
Interferons Are Pro-Inflammatory Cytokines in Sheared-Stressed Human Aortic Valve Endothelial Cells

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
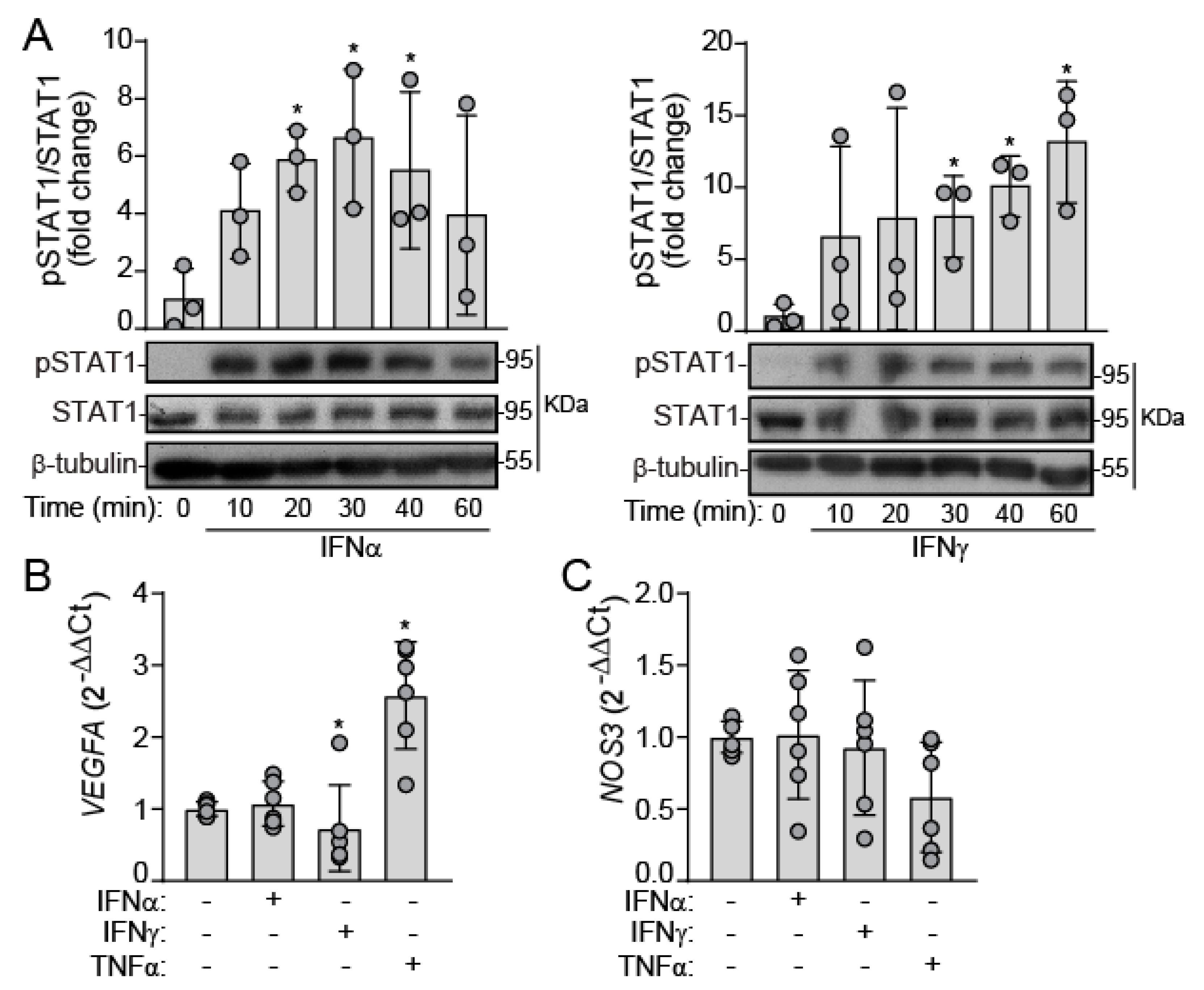
2.1. IFNs Activate JAK/STAT Signaling in Human VEC under Static Conditions
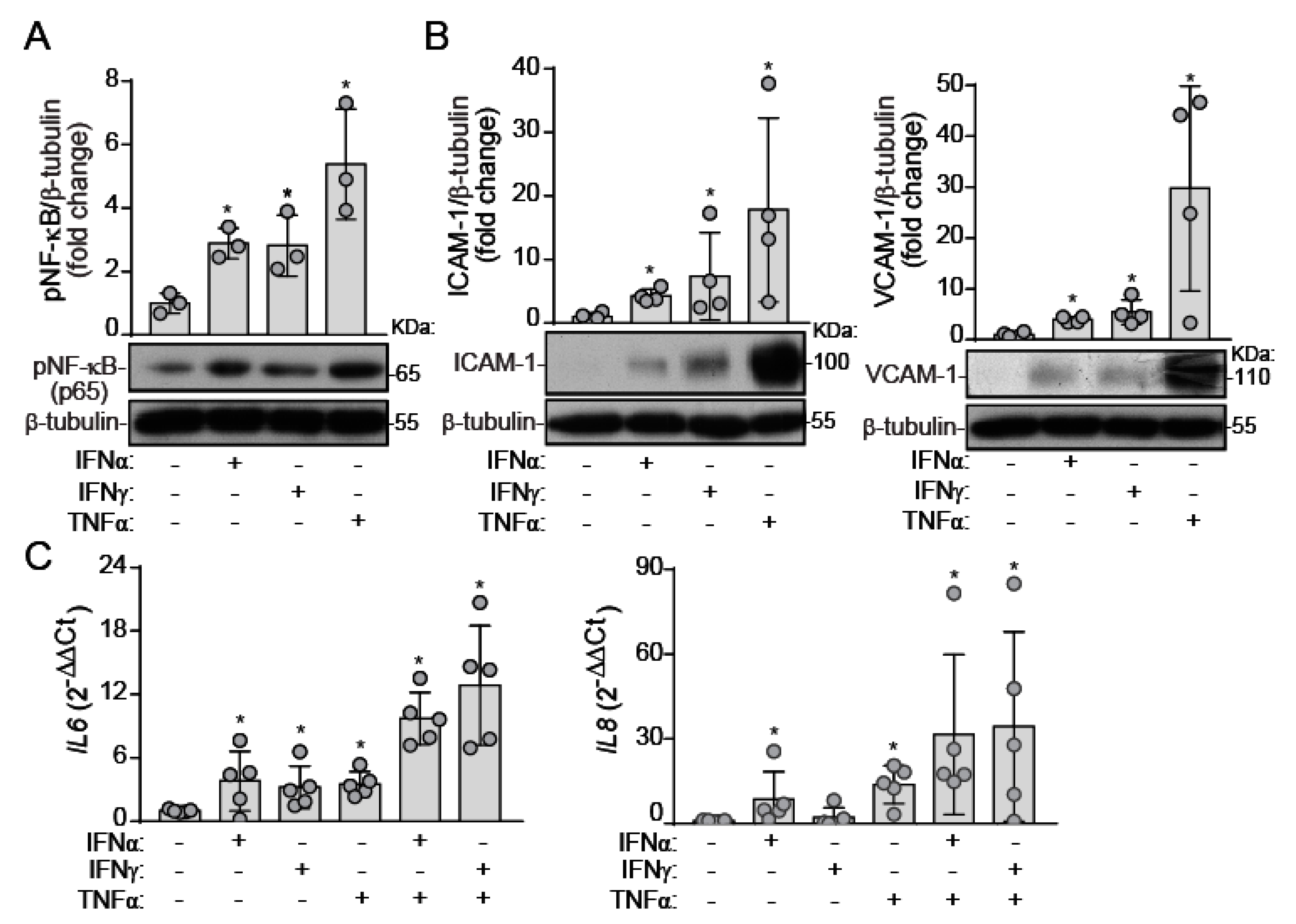
2.2. IFNs Induce NF-κB Activation and Inflammatory Molecule Expression under Static Conditions
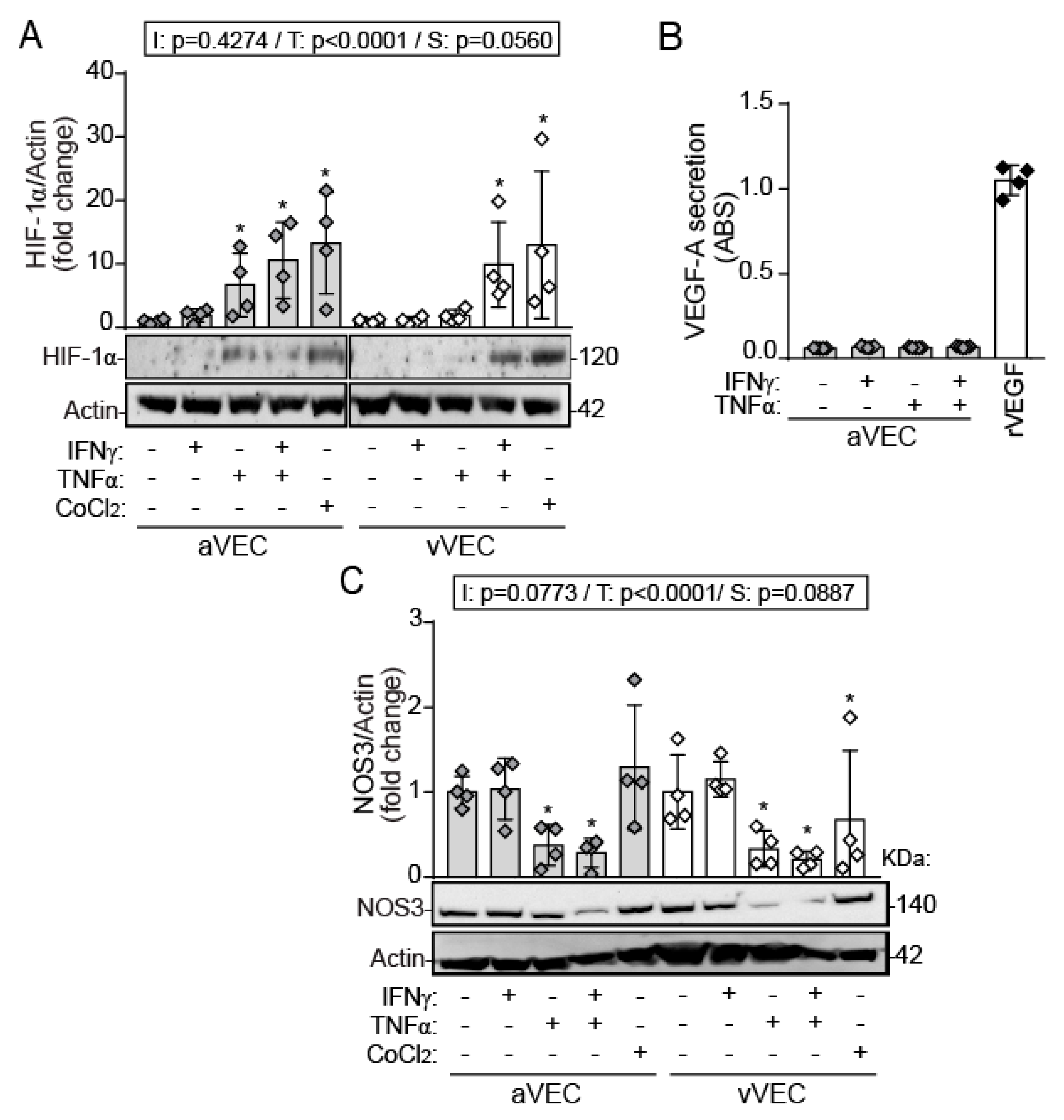
2.3. TNF-α, but Not IFN-γ, Induces HIF-1α Expression and Downregulation of NOS3 in Side-Specific VEC under Shear Stress
2.4. Differential Effects of IFN-γ and TNF-α on VEC Migration under Multiaxial Flow
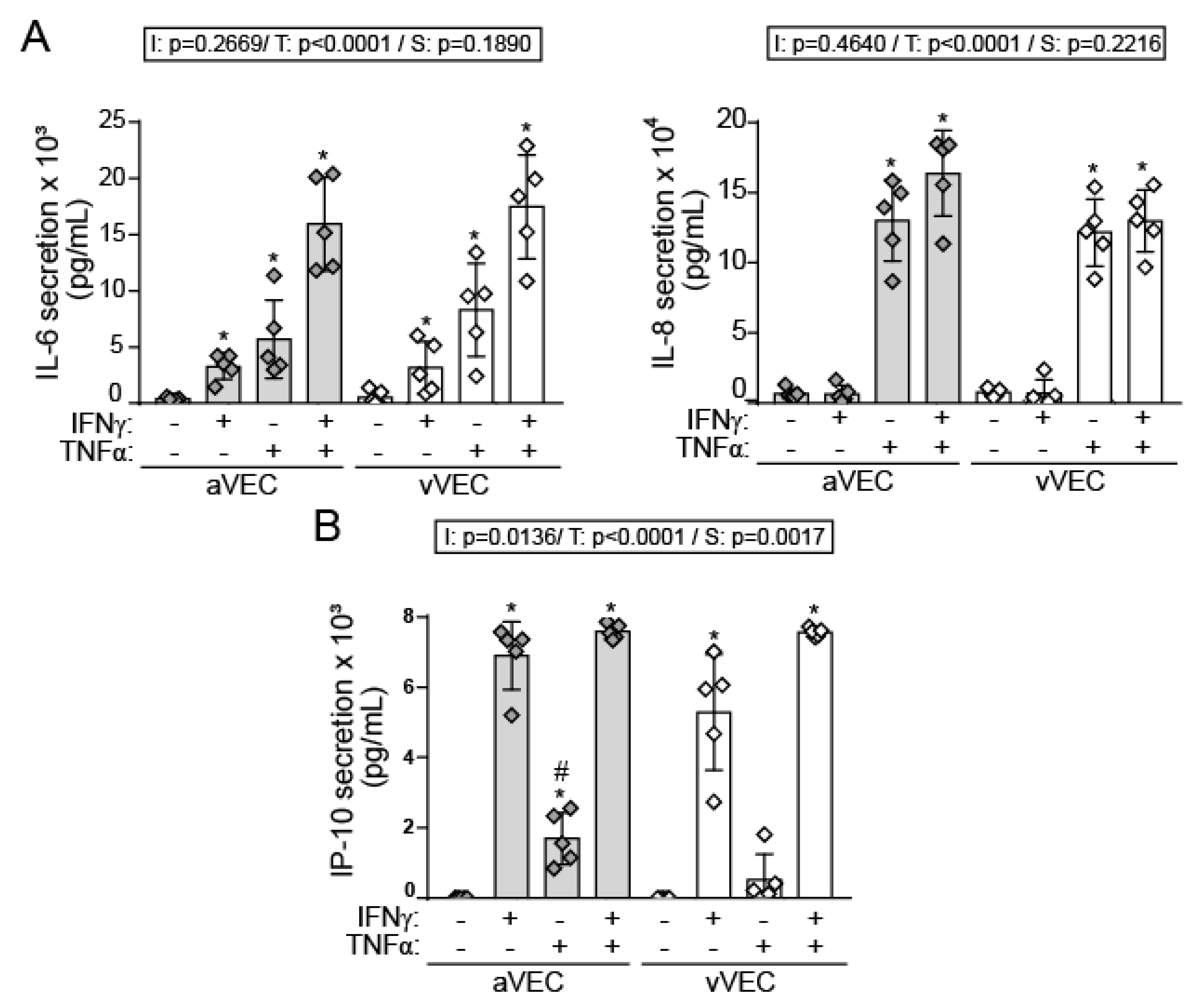
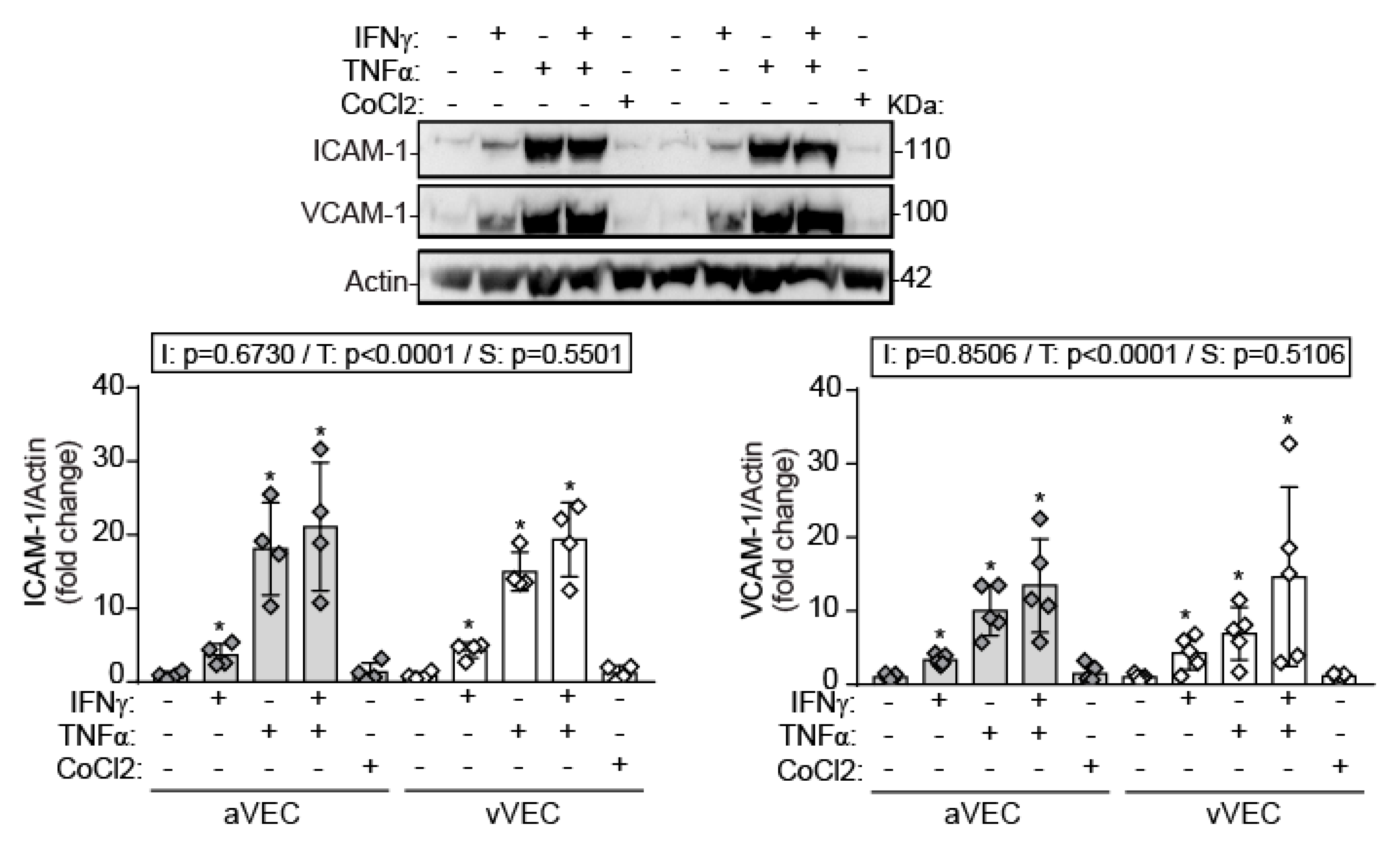
2.5. IFN-γ Induces Adhesion Molecule Expression and Cytokine Secretion under Shear Stress Conditions
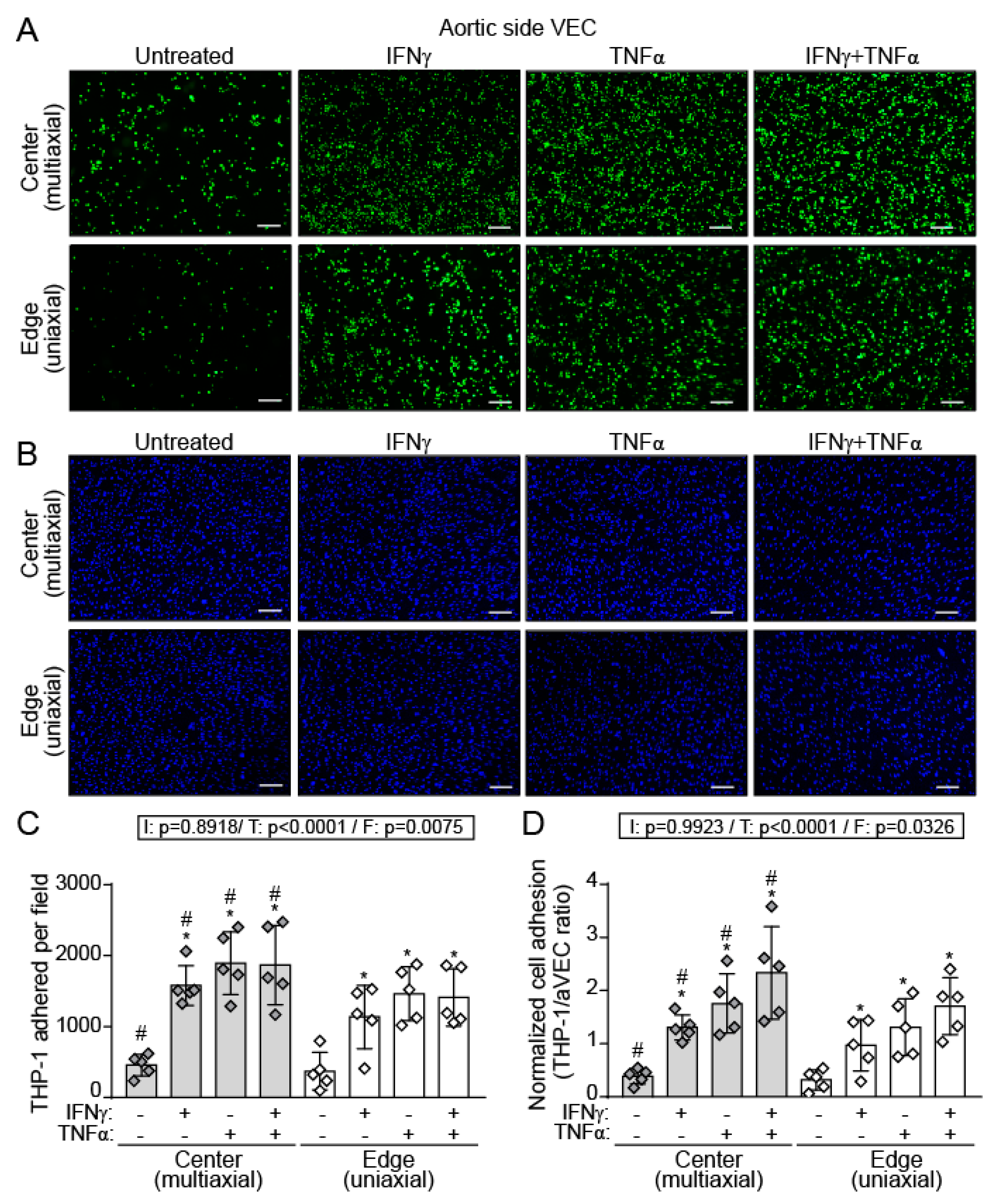
2.6. IFN-γ and TNF-α Increased THP-1 Adhesion to a Higher Extent in aVEC Cultured under Multiaxial Flow Conditions
3. Discussion
4. Materials and Methods
4.1. Endothelial Valve Cell Isolation
4.2. Cell Culture
4.3. Cell Activation under Static and Shear Stress Conditions
4.4. Protein Analysis by Immunoblot
4.5. Reverse Transcriptase Quantitative PCR (RT-qPCR)
4.6. Cell Migration Assay
4.7. Cytokine Secretion by ELISA
4.8. Monocyte-VEC Monolayer Adhesion Assay
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aVEC: | aortic-side valvular endothelial cells |
| CAVD: | calcific aortic valve disease |
| DAPI: | 4′,6-diamidino-2-phenylindole |
| EGM-2: | endothelial growth medium-2 |
| ELISA: | enzyme linked immunosorbent assay |
| NO: | nitric oxide |
| NOS3: | endothelial nitric oxide synthase |
| FBSi: | inactivated fetal bovine serum |
| HIF-1α: | hypoxia inducible factor 1α |
| ICAM-1: | intercellular cell adhesion molecule 1 |
| IFNs: | interferons |
| IL: | interleukin |
| IP-10/CXCL10: | interferon gamma-induced protein 10 |
| NF-κB: | nuclear factor κ-light-chain-enhancer of activated B cells |
| qPCR: | quantitative polymerase chain reaction |
| STAT: | signal transducers and activators of transcription |
| TNF-α: | tumor necrosis factor-α |
| VCAM-1: | vascular cell adhesion molecule 1 |
| VEC: | valvular endothelial cells |
| VEGF-A: | vascular endothelial growth factor A |
| vVEC: | ventricular-side aortic valve endothelial cells |
| VIC: | valvular interstitial cells |
References
- O’Brien, K.D. Pathogenesis of calcific aortic valve disease: A disease process comes of age (and a good deal more). Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1721–1728. [Google Scholar] [CrossRef] [Green Version]
- Helske, S.; Kupari, M.; Lindstedt, K.A.; Kovanen, P.T. Aortic valve stenosis: An active atheroinflammatory process. Curr. Opin. Lipidol. 2007, 18, 483–491. [Google Scholar] [CrossRef]
- New, S.E.; Aikawa, E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ. Res. 2011, 108, 1381–1391. [Google Scholar] [CrossRef] [Green Version]
- Pawade, T.A.; Newby, D.E.; Dweck, M.R. Calcification in Aortic Stenosis: The Skeleton Key. J. Am. Coll. Cardiol. 2015, 66, 561–577. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, P.; Bouchareb, R.; Boulanger, M.C. Innate and Adaptive Immunity in Calcific Aortic Valve Disease. J. Immunol. Res. 2015, 2015, 851945. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rodriguez, C.; Parra-Izquierdo, I.; Castaños-Mollor, I.; Lopez, J.; San Roman, J.A.; Sanchez Crespo, M. Toll-Like Receptors, Inflammation, and Calcific Aortic Valve Disease. Front. Physiol. 2018, 9, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, K.D.; Reichenbach, D.D.; Marcovina, S.M.; Kuusisto, J.; Alpers, C.E.; Otto, C.M. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 523–532. [Google Scholar] [CrossRef]
- Mohty, D.; Pibarot, P.; Despres, J.P.; Cote, C.; Arsenault, B.; Cartier, A.; Cosnay, P.; Couture, C.; Mathieu, P. Association between plasma LDL particle size, valvular accumulation of oxidized LDL, and inflammation in patients with aortic stenosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Cote, N.; Mahmut, A.; Fournier, D.; Boulanger, M.C.; Couture, C.; Despres, J.P.; Trahan, S.; Bosse, Y.; Page, S.; Pibarot, P.; et al. Angiotensin receptor blockers are associated with reduced fibrosis and interleukin-6 expression in calcific aortic valve disease. Pathobiology 2014, 81, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Farrar, E.J.; Huntley, G.D.; Butcher, J. Endothelial-derived oxidative stress drives myofibroblastic activation and calcification of the aortic valve. PLoS ONE 2015, 10, e0123257. [Google Scholar] [CrossRef]
- Mahler, G.J.; Farrar, E.J.; Butcher, J.T. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrar, E.J.; Butcher, J.T. Heterogeneous susceptibility of valve endothelial cells to mesenchymal transformation in response to TNFalpha. Ann. Biomed. Eng. 2014, 42, 149–161. [Google Scholar] [CrossRef]
- Parra-Izquierdo, I.; Castaños-Mollor, I.; López, J.; Gómez, C.; San Román, J.A.; Sánchez Crespo, M.; García-Rodríguez, C. Calcification induced by type I interferon in human aortic valve interstitial cells is larger in males and blunted by a Janus Kinase inhibitor. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2148–2159. [Google Scholar] [CrossRef] [Green Version]
- Parra-Izquierdo, I.; Castaños-Mollor, I.; Lopez, J.; Gomez, C.; San Roman, J.A.; Sanchez Crespo, M.; Garcia-Rodriguez, C. Lipopolysaccharide and interferon-gamma team up to activate HIF-1alpha via STAT1 in normoxia and exhibit sex differences in human aortic valve interstitial cells. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2168–2179. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashkiv, L.B. IFNgamma: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- Nagy, E.; Lei, Y.; Martinez-Martinez, E.; Body, S.C.; Schlotter, F.; Creager, M.; Assmann, A.; Khabbaz, K.; Libby, P.; Hansson, G.K.; et al. Interferon-gamma Released by Activated CD8(+) T Lymphocytes Impairs the Calcium Resorption Potential of Osteoclasts in Calcified Human Aortic Valves. Am. J. Pathol. 2017, 187, 1413–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mongkoldhumrongkul, N.; Yacoub, M.H.; Chester, A.H. Valve Endothelial Cells—Not Just Any Old Endothelial Cells. Curr. Vasc. Pharmacol. 2016, 14, 146–154. [Google Scholar] [CrossRef]
- Kennedy, J.A.; Hua, X.; Mishra, K.; Murphy, G.A.; Rosenkranz, A.C.; Horowitz, J.D. Inhibition of calcifying nodule formation in cultured porcine aortic valve cells by nitric oxide donors. Eur. J. Pharmacol. 2009, 602, 28–35. [Google Scholar] [CrossRef]
- El-Hamamsy, I.; Balachandran, K.; Yacoub, M.H.; Stevens, L.M.; Sarathchandra, P.; Taylor, P.M.; Yoganathan, A.P.; Chester, A.H. Endothelium-dependent regulation of the mechanical properties of aortic valve cusps. J. Am. Coll. Cardiol. 2009, 53, 1448–1455. [Google Scholar] [CrossRef]
- Richards, J.; El-Hamamsy, I.; Chen, S.; Sarang, Z.; Sarathchandra, P.; Yacoub, M.H.; Chester, A.H.; Butcher, J.T. Side-specific endothelial-dependent regulation of aortic valve calcification: Interplay of hemodynamics and nitric oxide signaling. Am. J. Pathol. 2013, 182, 1922–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacks, M.S.; David Merryman, W.; Schmidt, D.E. On the biomechanics of heart valve function. J. Biomech. 2009, 42, 1804–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef] [PubMed]
- Miragoli, M.; Yacoub, M.H.; El-Hamamsy, I.; Sanchez-Alonso, J.L.; Moshkov, A.; Mongkoldhumrongkul, N.; Padala, M.; Paramagurunathan, S.; Sarathchandra, P.; Korchev, Y.E.; et al. Side-specific mechanical properties of valve endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H15–H24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, C.A.; Grant, G.R.; Manduchi, E.; Davies, P.F. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ. Res. 2005, 96, 792–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaden, J.J.; Dempfle, C.E.; Grobholz, R.; Fischer, C.S.; Vocke, D.C.; Kilic, R.; Sarikoc, A.; Pinol, R.; Hagl, S.; Lang, S.; et al. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc. Pathol. 2005, 14, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Boshuizen, M.C.; de Winther, M.P. Interferons as Essential Modulators of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1579–1588. [Google Scholar] [CrossRef] [Green Version]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.; Fernandez-Pisonero, I.; Duenas, A.I.; Maeso, P.; Roman, J.A.; Crespo, M.S.; Garcia-Rodriguez, C. Viral and bacterial patterns induce TLR-mediated sustained inflammation and calcification in aortic valve interstitial cells. Int. J. Cardiol. 2012, 158, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.A.; Kim, E.K.; Now, H.; Nguyen, N.T.; Kim, W.J.; Yoo, J.Y.; Lee, J.; Jeong, Y.M.; Kim, C.H.; Kim, O.H.; et al. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am. J. Hum. Genet. 2015, 96, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Buers, I.; Nitschke, Y.; Rutsch, F. Novel interferonopathies associated with mutations in RIG-I like receptors. Cytokine Growth Factor Rev. 2016, 29, 101–107. [Google Scholar] [CrossRef]
- Knight, R.A.; Scarabelli, T.M.; Stephanou, A. STAT transcription in the ischemic heart. JAKSTAT 2012, 1, 111–117. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef]
- Raddatz, M.A.; Huffstater, T.; Bersi, M.R.; Reinfeld, B.I.; Madden, M.Z.; Booton, S.E.; Rathmell, W.K.; Rathmell, J.C.; Lindman, B.R.; Madhur, M.S.; et al. Macrophages Promote Aortic Valve Cell Calcification and Alter STAT3 Splicing. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e153–e165. [Google Scholar] [CrossRef]
- Kuo, H.P.; Lee, D.F.; Xia, W.; Wei, Y.; Hung, M.C. TNFalpha induces HIF-1alpha expression through activation of IKKbeta. Biochem. Biophys. Res. Commun. 2009, 389, 640–644. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Bowden, N.; Fragiadaki, M.; Souilhol, C.; Hsiao, S.; Mahmoud, M.; Allen, S.; Pirri, D.; Ayllon, B.T.; Akhtar, S.; et al. Mechanical Activation of Hypoxia-Inducible Factor 1alpha Drives Endothelial Dysfunction at Atheroprone Sites. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2087–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez Esmerats, J.; Villa-Roel, N.; Kumar, S.; Gu, L.; Salim, M.T.; Ohh, M.; Taylor, W.R.; Nerem, R.M.; Yoganathan, A.P.; Jo, H. Disturbed Flow Increases UBE2C (Ubiquitin E2 Ligase C) via Loss of miR-483-3p, Inducing Aortic Valve Calcification by the pVHL (von Hippel-Lindau Protein) and HIF-1alpha (Hypoxia-Inducible Factor-1alpha) Pathway in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 467–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helotera, H.; Alitalo, K. The VEGF family, the inside story. Cell 2007, 130, 591–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamas, S.; Michel, T.; Collins, T.; Brenner, B.M.; Marsden, P.A. Effects of interferon-gamma on nitric oxide synthase activity and endothelin-1 production by vascular endothelial cells. J. Clin. Investig. 1992, 90, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Poggianti, E.; Venneri, L.; Chubuchny, V.; Jambrik, Z.; Baroncini, L.A.; Picano, E. Aortic valve sclerosis is associated with systemic endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 41, 136–141. [Google Scholar] [CrossRef] [Green Version]
- Butcher, J.T.; Tressel, S.; Johnson, T.; Turner, D.; Sorescu, G.; Jo, H.; Nerem, R.M. Transcriptional profiles of valvular and vascular endothelial cells reveal phenotypic differences: Influence of shear stress. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Yang, Y.; Luo, X.; Cheng, Y.; Zhang, M.; Wang, K.; Ge, C. Inhibition of tumor angiogenesis by interferon-gamma by suppression of tumor-associated macrophage differentiation. Oncol. Res. 2014, 21, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Cai, C.; Zhao, G.; Qiu, X.; Zhao, H.; Ma, Q.; Tian, L.; Li, X.; Hu, Y.; Liao, B.; et al. Hypoxia promotes migration and induces CXCR4 expression via HIF-1alpha activation in human osteosarcoma. PLoS ONE 2014, 9, e90518. [Google Scholar]
- Hu, Y.; Hu, X.; Boumsell, L.; Ivashkiv, L.B. IFN-gamma and STAT1 arrest monocyte migration and modulate RAC/CDC42 pathways. J. Immunol. 2008, 180, 8057–8065. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, G.; Wu, X.; Jiang, Z.; Kasman, I.; Yao, J.; Guan, Y.; Oeh, J.; Modrusan, Z.; Bais, C.; Sampath, D.; et al. Tumour-secreted miR-9 promotes endothelial cell migration and angiogenesis by activating the JAK-STAT pathway. EMBO J. 2012, 31, 3513–3523. [Google Scholar] [CrossRef]
- Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial-Mesenchymal Transition. Cells 2020, 9, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, Z.; Gerritsen, M.E.; Carley, W.W.; Strieter, R.M.; Kunkel, S.L.; Westwick, J. Chemokine gene expression and secretion by cytokine-activated human microvascular endothelial cells. Differential regulation of monocyte chemoattractant protein-1 and interleukin-8 in response to interferon-gamma. Am. J. Pathol. 1994, 145, 913–921. [Google Scholar]
- Heller, E.A.; Liu, E.; Tager, A.M.; Yuan, Q.; Lin, A.Y.; Ahluwalia, N.; Jones, K.; Koehn, S.L.; Lok, V.M.; Aikawa, E.; et al. Chemokine CXCL10 promotes atherogenesis by modulating the local balance of effector and regulatory T cells. Circulation 2006, 113, 2301–2312. [Google Scholar] [CrossRef]
- Raddatz, M.A.; Madhur, M.S.; Merryman, W.D. Adaptive immune cells in calcific aortic valve disease. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H141–H155. [Google Scholar] [CrossRef]
- Grim, J.C.; Aguado, B.A.; Vogt, B.J.; Batan, D.; Andrichik, C.L.; Schroeder, M.E.; Gonzalez-Rodriguez, A.; Yavitt, F.M.; Weiss, R.M.; Anseth, K.S. Secreted Factors from Proinflammatory Macrophages Promote an Osteoblast-Like Phenotype in Valvular Interstitial Cells. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e296–e308. [Google Scholar] [CrossRef]
- Fernandez Esmerats, J.; Heath, J.; Jo, H. Shear-Sensitive Genes in Aortic Valve Endothelium. Antioxid. Redox Signal. 2016, 25, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, H.; Mochizuki, N. Flow pattern-dependent endothelial cell responses through transcriptional regulation. Cell Cycle 2017, 16, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Ghim, M.; Pang, K.T.; Arshad, M.; Wang, X.; Weinberg, P.D. A novel method for segmenting growth of cells in sheared endothelial culture reveals the secretion of an anti-inflammatory mediator. J. Biol. Eng. 2018, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Butcher, J.T.; Penrod, A.M.; Garcia, A.J.; Nerem, R.M. Unique morphology and focal adhesion development of valvular endothelial cells in static and fluid flow environments. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1429–1434. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, K.A.; Finnegan, A. Regulation of intercellular adhesion molecule-1 (CD54) gene expression. J. Leukoc. Biol. 1999, 66, 876–888. [Google Scholar] [CrossRef]
- Jaruga, B.; Hong, F.; Kim, W.H.; Gao, B. IFN-gamma/STAT1 acts as a proinflammatory signal in T cell-mediated hepatitis via induction of multiple chemokines and adhesion molecules: A critical role of IRF-1. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1044–G1052. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, Y.; Chen, H.; Zhang, J.; Xu, C.; Li, J.; Li, M. Enhancement of ICAM-1 via the JAK2/STAT3 signaling pathway in a rat model of severe acute pancreatitis-associated lung injury. Exp. Ther. Med. 2016, 11, 788–796. [Google Scholar] [CrossRef] [Green Version]
- Park, K.H.; Lee, T.H.; Kim, C.W.; Kim, J. Enhancement of CCL15 expression and monocyte adhesion to endothelial cells (ECs) after hypoxia/reoxygenation and induction of ICAM-1 expression by CCL15 via the JAK2/STAT3 pathway in ECs. J. Immunol. 2013, 190, 6550–6558. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, L.M. The role of nuclear factor kappaB in the interferon response. J. Interferon Cytokine Res. 2011, 31, 553–559. [Google Scholar] [CrossRef] [Green Version]
- Collins, T.; Read, M.A.; Neish, A.S.; Whitley, M.Z.; Thanos, D.; Maniatis, T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995, 9, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Q.; Jin, C.; Ao, L.; Cleveland, J.C., Jr.; Song, R.; Xu, D.; Fullerton, D.A.; Meng, X. Cross-talk between the Toll-like receptor 4 and Notch1 pathways augments the inflammatory response in the interstitial cells of stenotic human aortic valves. Circulation 2012, 126 (Suppl. 1), S222–S230. [Google Scholar] [CrossRef] [Green Version]
- Amin, M.A.; Haas, C.S.; Zhu, K.; Mansfield, P.J.; Kim, M.J.; Lackowski, N.P.; Koch, A.E. Migration inhibitory factor up-regulates vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 via Src, PI3 kinase, and NFkappaB. Blood 2006, 107, 2252–2261. [Google Scholar] [CrossRef] [Green Version]
- Minhajuddin, M.; Bijli, K.M.; Fazal, F.; Sassano, A.; Nakayama, K.I.; Hay, N.; Platanias, L.C.; Rahman, A. Protein kinase C-delta and phosphatidylinositol 3-kinase/Akt activate mammalian target of rapamycin to modulate NF-kappaB activation and intercellular adhesion molecule-1 (ICAM-1) expression in endothelial cells. J. Biol. Chem. 2009, 284, 4052–4061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, C.M.; Lundberg, M.H.; Harrington, L.S.; Warboys, C.M.; Warner, T.D.; Berson, R.E.; Moshkov, A.V.; Gorelik, J.; Weinberg, P.D.; Mitchell, J.A. Role of shear stress in endothelial cell morphology and expression of cyclooxygenase isoforms. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 384–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, R.A.; Butcher, J.T. Isolation of valvular endothelial cells. J. Vis. Exp. 2010, 46, e2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mongkoldhumrongkul, N.; Latif, N.; Yacoub, M.H.; Chester, A.H. Effect of Side-Specific Valvular Shear Stress on the Content of Extracellular Matrix in Aortic Valves. Cardiovasc. Eng. Technol. 2018, 9, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parra-Izquierdo, I.; Sánchez-Bayuela, T.; López, J.; Gómez, C.; Pérez-Riesgo, E.; San Román, J.A.; Sánchez Crespo, M.; Yacoub, M.; Chester, A.H.; García-Rodríguez, C. Interferons Are Pro-Inflammatory Cytokines in Sheared-Stressed Human Aortic Valve Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 10605. https://doi.org/10.3390/ijms221910605
Parra-Izquierdo I, Sánchez-Bayuela T, López J, Gómez C, Pérez-Riesgo E, San Román JA, Sánchez Crespo M, Yacoub M, Chester AH, García-Rodríguez C. Interferons Are Pro-Inflammatory Cytokines in Sheared-Stressed Human Aortic Valve Endothelial Cells. International Journal of Molecular Sciences. 2021; 22(19):10605. https://doi.org/10.3390/ijms221910605
Chicago/Turabian StyleParra-Izquierdo, Iván, Tania Sánchez-Bayuela, Javier López, Cristina Gómez, Enrique Pérez-Riesgo, J. Alberto San Román, Mariano Sánchez Crespo, Magdi Yacoub, Adrian H. Chester, and Carmen García-Rodríguez. 2021. "Interferons Are Pro-Inflammatory Cytokines in Sheared-Stressed Human Aortic Valve Endothelial Cells" International Journal of Molecular Sciences 22, no. 19: 10605. https://doi.org/10.3390/ijms221910605
APA StyleParra-Izquierdo, I., Sánchez-Bayuela, T., López, J., Gómez, C., Pérez-Riesgo, E., San Román, J. A., Sánchez Crespo, M., Yacoub, M., Chester, A. H., & García-Rodríguez, C. (2021). Interferons Are Pro-Inflammatory Cytokines in Sheared-Stressed Human Aortic Valve Endothelial Cells. International Journal of Molecular Sciences, 22(19), 10605. https://doi.org/10.3390/ijms221910605