinion in Cell Biol. 2005, 17, 631–636. [Google Scholar] [CrossRef]
- Meng, X.; Bi, J.; Li, Y.; Yang, S.; Zhang, Y.; Li, M.; Liu, H.; Li, Y.; McDonald, M.; Thiel, K.; et al. AZD1775 Increases Sensitivity to Olaparib and Gemcitabine in Cancer Cells with p53 Mutations. Cancers 2018, 10, 149. [Google Scholar] [CrossRef]
- Langland, G.T.; Yannone, S.M.; Langland, R.A.; Nakao, A.; Guan, Y.; Long, S.B.; Vonguyen, L.; Chen, D.J.; Gray, J.W.; Chen, F. Radiosensitivity profiles from a panel of ovarian cancer cell lines exhibiting genetic alterations in p53 and disparate DNA-dependent protein kinase activities. Oncol. Rep. 2010, 23, 1021–1026. [Google Scholar] [CrossRef]
- Zhang, Z.; Alaee, M.; Danesh, G.; Pasdar, M. Plakoglobin Reduces the in vitro Growth, Migration and Invasion of Ovarian Cancer Cells Expressing N-Cadherin and Mutant p53. PLoS ONE 2016, 11, e0154323. [Google Scholar] [CrossRef]
- Patel, S.; Singh, N.; Kumar, L. Anticancer role of antidiabetic drug Metformin in ovarian cancer cells. Int. J. Cancer Ther. Oncol. 2016, 4, 427. [Google Scholar] [CrossRef][Green Version]
- Park, E.Y.; Woo, Y.; Kim, S.J.; Kim, D.H.; Lee, E.K.; De, U.; Kim, K.S.; Lee, J.; Jung, J.H.; Ha, K.-T.; et al. Anticancer Effects of a New SIRT Inhibitor, MHY2256, against Human Breast Cancer MCF-7 Cells via Regulation of MDM2-p53 Binding. Int. J. Biol. Sci. 2016, 12, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, K.; Zhu, Y.L.; Lin, Z.P.; Penketh, P.G.; Shyam, K.; Zhu, R.; Baumann, R.P.; Sartorelli, A.C.; Rutherford, T.J.; Ratner, E.S. Cataloging antineoplastic agents according to their effectiveness against platinum-resistant and platinum-sensitive ovarian carcinoma cell lines. J. Transl. Sci. 2016, 2, 117–124. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, M.; Tichet, M.; Abbe, P.; Ohanna, M.; Lehraiki, A.; Rouaud, F.; Allegra, M.; Giacchero, D.; Bahadoran, P.; Bertolotto, C.; et al. Metformin Blocks Melanoma Invasion and Metastasis Development in AMPK/p53-Dependent Manner. Mol. Cancer Ther. 2013, 12, 1605–1615. [Google Scholar] [CrossRef]
- Gu, J.J.; Zhang, Q.; Mavis, C.; Czuczman, M.S.; Hernandez-Ilizaliturri, F.J. Metformin Induces p53-Dependent Mitochondrial Stress in Therapy-Sensitive and -Resistant Lymphoma Pre-Clinical Model and Primary Patients Sample with B-Cell Non-Hodgkin Lymphoma (NHL). Blood 2015, 126, 4008. [Google Scholar] [CrossRef]
- Smeby, J.; Kryeziu, K.; Berg, K.C.G.; Eilertsen, I.A.; Eide, P.W.; Johannessen, B.; Guren, M.G.; Nesbakken, A.; Bruun, J.; Lothe, R.A.; et al. Molecular correlates of sensitivity to PARP inhibition beyond homologous recombination deficiency in pre-clinical models of colorectal cancer point to wild-type TP53 activity. EBioMedicine 2020, 59, 102923. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, J.; Zhou, J.; Liu, H.; Xu, C. Olaparib induced senescence under P16 or P53 dependent manner in ovarian cancer. J. Gynecol. Oncol. 2019, 30, e26. [Google Scholar] [CrossRef]
- Saini, N.; Yang, X. Metformin as an anti-cancer agent: Actions and mechanisms targeting cancer stem cells. Acta Biochim. Biophys. Sin. 2018, 50, 133–143. [Google Scholar] [CrossRef]
- Parsels, L.A.; Karnak, D.; Parsels, J.D.; Zhang, Q.; Velez-Padilla, J.; Reichert, Z.R.; Wahl, D.R.; Maybaum, J.; O’Connor, M.J.; Lawrence, T.S.; et al. PARP1 Trapping and DNA Replication Stress Enhance Radiosensitization with Combined WEE1 and PARP Inhibitors. Mol. Cancer Res. 2018, 16, 222–232. [Google Scholar] [CrossRef]
- Valdez, B.C.; Li, Y.; Murray, D.; Liu, Y.; Nieto, Y.; Champlin, R.E.; Andersson, B.S. The PARP inhibitor olaparib enhances the cytotoxicity of combined gemcitabine, busulfan and melphalan in lymphoma cells. Leuk Lymphoma 2017, 58, 2705–2716. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, E.; Lv, N.; Ma, L.; Yao, S.; Yan, M.; Zi, F.; Deng, G.; Liu, X.; He, J.; et al. Metformin and FTY720 Synergistically Induce Apoptosis in Multiple Myeloma Cells. Cell. Physiol. Biochem. 2018, 48, 785–800. [Google Scholar] [CrossRef]
- Patel, S.; Kumar, L.; Singh, N. Metformin and epithelial ovarian cancer therapeutics. Cell. Oncol. 2015, 38, 365–375. [Google Scholar] [CrossRef]
- Siraj, A.K.; Pratheeshkumar, P.; Parvathareddy, S.K.; Divya, S.P.; Al-Dayel, F.; Tulbah, A.; Ajarim, D.; Al-Kuraya, K.S. Overexpression of PARP is an independent prognostic marker for poor survival in Middle Eastern breast cancer and its inhibition can be enhanced with embelin co-treatment. Oncotarget 2018, 9, 37319–37332. [Google Scholar] [CrossRef] [PubMed]
- Passaro, C.; Volpe, M.; Botta, G.; Scamardella, E.; Perruolo, G.; Gillespie, D.; Libertini, S.; Portella, G. PARP inhibitor olaparib increases the oncolytic activity of dl922-947 in in vitro and in vivo model of anaplastic thyroid carcinoma. Mol. Oncol. 2015, 9, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, G.K.; Russell, W.K.; Garg, N.J.; Yin, Y.W. Poly(ADP-ribose) polymerase 1 regulates mitochondrial DNA repair in an NAD-dependent manner. J. Biol. Chem. 2021, 296, 100309. [Google Scholar] [CrossRef]
- Cseh, A.M.; Fabian, Z.; Quintana-Cabrera, R.; Szabo, A.; Eros, K.; Soriano, M.E.; Gallyas, F.; Scorrano, L.; Sumegi, B. PARP Inhibitor PJ34 Protects Mitochondria and Induces DNA-Damage Mediated Apoptosis in Combination With Cisplatin or Temozolomide in B16F10 Melanoma Cells. Front. Physiol. 2019, 10, 538. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; López-Ozuna, V.M.; Baloch, T.; Bithras, J.; Amin, O.; Kessous, R.; Kogan, L.; Laskov, I.; Yasmeen, A. Biguanides in combination with olaparib limits tumorigenesis of drug-resistant ovarian cancer cells through inhibition of Snail. Cancer Med. 2019, 9, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Guo, J.; Zhu, Y.; Huang, Z.; Liu, T.; Cai, J.; Yu, L.; Wang, Z. Metformin inhibits ovarian cancer via decreasing H3K27 trimethylation. Int. J. Oncol. 2018, 52, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.-s.; Kim, B.; Gwak, H.; Suh, D.H.; Song, Y.S. Autophagy and protein kinase RNA-like endoplasmic reticulum kinase (PERK)/eukaryotic initiation factor 2 alpha kinase (eIF2α) pathway protect ovarian cancer cells from metformin-induced apoptosis. Mol. Carcinog. 2016, 55, 346–356. [Google Scholar] [CrossRef]
- Dale Rein, I.; Solberg Landsverk, K.; Micci, F.; Patzke, S.; Stokke, T. Replication-induced DNA damage after PARP inhibition causes G2 delay, and cell line-dependent apoptosis, necrosis and multinucleation. Cell Cycle 2015, 14, 3248–3260. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhou, F.; Jiang, F.; Lu, H.; Wang, J.; Cheng, C. PARP inhibitor reduces proliferation and increases apoptosis in breast cancer cells. Chin. J. Cancer Res. 2014, 26, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhu, J.; Zhang, H.; Liu, Y.; Sun, H. Metformin inhibits ovarian cancer growth and migration in vitro and in vivo by enhancing cisplatin cytotoxicity. Am. J. Transl. Res. 2018, 10, 3086–3098. [Google Scholar] [PubMed]
- Sahin, I.D.; Jönsson, J.-M.; Hedenfalk, I. Crizotinib and PARP inhibitors act synergistically by triggering apoptosis in high-grade serous ovarian cancer. Oncotarget 2019, 10, 6981–6996. [Google Scholar] [CrossRef] [PubMed]
- Hijaz, M.; Chhina, J.; Dar, S.; Tebbe, C.; Al-Wahab, Z.; Hanna, R.K.; Rattan, R.; Munkarah, A.R. Synthetic lethality of PARP inhibitors and metformin in BRCA1 intact ovarian cancer. Gynecol. Oncol. 2015, 137, 2–91. [Google Scholar] [CrossRef]
- Musacchio, L.; Caruso, G.; Pisano, C.; Cecere, S.C.; Di Napoli, M.; Attademo, L.; Tambaro, R.; Russo, D.; Califano, D.; Palaia, I.; et al. PARP Inhibitors in Endometrial Cancer: Current Status and Perspectives. Cancer Manag. Res. 2020, 12, 6123–6135. [Google Scholar] [CrossRef]
- Han, Y.; Li, C.W.; Hsu, J.M.; Hsu, J.L.; Chan, L.C.; Tan, X.; He, G.J. Metformin reverses PARP inhibitors-induced epithelial-mesenchymal transition and PD-L1 upregulation in triple-negative breast cancer. Am. J. Cancer Res. 2019, 9, 800–815. [Google Scholar]
- Gralewska, P.; Gajek, A.; Marczak, A.; Mikula, M.; Ostrowski, J.; Sliwinska, A.; Rogalska, A. PARP Inhibition Increases the Reliance on ATR/CHK1 Checkpoint Signaling Leading to Synthetic Lethality-An Alternative Treatment Strategy for Epithelial Ovarian Cancer Cells Independent from HR Effectiveness. Int. J. Mol. Sci. 2020, 21, 9715. [Google Scholar] [CrossRef]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [Google Scholar]
- Lopez-Acevedo, M.; Grace, L.; Teoh, D.; Whitaker, R.; Adams, D.J.; Jia, J.; Nixon, A.B.; Secord, A.A. Dasatinib (BMS-35482) potentiates the activity of gemcitabine and docetaxel in uterine leiomyosarcoma cell lines. Gynecol. Oncol. Res. Pract. 2014, 1, 2. [Google Scholar] [CrossRef][Green Version]
- Rogalska, A.; Gajek, A.; Lukawska, M.; Oszczapowicz, I.; Marczak, A. Novel oxazolinoanthracyclines as tumor cell growth inhibitors-Contribution of autophagy and apoptosis in solid tumor cells death. PLoS ONE 2018, 13, e0201296. [Google Scholar] [CrossRef] [PubMed]
- Rogalska, A.; Marczak, A.; Gajek, A.; Szwed, M.; Sliwinska, A.; Drzewoski, J.; Jozwiak, Z. Induction of apoptosis in human ovarian cancer cells by new anticancer compounds, epothilone A and B. Toxicol. In Vitro 2013, 27, 239–249. [Google Scholar] [CrossRef]
- Rogalska, A.; Gajek, A.; Marczak, A. Suppression of autophagy enhances preferential toxicity of epothilone A and epothilone B in ovarian cancer cells. Phytomedicine 2019, 61, 152847. [Google Scholar] [CrossRef]
- Rogalska, A.; Gajek, A.; Marczak, A. Epothilone B induces extrinsic pathway of apoptosis in human SKOV-3 ovarian cancer cells. Toxicol. In Vitro 2014, 28, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Gajek, A.; Poczta, A.; Łukawska, M.; Cecuda- Adamczewska, V.; Tobiasz, J.; Marczak, A. Chemical modification of melphalan as a key to improving treatment of haematological malignancies. Sci. Rep. 2020, 10, 4479. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gralewska, P.; Gajek, A.; Marczak, A.; Rogalska, A. Metformin Affects Olaparib Sensitivity through Induction of Apoptosis in Epithelial Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2021, 22, 10557. https://doi.org/10.3390/ijms221910557
Gralewska P, Gajek A, Marczak A, Rogalska A. Metformin Affects Olaparib Sensitivity through Induction of Apoptosis in Epithelial Ovarian Cancer Cell Lines. International Journal of Molecular Sciences. 2021; 22(19):10557. https://doi.org/10.3390/ijms221910557
Chicago/Turabian StyleGralewska, Patrycja, Arkadiusz Gajek, Agnieszka Marczak, and Aneta Rogalska. 2021. "Metformin Affects Olaparib Sensitivity through Induction of Apoptosis in Epithelial Ovarian Cancer Cell Lines" International Journal of Molecular Sciences 22, no. 19: 10557. https://doi.org/10.3390/ijms221910557
APA StyleGralewska, P., Gajek, A., Marczak, A., & Rogalska, A. (2021). Metformin Affects Olaparib Sensitivity through Induction of Apoptosis in Epithelial Ovarian Cancer Cell Lines. International Journal of Molecular Sciences, 22(19), 10557. https://doi.org/10.3390/ijms221910557