
Inhibition of NHE-1 Increases Smoke-Induced Proliferative Activity of Barrett’s Esophageal Cell Line

 ,
,  , ,
, ,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
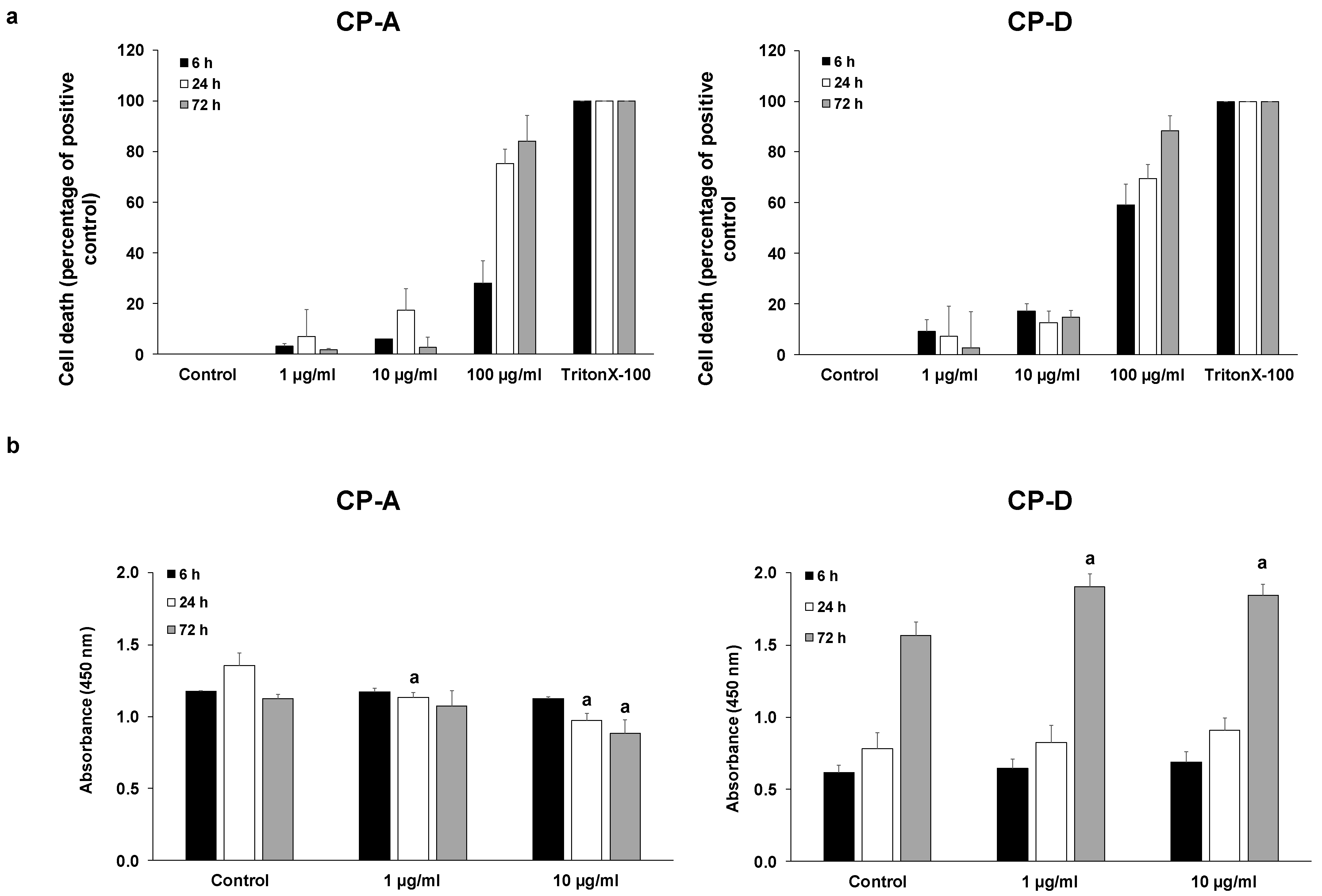
2.1. Effect of CSE on Esophageal Epithelial Cell Proliferation
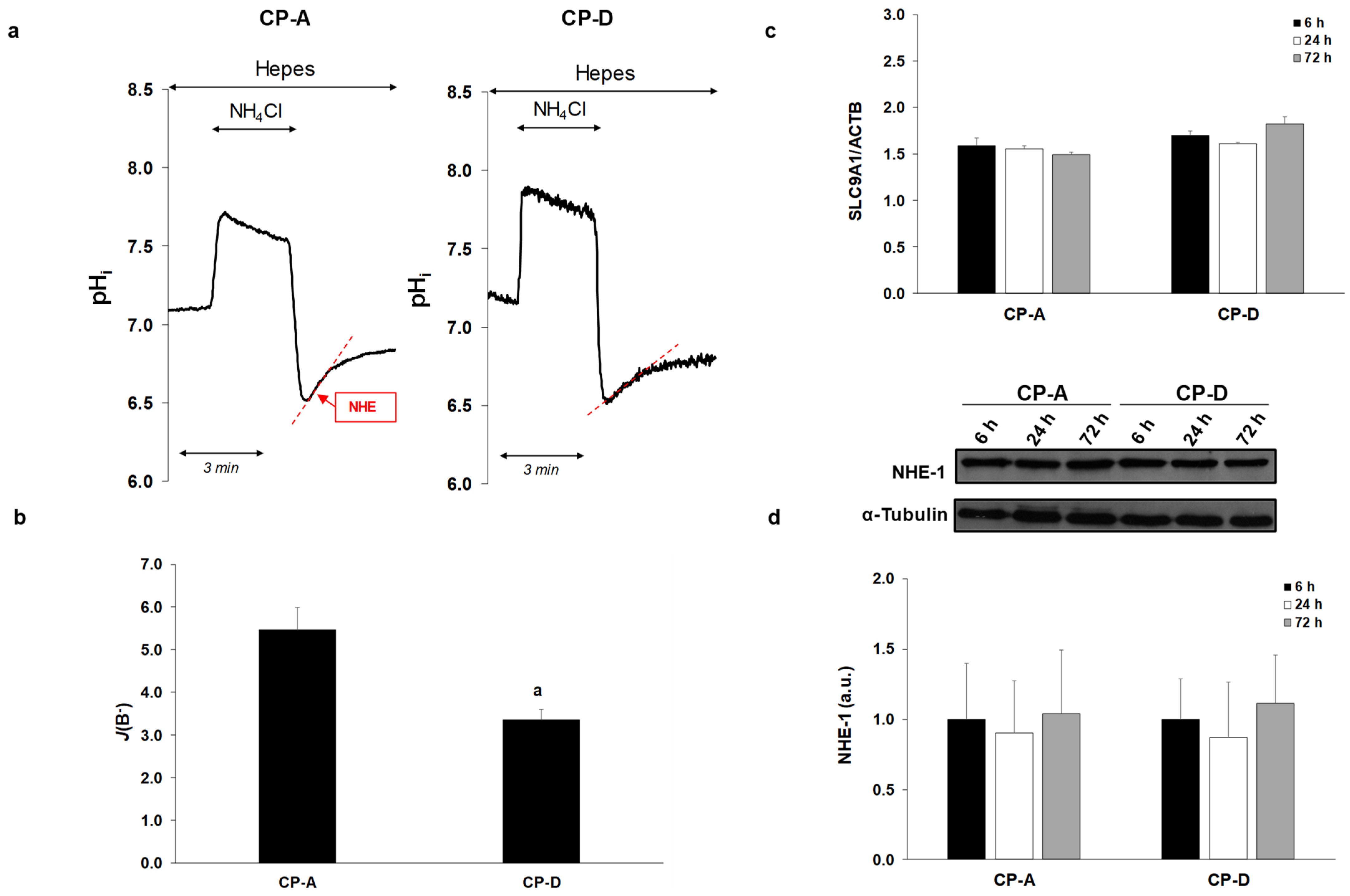
2.2. Activity and Expression of NHE-1 in the Metaplastic and Dysplastic Cells
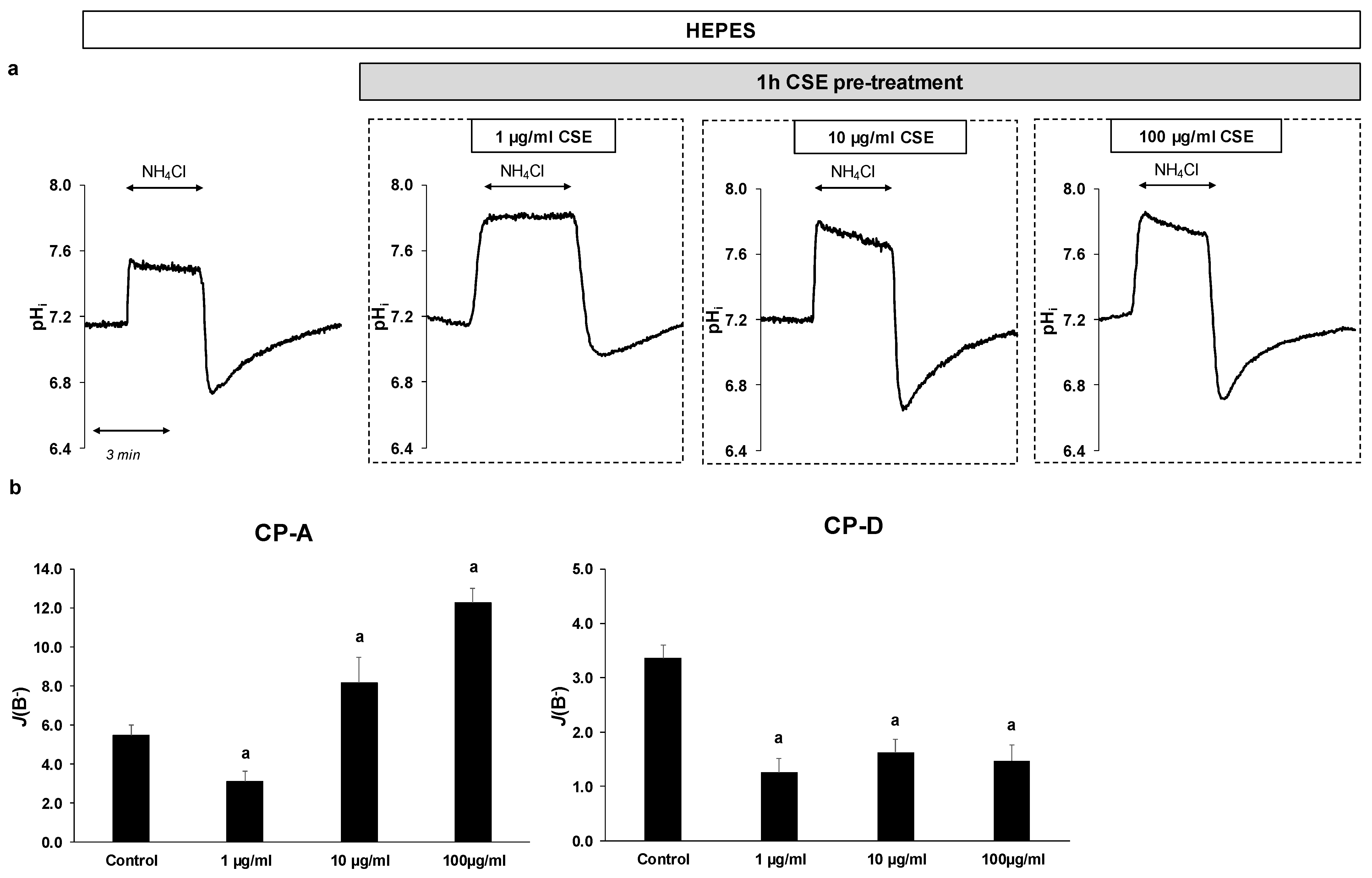
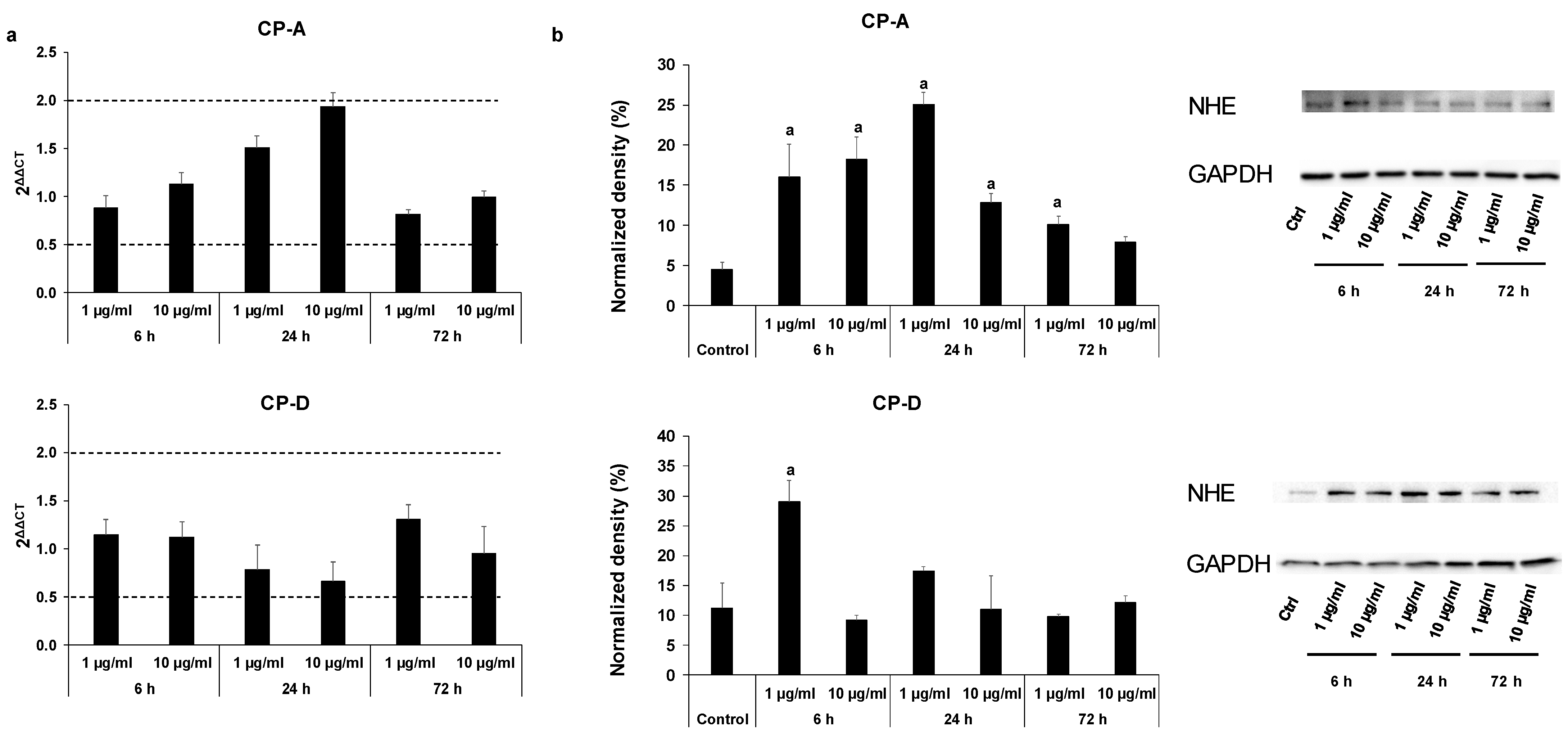
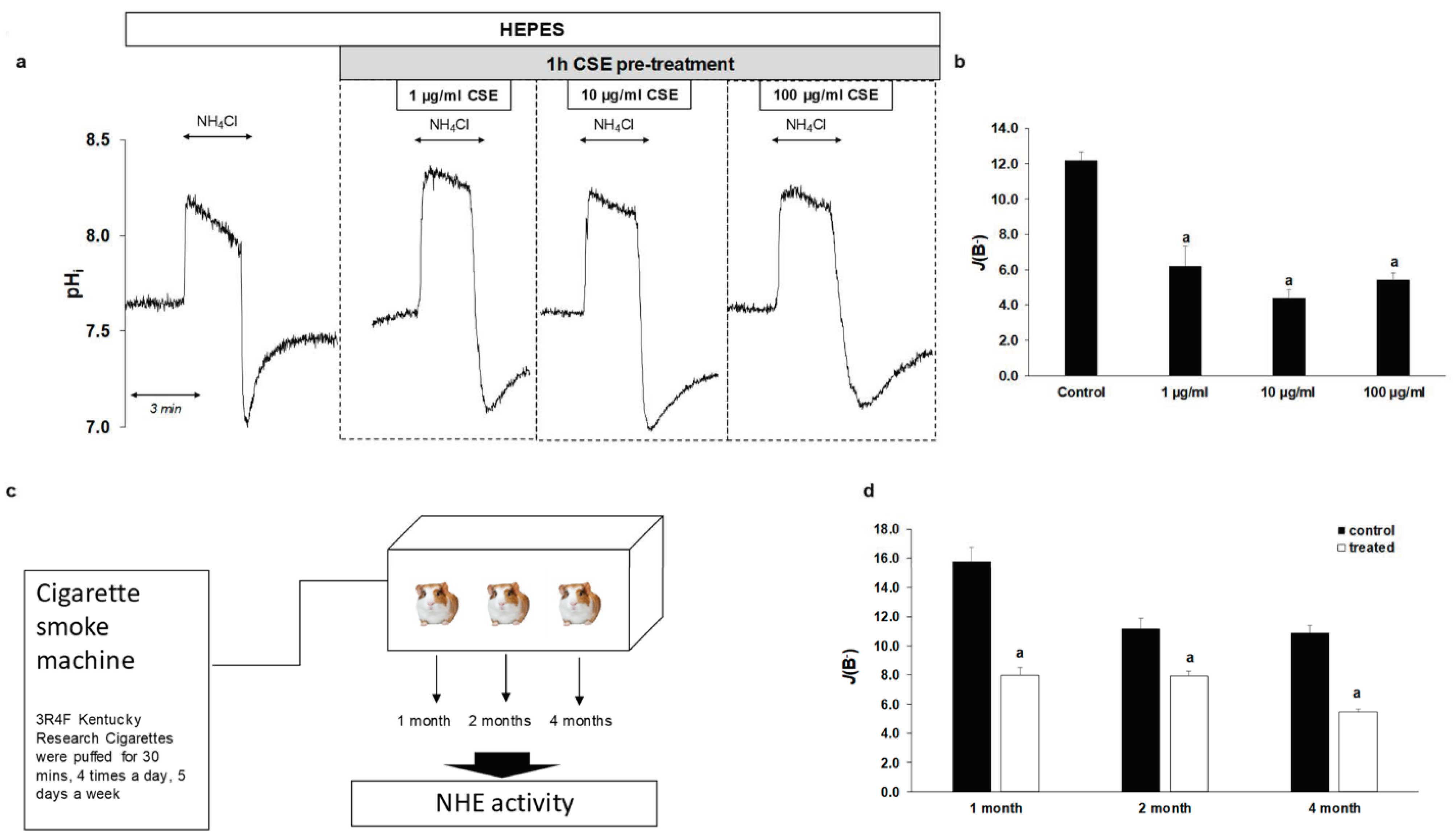
2.3. Effect of CSE on The Activity and Expression of NHE-1
2.4. Smoking Decreases NHE-1 Activity on Normal Esophageal Epithelial Cells
2.5. Effect of Smoking on NHE-1 Protein Expression in Human Esophageal Samples
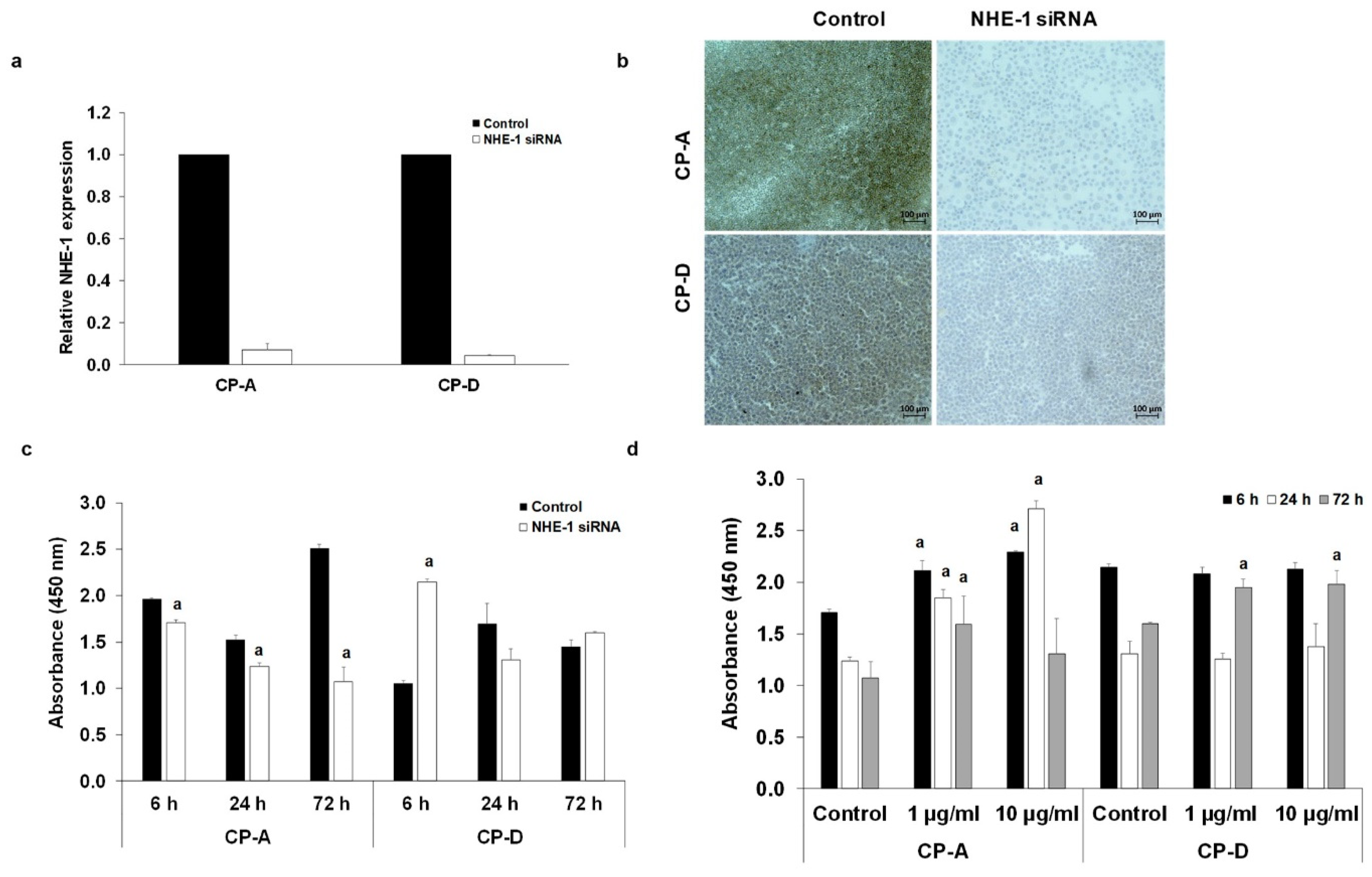
2.6. Role of NHE-1 in The CSE-Induced Proliferation
3. Discussion
4. Materials and Methods
4.1. Chemicals and Solutions
4.2. Animals
4.3. Patients
4.4. Cell Cultures
4.5. Preparation of Cigarette-Smoke Extract
4.6. Cigarette-Smoke Exposure
4.7. Isolation of Guinea Pig Esophageal Epithelial Cells
4.8. Immunohistochemistry
4.9. Quantitative Real-Time PCR Analysis
4.10. Western Blot
4.11. Measurement of Intracellular pH
4.12. Determination of Buffering Capacity
4.13. Measurement of Na+/H+ Exchanger Activity
4.14. SLC9A1 Gene Silencing
4.15. Proliferation
4.16. Cytotoxicity Assay
4.17. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Cook, M.B.; Kamangar, F.; Whiteman, D.C.; Freedman, N.D.; Gammon, M.D.; Bernstein, L.; Brown, L.M.; Risch, H.A.; Ye, W.; Sharp, L.; et al. Cigarette smoking and adenocarcinomas of the esophagus and esophagogastric junction: A pooled analysis from the international BEACON consortium. J. Natl. Cancer Inst. 2010, 102, 1344–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, M.B.; Shaheen, N.J.; Anderson, L.A.; Giffen, C.; Chow, W.H.; Vaughan, T.L.; Whiteman, D.C.; Corley, D.A. Cigarette smoking increases risk of Barrett’s esophagus: An analysis of the Barrett’s and Esophageal Adenocarcinoma Consortium. Gastroenterology 2012, 142, 744–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, J.J.; Jiang, Z.M.; Chen, Y.X.; Ye, W.P.; Yang, Q.; Wang, H.Z.; Xie, D.R. Smoking Exposure and Survival of Patients with Esophagus Cancer: A Systematic Review and Meta-Analysis. Gastroenterol. Res. Pract. 2016, 2016, 7682387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.L.; Xie, S.H.; Li, W.T.; Lagergren, J. Smoking Cessation and Risk of Esophageal Cancer by Histological Type: Systematic Review and Meta-analysis. J. Natl. Cancer Inst. 2017, 109, djx115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, H.G.; Bhat, S.; Johnston, B.T.; McManus, D.; Gavin, A.T.; Murray, L.J. Tobacco smoking increases the risk of high-grade dysplasia and cancer among patients with Barrett’s esophagus. Gastroenterology 2012, 142, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, R.C.; Bryson, J.C.; Powell, D.W. Effect of cigarette smoke on esophageal epithelium of the rabbit. Gastroenterology 1986, 91, 1536–1542. [Google Scholar] [CrossRef]
- Becskehazi, E.; Korsos, M.M.; Eross, B.; Hegyi, P.; Venglovecz, V. OEsophageal Ion Transport Mechanisms and Significance Under Pathological Conditions. Front. Physiol. 2020, 11, 855. [Google Scholar] [CrossRef]
- Laczko, D.; Rosztoczy, A.; Birkas, K.; Katona, M.; Rakonczay, Z., Jr.; Tiszlavicz, L.; Roka, R.; Wittmann, T.; Hegyi, P.; Venglovecz, V. Role of ion transporters in the bile acid-induced esophageal injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G16–G31. [Google Scholar] [CrossRef] [Green Version]
- Demaurex, N.; Grinstein, S. Na+/H+ antiport: Modulation by ATP and role in cell volume regulation. J. Exp. Biol. 1994, 196, 389–404. [Google Scholar] [CrossRef]
- Grinstein, S.; Rotin, D.; Mason, M.J. Na+/H+ exchange and growth factor-induced cytosolic pH changes. Role in cellular proliferation. Biochim. Biophys. Acta 1989, 988, 73–97. [Google Scholar] [CrossRef]
- Stock, C.; Schwab, A. Role of the Na/H exchanger NHE1 in cell migration. Acta Physiol. 2006, 187, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Shallat, S.; Schmidt, L.; Reaka, A.; Rao, D.; Chang, E.B.; Rao, M.C.; Ramaswamy, K.; Layden, T.J. NHE-1 isoform of the Na+/H+ antiport is expressed in the rat and rabbit esophagus. Gastroenterology 1995, 109, 1421–1428. [Google Scholar] [CrossRef]
- Ariyoshi, Y.; Shiozaki, A.; Ichikawa, D.; Shimizu, H.; Kosuga, T.; Konishi, H.; Komatsu, S.; Fujiwara, H.; Okamoto, K.; Kishimoto, M.; et al. Na+/H+ exchanger 1 has tumor suppressive activity and prognostic value in esophageal squamous cell carcinoma. Oncotarget 2017, 8, 2209–2223. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, Y.; Higuchi, K.; Tominaga, K.; Watanabe, T.; Oshitani, N.; Arakawa, T. Functional oesophageal epithelial defense against acid. Inflammopharmacology 2005, 13, 1–13. [Google Scholar] [CrossRef]
- Goldman, A.; Shahidullah, M.; Goldman, D.; Khailova, L.; Watts, G.; Delamere, N.; Dvorak, K. A novel mechanism of acid and bile acid-induced DNA damage involving Na+/H+ exchanger: Implication for Barrett’s oesophagus. Gut 2010, 59, 1606–1616. [Google Scholar] [CrossRef] [Green Version]
- Guan, B.; Hoque, A.; Xu, X. Amiloride and guggulsterone suppression of esophageal cancer cell growth in vitro and in nude mouse xenografts. Front. Biol. 2014, 9, 75–81. [Google Scholar] [CrossRef]
- Tobey, N.A.; Koves, G.; Orlando, R.C. Human esophageal epithelial cells possess an Na+/H+ exchanger for H+ extrusion. Am. J. Gastroenterol. 1998, 93, 2075–2081. [Google Scholar] [CrossRef]
- Goldman, A.; Chen, H.; Khan, M.R.; Roesly, H.; Hill, K.A.; Shahidullah, M.; Mandal, A.; Delamere, N.A.; Dvorak, K. The Na+/H+ exchanger controls deoxycholic acid-induced apoptosis by a H+-activated, Na+-dependent ionic shift in esophageal cells. PLoS ONE 2011, 6, e23835. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, B.; Liu, X.; Lu, L.; Luo, F.; Lu, X.; Shi, L.; Xu, W.; Liu, Q. Epigenetic silencing of p21 by long non-coding RNA HOTAIR is involved in the cell cycle disorder induced by cigarette smoke extract. Toxicol. Lett. 2016, 240, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, Y.; Li, Y.; Xu, W.; Luo, F.; Wang, B.; Pang, Y.; Xiang, Q.; Zhou, J.; Wang, X.; et al. NF-kappaB-mediated inflammation leading to EMT via miR-200c is involved in cell transformation induced by cigarette smoke extract. Toxicol. Sci. 2013, 135, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Nakagawa, H.; Isariyawongse, B.K.; Funakoshi, S.; Silberg, D.G.; Rustgi, A.K.; Lynch, J.P. Induction of intestinalization in human esophageal keratinocytes is a multistep process. Carcinogenesis 2009, 30, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Boedtkjer, E. Ion Channels, Transporters, and Sensors Interact with the Acidic Tumor Microenvironment to Modify Cancer Progression. Rev. Physiol. Biochem. Pharmacol. 2021, 1–46. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, M.; Ma, Z.; Yuan, D.; Zhu, J.; Tuo, B.; Li, T.; Liu, X. Alteration and dysfunction of ion channels/transporters in a hypoxic microenvironment results in the development and progression of gastric cancer. Cell Oncol. 2021, 44, 739–749. [Google Scholar] [CrossRef]
- Lu, C.; Ma, Z.; Cheng, X.; Wu, H.; Tuo, B.; Liu, X.; Li, T. Pathological role of ion channels and transporters in the development and progression of triple-negative breast cancer. Cancer Cell Int. 2020, 20, 377. [Google Scholar] [CrossRef]
- Stock, C. How Dysregulated Ion Channels and Transporters Take a Hand in Esophageal, Liver, and Colorectal Cancer. Rev. Physiol. Biochem. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Zhang, M.; Li, T.; Zhu, J.; Tuo, B.; Liu, X. Physiological and pathophysiological role of ion channels and transporters in the colorectum and colorectal cancer. J. Cell Mol. Med. 2020, 24, 9486–9494. [Google Scholar] [CrossRef] [PubMed]
- Arcangeli, A.; Becchetti, A. New Trends in Cancer Therapy: Targeting Ion Channels and Transporters. Pharmaceuticals 2010, 3, 1202–1224. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.; Garcia-Quiroz, J.; Aguilar-Eslava, L.; Sanchez-Perez, Y.; Camacho, J. Novel Therapeutic Approaches of Ion Channels and Transporters in Cancer. Rev. Physiol. Biochem. Pharmacol. 2020, 1–57. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Philos Trans. R Soc. Lond B Biol. Sci. 2014, 369, 20130099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisson, L.; Driffort, V.; Benoist, L.; Poet, M.; Counillon, L.; Antelmi, E.; Rubino, R.; Besson, P.; Labbal, F.; Chevalier, S.; et al. NaV1.5 Na(+) channels allosterically regulate the NHE-1 exchanger and promote the activity of breast cancer cell invadopodia. J. Cell Sci. 2013, 126, 4835–4842. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Bao, P.; Li, Z.; Ouyang, H.; Wu, C.; Qian, G. Inhibition of proliferation and apoptosis induced by a Na+/H+ exchanger-1 (NHE-1) antisense gene on drug-resistant human small cell lung cancer cells. Oncol. Rep. 2009, 21, 1243–1249. [Google Scholar] [CrossRef] [Green Version]
- Serafino, A.; Moroni, N.; Psaila, R.; Zonfrillo, M.; Andreola, F.; Wannenes, F.; Mercuri, L.; Rasi, G.; Pierimarchi, P. Anti-proliferative effect of atrial natriuretic peptide on colorectal cancer cells: Evidence for an Akt-mediated cross-talk between NHE-1 activity and Wnt/beta-catenin signaling. Biochim. Biophys. Acta 2012, 1822, 1004–1018. [Google Scholar] [CrossRef]
- Vaish, V.; Sanyal, S.N. Role of Sulindac and Celecoxib in chemoprevention of colorectal cancer via intrinsic pathway of apoptosis: Exploring NHE-1, intracellular calcium homeostasis and Calpain 9. Biomed. Pharm. 2012, 66, 116–130. [Google Scholar] [CrossRef]
- Reshkin, S.J.; Cardone, R.A.; Harguindey, S. Na+-H+ exchanger, pH regulation and cancer. Recent Pat. Anticancer Drug Discov. 2013, 8, 85–99. [Google Scholar] [CrossRef]
- Loo, S.Y.; Chang, M.K.; Chua, C.S.; Kumar, A.P.; Pervaiz, S.; Clement, M.V. NHE-1: A promising target for novel anti-cancer therapeutics. Curr. Pharm. Des. 2012, 18, 1372–1382. [Google Scholar] [CrossRef]
- Venglovecz, V.; Rakonczay, Z., Jr.; Ozsvari, B.; Takacs, T.; Lonovics, J.; Varro, A.; Gray, M.A.; Argent, B.E.; Hegyi, P. Effects of bile acids on pancreatic ductal bicarbonate secretion in guinea pig. Gut 2008, 57, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Bracke, K.R.; D’Hulst, A.I.; Maes, T.; Demedts, I.K.; Moerloose, K.B.; Kuziel, W.A.; Joos, G.F.; Brusselle, G.G. Cigarette smoke-induced pulmonary inflammation, but not airway remodelling, is attenuated in chemokine receptor 5-deficient mice. Clin. Exp. Allergy 2007, 37, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- D’Hulst, A.I.; Vermaelen, K.Y.; Brusselle, G.G.; Joos, G.F.; Pauwels, R.A. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur. Respir. J. 2005, 26, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, C.S.; Docx, C.; Webster, R.; Battram, C.; Hynx, D.; Giddings, J.; Cooper, P.R.; Chakravarty, P.; Rahman, I.; Marwick, J.A.; et al. Comprehensive gene expression profiling of rat lung reveals distinct acute and chronic responses to cigarette smoke inhalation. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1183–L1193. [Google Scholar] [CrossRef] [Green Version]
- Stock, C.; Pedersen, S.F. Roles of pH and the Na(+)/H(+) exchanger NHE1 in cancer: From cell biology and animal models to an emerging translational perspective? Semin. Cancer Biol. 2017, 43, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Higuchi, K.; Takashima, T.; Hamaguchi, M.; Hayakawa, T.; Tominaga, K.; Watanabe, T.; Oshitani, N.; Shimada, Y.; Arakawa, T. Roles of epidermal growth factor and Na+/H+ exchanger-1 in esophageal epithelial defense against acid-induced injury. Am. J. Physiol. Gastrointest Liver Physiol. 2006, 290, G665–G673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalabis, J.; Wong, G.S.; Vega, M.E.; Natsuizaka, M.; Robertson, E.S.; Herlyn, M.; Nakagawa, H.; Rustgi, A.K. Isolation and characterization of mouse and human esophageal epithelial cells in 3D organotypic culture. Nat. Protoc. 2012, 7, 235–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegyi, P.; Gray, M.A.; Argent, B.E. Substance P inhibits bicarbonate secretion from guinea pig pancreatic ducts by modulating an anion exchanger. Am. J. Physiol. Cell Physiol. 2003, 285, C268–C276. [Google Scholar] [CrossRef] [Green Version]
- Weintraub, W.H.; Machen, T.E. pH regulation in hepatoma cells: Roles for Na-H exchange, Cl-HCO3 exchange, and Na-HCO3 cotransport. Am. J. Physiol. 1989, 257, G317–G327. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becskeházi, E.; Korsós, M.M.; Gál, E.; Tiszlavicz, L.; Hoyk, Z.; Deli, M.A.; Köhler, Z.M.; Keller-Pintér, A.; Horváth, A.; Csekő, K.; et al. Inhibition of NHE-1 Increases Smoke-Induced Proliferative Activity of Barrett’s Esophageal Cell Line. Int. J. Mol. Sci. 2021, 22, 10581. https://doi.org/10.3390/ijms221910581
Becskeházi E, Korsós MM, Gál E, Tiszlavicz L, Hoyk Z, Deli MA, Köhler ZM, Keller-Pintér A, Horváth A, Csekő K, et al. Inhibition of NHE-1 Increases Smoke-Induced Proliferative Activity of Barrett’s Esophageal Cell Line. International Journal of Molecular Sciences. 2021; 22(19):10581. https://doi.org/10.3390/ijms221910581
Chicago/Turabian StyleBecskeházi, Eszter, Marietta Margaréta Korsós, Eleonóra Gál, László Tiszlavicz, Zsófia Hoyk, Mária A. Deli, Zoltán Márton Köhler, Anikó Keller-Pintér, Attila Horváth, Kata Csekő, and et al. 2021. "Inhibition of NHE-1 Increases Smoke-Induced Proliferative Activity of Barrett’s Esophageal Cell Line" International Journal of Molecular Sciences 22, no. 19: 10581. https://doi.org/10.3390/ijms221910581
APA StyleBecskeházi, E., Korsós, M. M., Gál, E., Tiszlavicz, L., Hoyk, Z., Deli, M. A., Köhler, Z. M., Keller-Pintér, A., Horváth, A., Csekő, K., Helyes, Z., Hegyi, P., & Venglovecz, V. (2021). Inhibition of NHE-1 Increases Smoke-Induced Proliferative Activity of Barrett’s Esophageal Cell Line. International Journal of Molecular Sciences, 22(19), 10581. https://doi.org/10.3390/ijms221910581