Abstract
Cardiovascular disease (CVD) is a common disease caused by many factors, including atherosclerosis, congenital heart disease, heart failure, and ischemic cardiomyopathy. CVD has been regarded as one of the most common diseases and has a severe impact on the life quality of patients. The main features of CVD include high morbidity and mortality, which seriously threaten human health. SUMO proteins covalently conjugate lysine residues with a large number of substrate proteins, and SUMOylation regulates the function of target proteins and participates in cellular activities. Under certain pathological conditions, SUMOylation of proteins related to cardiovascular development and function are greatly changed. Numerous studies have suggested that SUMOylation of substrates plays critical roles in normal cardiovascular development and function. We reviewed the research progress of SUMOylation in cardiovascular development and function, and the regulation of protein SUMOylation may be applied as a potential therapeutic strategy for CVD treatment.
1. Introduction
Cardiovascular disease (CVD) is a generic term of heart disease caused by a series of factors and is one of the major causes of human death. CVD is a serious threat to human life and has become a hot topic in modern medical research. SUMOylation acts as a dynamic equilibrium protein modification and is ubiquitous in human cells. It has been found that SUMOylation plays important roles in maintaining normal cell functions, and dysregulation of SUMOylation has been observed in the pathogenesis of various heart diseases. There are a large number of target proteins modified by SUMO in the heart, which affect cell proliferation, apoptosis, and metabolism. The imbalance of SUMOylation leads to abnormal cardiac function, resulting in the occurrence of CVD. However, the molecular mechanism of SUMOylation in CVD is still not completely clear. This review describes the latest research of SUMOylation in CVD and aims to provide new ideas and potential therapeutic targets for the treatment of CVD.
1.1. CVD
CVD is a group of diseases caused by many factors, including atherosclerosis, congenital heart disease (CHD), heart failure, and ischemic cardiomyopathy, with a high prevalence, morbidity, and mortality [1]. Atherosclerosis is a progressive inflammatory cardiovascular disease characterized by lipid plaque formation in the artery [2]. The formation and rupture of lipid plaques is one important cause of clinical cardiovascular events, including stroke and myocardial infarction, accounting for more than one-quarter of global deaths [3]. CHD is a congenital cardiac structural abnormality, and it is a common congenital defect in infants, accounting for about 1% of all newborns [4]. Atrial septal defects (ASDs) and ventricular septal defects (VSDs) are the most common CHDs, which are characterized by an existing connection between atria and ventricles [5]. Heart failure is a complex disease caused by valve abnormality, myocardial ischemia, coronary artery disease, hypertension, and other pathological changes [6]. Cardiac energy metabolism and mitochondrial biogenesis defects lead to heart failure and cardiac hypertrophy [7]. Acute myocardial infarction (AMI) is an ischemic cardiac disease with acute hypoxia in the myocardial tissue and is one of the leading causes of death. Ischemia/reperfusion (I/R) is an important treatment for rescuing ischemic myocardium from necrosis [8]. Readjustment of blood flow restores oxygen and nutrient supply but may damage the cardiac muscle and induce myocardial I/R injury. Advances in pharmacology and interventional therapy have improved CVD treatment but have not solved the problem from the source. Extensive proteins involved in the pathology of CVD are modified by SUMO proteins, indicating that SUMOylation of substrates plays important roles in the progression of CVD.
1.2. SUMOylation
SUMO (small ubiquitin-like modifier) is ubiquitous in eukaryotes, and the SUMO family contains SUMO1, SUMO2, SUMO3, and SUMO4 [9,10]. SUMO1, SUMO2, and SUMO3 are commonly found [11,12], while SUMO4 is only expressed in the spleen, kidney, and other organs [13]. The identical sequences of SUMO2 and SUMO3 are 97%, so they are difficult to distinguish and commonly referred to as SUMO2/3, while only 47% of the homologous sequences are between SUMO1 and SUMO2/3 [14,15]. SUMO4 was initially shown to be associated with the pathogenesis of type 1 diabetes, but it was later reported that SUMO4 was ineffective in covalent coupling with the substrate due to the presence of proline-90 amino acid residues that prevent maturation of SUMO4 [16].
SUMOylation is accomplished via maturation, activation, conjugation, and ligation with activating enzyme E1 (SAE1/SAE2), conjugating enzyme E2 (UBC9), and ligating enzyme E3 [15]. The C-terminal of SUMO forms a covalent bond with the lysine residues of target proteins through an isopeptide bond. The SUMO precursor is processed to expose the C-terminal di-glycine motif, then the cysteine residue of the E1 subunit forms a high-energy thioester bond with SUMO. The activated SUMO was transferred to the E2-binding enzyme, and linked to the cystine of UBC9 to form the SUMO-E2 intermediate. UBC9 transferred SUMO to the substrate, and the C-terminal glycine of SUMO bound to the lysine residue of the substrate to form the isopeptide bond [17]. Although E1 and E2 are sufficient to complete SUMOylation, the E3 is still required to enhance the specificity of SUMO isoforms and substrates, activate the SUMO binding site, and promote the formation of poly-SUMO chains.
The deSUMOylation is achieved by a family of SENP (Sentrin/SUMO-specific protease) [10,18], which deconjugate the SUMO from target proteins and catalyze the maturation of the SUMO proteins [7]. The SENP family in mammalian cells has six members, which can be divided into three groups. The first group is SENP1 and SENP2, and both have broad substrate specificity [19,20,21,22,23,24]. The second group is SENP3 and SENP5, and both in the nucleus, but mainly in the nucleolus, tend to deSUMOylation of SUMO2/3. The third group includes SENP6 and SENP7, both with additional ring structures in their catalytic zone. SUMO and SENP2 dynamically regulate the SUMOylation level of target proteins (Figure 1), and SUMOylation is an important dynamic reversible post-translational modification (PTM) to regulate protein stability, subcellular localization, and function [25]. Disorders of SUMOylation affect the normal function of target proteins and lead to the damage of important physiological processes and an abnormal intracellular environment [26].
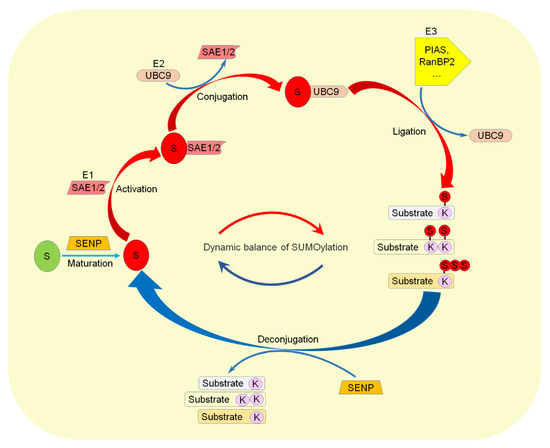
Figure 1.
Dynamic regulation of the SUMOylation pathway. The SUMO precursor protein is matured by SENP and then activated by E1, conjugated by E2, and ligated to substrates by E3. On the other hand, SENP deconjugates SUMO from target proteins. S: SUMO. K: Lysine.
Protein homeostasis is indispensable, especially in cells with low mitotic activity, such as neurons and cardiomyocytes, to maintain cellular function [27,28]. Under certain pathological conditions, SUMOylation-regulated signaling pathways related to cardiovascular development and function are changed [29,30]. SUMOylation affects the function of cardiovascular proteins, emerging as key factors of various CVD progression [29]. In recent years, numerous new SUMOylation targets have emerged, and the important roles of SUMOylation in CVD progression have been continuously discovered [31,32].
2. SUMO Machinery and CVD
2.1. UBC9
UBC9 is the only E2 enzyme responsible for the function of GATA4 in cardiac development and function. UBC9 enhanced the transcriptional activity and altered the nuclear localization of GATA4 [33]. UBC9-mediated SUMOylation of GATA4 on lysine residue 366 (K366) promoted specific gene expression in cardiac pluripotent cells. Knockout of UBC9 in mice stimulated early embryonic death at embryonic day 3.5 [34]. UBC9-mediated SUMOylation induced a higher level of autophagy and increased autophagy flux and the incidence of cardiac development [35]. These studies indicated an important role of UBC9 in cardiac development (Figure 2).
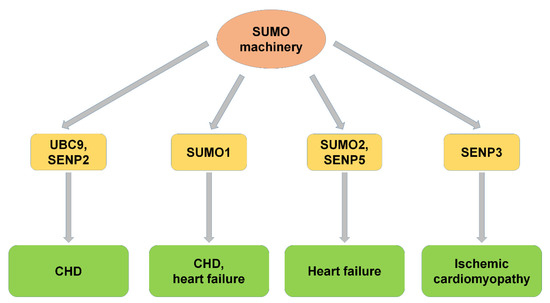
Figure 2.
SUMO machinery and CVD. SUMO machinery, including UBC9, SUMO1, SUMO2, SENP2, SENP3, and SENP5, participated in the progression of CVDs, including congenital heart disease, heart failure, and ischemic cardiomyopathy. CHD: congenital heart disease.
2.2. SUMO1
The SUMOylation was mediated by the SUMO family, and SUMO1 was one of the most important members. SUMO1 plays a critical role in early heart development. Mice with SUMO1 knockout showed the CHD phenotype. Heterozygous and homozygous mice with higher ASD and VSD mortality due to SUMO1 knockout were rescued by cardiac expression of SUMO1 [36]. Thus, SUMO1 played a key role in normal cardiac development, and decreased SUMOylation activity induces cardiac pathology. The changed SUMOylation levels affect a variety of transcription factors and enzymes that maintain normal cardiac function.
SUMO1 was also found to improve the oxidative stress response during increased cardiac hypertrophy [37]. SUMO1 transfection with adenovirus after TAC, a model of heart failure induced by stress overload, prevented the occurrence of cardiac hypertrophy, blocked the negative effect of hydrogen peroxide on SERCA2a, and decreased the oxidative stress index in mice [38]. These studies indicate that SUMO1 plays important roles in normal cardiac function and suggest that SUMO1 prevents heart failure in a toxic environment (Table 1).

Table 1.
SUMO machinery and CVD.
2.3. SUMO2
In previous studies, little is known about whether and how SUMO2/3 functions in CVD. It was reported that endogenous SUMO2 was important in maintaining normal endothelium-dependent vascular function [40], and SUMO2 upregulation disrupts the balance of the vascular environment and leads to endothelial dysfunction due to cholesterolemia [41]. Increased SUMO2 binding protein was found in the heart of SUMO2 transgenic mice, which showed the cardiomyopathy phenotype [41]. These results indicated that SUMO2-mediated SUMOylation maintains a great balance in normal conditions, and the imbalance of SUMOylation leads to certain pathological conditions, including heart failure.
2.4. SENP2
Studies have shown that SENP2 is associated with CHD, cardiac dysfunction, and embryonic development and is a key regulatory factor in early cardiac morphosis [13]. In transgenic mice with cardiac-specific SENP2 expression, overexpressed SENP2 leads to ASDs and VSDs, reduced proliferation of cardiomyocytes, and induced cardiomyopathy with aging [13]. SENP2 deconjugated SUMO from many transcriptional factors that played important roles in cardiac development [36]. SENP2 inhibits the transcriptional activity of NKX2-5 by promoting the uncoupling of SUMO1, affecting cardiac development and function [44]. These results suggested a crucial role of SENP2 in the pathogenesis of CHD.
2.5. SENP3
Interruption of blood supply to the heart is one of the leading causes of disability and death. However, little is known about the molecular events during cardiac ischemia [39] and how these changes function during cell death after reperfusion [45]. SUMOylation of proteins protects cells from several stresses, including ischemia-reperfusion. SENP3 has been reported to be an important determinant factor of cell survival after ischemic infarction [42]. It was found that ischemia-reperfusion significantly reduced the cytoplasmic level of SENP3 and accelerated cell death. It was suggested that SENP3-mediated deSUMOylation plays important roles in regulating myocardial cell survival after ischemic injury.
2.6. SENP5
SENP5 exhibits both endopeptidase and isopeptidase activities [46] and plays important roles in cell proliferation and survival [47,48]. Studies have shown that the level of SENP5, which mainly deconjugates SUMO2/3, is elevated in heart failure [43]. In mitochondria of the normal heart, SUMO1 modified Drp1 to increase its stability [47]. However, in a failing heart, the SENP5 level was increased. The increase of mitochondrial division mediated by Drp1 leads to changes in the morphology and structure of cardiac mitochondria, resulting in mitochondrial dysfunction, thereby inhibiting the proliferation of cardiomyocytes and aggravating apoptosis [43]. These results indicated that SENP5 plays important roles in heart failure and is a potential target for the treatment of heart failure.
3. SUMOylation of Proteins and CVD
3.1. Atherosclerosis
Atherosclerosis is a progressive inflammatory cardiovascular disease characterized by lipid plaque formation in the artery [2,49]. Endothelial cells form the main barrier to vascular permeability, preventing the formation of atherosclerosis [50]. Dysfunction of endothelial cells increases vascular wall permeability, lipid accumulation, and immune cell infiltration in large- and medium-sized artery walls, and subendothelial LDL (low-density lipoprotein) accumulation increases the rate of plaque formation [51,52]. Increasing evidence has shown that SUMOylation plays an important role in regulating inflammation, migration, actin filament remodeling, and apoptosis during the formation of atherosclerosis (Figure 3).
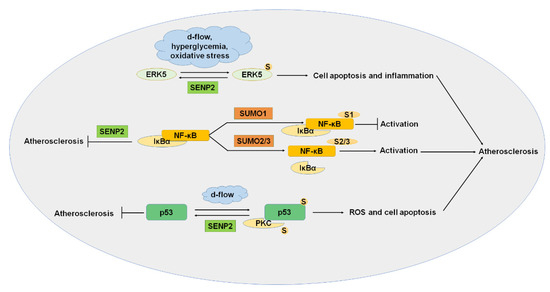
Figure 3.
SUMOylation and atherosclerosis. ERK5, NF-κB, p53, and PKC were SUMOylated, and SUMOylation of these proteins played important roles in the progression of atherosclerosis. ERK5, extracellular signal-regulated kinase 5; PKC, protein kinase C. S: SUMO.
3.1.1. ERK5 (Extracellular Signal-Regulated Kinase 5)
MAPK (mitogen-activated protein kinases) is a family of serine/threonine kinase composed of four major subfamilies that regulate cellular activities, including cell differentiation, proliferation, and apoptosis. ERK5 (or MAPK7) is one of the most important members of MAPK. ERK5 is not SUMOylated in normal conditions. However, the SUMOylation of ERK5 was induced by d-flow, hyperglycemia, and oxidative stress, and ERK5 SUMOylation induced cell apoptosis and inflammation. SENP2 protects endothelial cells by deSUMOylation of ERK5 to inhibit the pathological change [3]. It has shown that ERK5 activation plays an anti-inflammatory role in the progression of atherosclerosis [53]. It was reported that SUMOylation of ERK5 induced by H2O2 significantly inhibited the transcriptional activity of ERK5, causing inflammation in the onset and promoting the progression of atherosclerosis [54]. These results suggest that the SUMOylation of ERK5 is one of the most meaningful causes of atherosclerosis, and maintaining the balance of ERK5 SUMOylation is a potential treatment for atherosclerosis.
3.1.2. NF-κB
NF-κB is a nuclear transcription factor and regulates cytokines and adhesion molecules released by leukocyte accumulation [55]. NF-κB in the cytoplasm binds to the inhibitor of κB (IκB) during the resting state. It was reported that endothelium-specific knockout of NEMO (NF-κB essential modulator) or dominant-negative IκBα inhibits atherosclerotic plaque formation [56]. NF-κB was SUMOylated, and SUMOylation of NF-κB plays a critical role in the pathogenesis of atherosclerosis. SUMO1-mediated SUMOylation of NF-κB inhibits ubiquitination-mediated degradation of IκBα and downregulates NF-κB activation [57]. The modification by SUMO2/3 promoted the detachment of IκBα from NF-κB and upregulated NF-κB activation, ultimately inducing atherosclerosis [57,58]. On the other hand, SENP2 negatively regulates inflammation through deSUMOylation of NEMO induced by genotoxic stress and ultimately inhibits atherosclerosis [58]. These results indicated that NF-κB plays important roles in atherosclerosis, and the regulation of NF-κB SUMOylation has become a potential therapeutic target for atherosclerosis.
3.1.3. p53
p53 is an important tumor suppressor gene that determines cell response to stress conditions, such as apoptosis and cell cycle arrest [59]. When cells are stimulated, nuclear p53 is increased, and cell cycle regulators and pro-apoptotic genes are upregulated. The role of p53 in atherosclerotic endothelial cells is complicated [60]. In d-flow, PKC mediates the SUMOylation of p53 and induces reactive oxygen species (ROS), leading to the transfer of p53 from the nucleus to the cytoplasm of endothelial cells. Combined with the anti-apoptotic factor Bax and Bcl-2, SUMOylated p53 inhibited the anti-apoptotic and led to apoptosis, ultimately causing the occurrence of atherosclerosis [59]. On the other hand, SENP2 deSUMOylated p53 and inhibited atherosclerosis [3]. p90 ribosomal S6 kinase (p90RSK) is a serine/threonine kinase with two kinase functional domains. D-flow activation of p90RSK resulted in the extranuclear output of SENP2 and increasing SUMOylation of p53 and ERK5 [61]. These results indicated that deSUMOylation of p53 by SENP2 may be an effective strategy to prevent atherosclerosis.
3.1.4. PKC (Protein Kinase C)
PKC isoforms consist of a series of kinases chronically activated by hyperglycemia in diabetic patients and are essential in cell contractility and permeability. PKC is critical in regulating insulin resistance, gluconeogenesis, liver lipid metabolism, and key components of vascular homeostasis and is necessary for atherosclerosis. Changes in the PKC signaling pathway in diabetic patients may lead to various pathological results, thus accelerating DM-mediated atherosclerosis. PKC gene knockout reduced the size of plaques and reduced the production of inflammatory factors [62]. PKC was modified by SUMO1 at K465 and was deSUMOylated by SENP1 [62]. SUMOylation inhibited the function of PKC and played critical roles in the progression of atherosclerosis. The regulation of PKC SUMOylation may become a potential therapeutic target for DM-mediated atherosclerosis.
3.2. CHD (Congenital Heart Disease)
The development of the vertebrate heart begins at the later stage of fetal development and involves a series of complex morphogenetic events, which are strictly regulated by the specific expression of various genes (Table 2) [29]. CHD is a congenital cardiac structural abnormality, and it is a common congenital defect in infants, accounting for about 1% of all newborns [4]. ASDs and VSDs are the most common CHDs, which are characterized by an existing connection between the atria and ventricles [5]. The interaction between different kinds of proteins is the basis of normal cardiac development, and the imbalance of their expression regulated by SUMOylation may lead to changes in protein function and the occurrence of CVD (Figure 4).
Table 2.
The roles of SUMOylation of substrates in CVD.
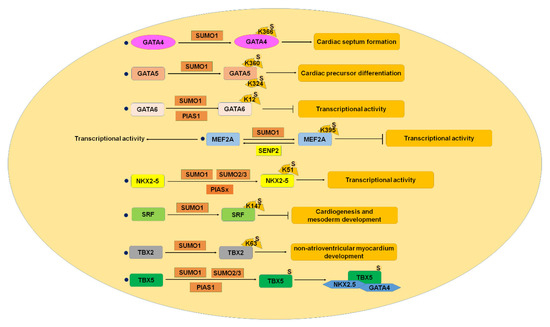
Figure 4.
SUMOylation and CHD. GATA4-6, MEF2, NKX2-5, SRF, TBX2, and TBX5 were SUMOylated, and SUMOylation of these proteins played critical roles in cardiac development, whereas imbalance of SUMOylation caused congenital heart disease. CHD, congenital heart disease; MEF2, myocyte enhancer factor 2; SRF, serum responsive factor. S: SUMO.
3.2.1. GATA4
GATA4 is a transcription factor and one of the most important members of the GATA superfamily of DNA-binding transcription factors, which is reported to be one of the primary causes of CHD [85]. The GATA family plays important roles in regulating cardiac early stage and end-stage development, proliferation and differentiation of cardiac progenitor cells, and cardiac-specific gene expression [86]. GATA4 plays a key role in early heart development as one of the early markers of cardiomyocytes in embryonic development. GATA4 was SUMOylated, and the SUMOylation of GATA4 results in enhanced transcriptional activity and altered nuclear localization [87]. SUMO1 modification of GATA4 on lysine 366 promoted specific gene expression in cardiac pluripotent cells. The SUMOylation of GATA4 may play a role in a complex that regulates a set of genes needed to form cardiac septum. GATA4 mutation was found to be associated with non-syndromic cardiac septal defect [63]. Multiple GATA4 mutations were also found in patients with ASDs and VSDs [88]. Histological analysis suggests that the SUMOylation of GATA4 is essential in the early development of the heart, indicating that the regulation of GATA4 SUMOylation is a potential therapeutic target for CHD treatment.
3.2.2. GATA5
GATA5 was found to be an important transcriptional factor in zebrafish heart development. GATA5 is reported to be modified by SUMO1 at lysine 324 (K324) and lysine 360 (K360). The SUMOylation sites’ mutation affected the transcriptional activity of GATA5 but did not affect the subcellular localization. Studies have shown that K324R mutant GATA5 could rescue the damaged cardiac precursor differentiation, while K360R mutant GATA5 cannot. In addition, the SUMOylation of GATA5 could restore cardiac insufficiency caused by cardiac dysplasia [64]. These results suggest that SUMOylation of GATA5 plays important roles in cardiac development.
3.2.3. GATA6
GATA6 is one of the most important members of the DNA binding transcription factor in the GATA superfamily and is an important factor in the regulation of cardiac development [86]. GATA6 was modified by SUMO1 at lysine 12 (K12), and the specific E3 ligase PIAS1 preferentially promoted the conjugation of SUMO1 to GATA6 via its RING finger domain [20,65]. The SUMOylation of GATA6 inhibited its transcriptional activity but did not affect the subcellular localization [65]. The SUMOylation provides a new regulated mechanism for the modification of GATA-6 activity in cardiac development.
3.2.4. MEF2 (Myocyte Enhancer Factor 2)
The MEF2 family is transcription factor that regulates muscle-specific genes involved in cardiac development, including MEF2A, MEF2B, MEF2C, and MEF2D [66]. Reducing the activity of MEF2 inhibits cardiomyocyte differentiation and cardiac development [71]. All these four transcription factors have the SUMO consensus sequence. Previous studies have shown that MEF2A, MEF2C, and MEF2D were modified by SUMO [89]. MEF2A can be modified by SUMO1 in vivo and in vitro. The SUMOylation of MEF2A inhibited its transcriptional activity, but the mutation of lysine 395 to arginine (K395A) in MEF2A enhanced the transcriptional activity of MEF2A [66]. SENP2 enhanced the transcriptional activity of MEF2A in activity dependent-regulation of MEF2A deSUMOylation. Taken together, these results showed that MEF2 played critical roles in embryonic cardiac development.
3.2.5. NKX2-5
NKX2-5 is a cardiac-specific gene that regulates cardiac progenitor cell differentiation, mature and atrioventricular conduction system, and the formation of the atrioventricular outflow [90]. NKX2-5 was SUMOylated by SUMO1 on lysine 51, and SUMO E3 ligase PIASx enhanced the attachment between them [44]. NKX2-5 is modified by SUMO1 and can be modified by SUMO2/3 but to a much lesser degree than SUMO1, and SUMOylation promoted the transcription activity of NKX2-5 [91,92]. SUMOylation regulates NKX2-5 function by changing the interaction between NKX2-5 and related proteins and promotes stable complexes containing NKX2-5. The SUMOylation of NKX2-5 is necessary for normal cardiac development, whereas NKX2-5 mutation was associated with forming a variety of CHD [92]. These results indicate that NKX2-5 SUMOylation plays an important role in CHD and becomes a therapeutic target for CHD.
3.2.6. SRF (Serum Responsive Factor)
SRF is a DNA binding protein family known as MADS Box, which has a highly conserved DNA binding/dimerization [93]. In vertebrates, SRF is mainly expressed in mesoderm and neuroectoderm tissues. SRF plays an important role during cardiac development, particularly in cardiogenesis and mesoderm development [94]. Serum stimulation activates SRF-mediated early gene transcription through various signal transduction pathways, such as newborn heart and smooth muscle-related genes [95]. SRF deficiency decreased cell proliferation and survival rate and increased cell apoptosis [96]. Studies have shown that SRF was SUMOylated by SUMO1 on its lysine residue 147 (K147), and the SUMOylation of SRF enhanced activation of the c-fos promoter but inhibited cardiac α-actin promoter [95]. These results suggest that the regulation of SRF SUMOylation plays critical roles in cardiac development.
3.2.7. TBX2
TBX2 is a member of the T-Box transcription factors family and plays vital roles in cardiac cushion development [97]. Changes in TBX2 activity are associated with a variety of human congenital diseases and cancers. Changes in numerous factors associated with CHD are accompanied by changes in TBX2 [98]. It was reported that common genetic variation in the TBX2 promoter region is strongly associated with the cardiac development and occurrence of CHD [99]. TBX2 is associated with the non-atrioventricular myocardium in the atrioventricular and outflow canals during cardiac development. TBX2 was identified to be modified by SUMO1, and TBX2 SUMOylation plays an important role in cardiac development, especially non-atrioventricular myocardium in the atrioventricular and outflow canals [100]. Therefore, the SUMOylation of TBX2 plays an important role in cardiac development.
3.2.8. TBX5
TBX5 is a member of the T-Box transcription factor family and plays a critical role in cardiac development [101]. In the early development of the heart, TBX5 acts as an upstream transcriptional activator to promote cardiomyocyte maturation [102]. In the late stage of heart development, TBX5 acts on the heart conduction system and maintains the function of mature cardiomyocytes [70,103]. TBX5 was SUMOylated by SUMO1, and SUMOylation enhanced the transcriptional activity of TBX5 [104]. The E3 ligase PIAS1 specifically enhanced the SUMOylation of TBX5 and enhanced the coordinate regulation with NKX2-5 and GATA4 [104]. These studies suggest the important roles of TBX5 in the progression of cardiac development.
3.3. HF (Heart Failure)
HF is a complex disease caused by valve abnormality, coronary artery disease, hypertension, and other pathological changes [6]. Cardiac energy metabolism and mitochondrial biogenesis defects lead to HF and cardiac hypertrophy [7]. Cardiac hypertrophy is a transitional stage of HF that initiates a process of molecular and structural changes and lead to ventricular remodeling. Initially, persistent cardiac hypertrophy is an adaptive response and increases the risk of HF and sudden death [105]. Treatment with drugs for CVD is effective in reducing left ventricular hypertrophy and related mortality [106], and the SUMOylation pathway is involved in the development of HF and progression of cardiac hypertrophy (Figure 5).

Figure 5.
SUMOylation and HF. HSF2, myocardin, PARIS, PPARγ1, and SERCA2a were SUMOylated, and SUMOylation of these proteins played crucial roles in the progression of heart failure. HF, heart failure; HSF2, heat shock transcription factor 2; PARIS, parkin-interacting substrate; PPARγ1, peroxisome proliferation activated receptor γ1; SERCA2a, sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 2a. S: SUMO.
3.3.1. HSF2 (Heat Shock Transcription Factor 2)
Cardiac hypertrophy is a major feature of early hypertensive heart failure. Insulin-like growth factor II receptor (IGF-IIR) plays key roles in cardiomyocyte hypertrophy induced by hypertensive angiotensin II [72]. IGF-IIR is regulated by HSF2, which is modified by SUMO1 during HF [72]. PGCF2 (polycomb group ring finger 2) significantly reduced the SUMOylation of HSF2 in spontaneously hypertensive rat hearts. Inhibition of HSF2 SUMOylation induced cardiac hypertrophy in hypertensive rats. The treatment by angiotensin-receptor I blocker (ARB) restored the SUMOylation of HSF2 and alleviated cardiac defects in spontaneously hypertensive rats. The studies showed that the PGCF2-SUMO1-HSF2-IGF-IIR pathway had a profound effect on cardiac hypertrophy in patients with hypertensive HF [72]. These results suggested that the regulation of HSF2 SUMOylation is a potential target for the treatment of cardiac hypertrophy.
3.3.2. Myocardin
Myocardin belongs to the SAP superfamily and is only expressed in cardiac smooth muscle during embryonic development and plays a key role in cardiomyocyte hypertrophy [107]. Myocardin affects the growth of cardiomyocytes by regulating the stability and metabolic activity of cardiomyocytes [108]. Myocardin promotes smooth muscle cell differentiation with a coactivator of SRF [109]. Loss of function of myocardin leads to serious defects in vascular smooth muscle development [110]. Abnormal expression of myocardin leads to hypertrophic cardiomyopathy and HF, and its mutation may lead to cardiovascular abnormalities in humans. It was reported that myocardin was SUMOylated by SUMO1 in K445, and SUMOylation of myocardin by SUMO1 and E3 PIAS1 reversed the expression of myocardial genes and enhanced the cardiac hypertrophy of primary neonatal cardiomyocytes [74]. These results indicated the critical roles of myocardin SUMOylation in the development of cardiac hypertrophy.
3.3.3. PARIS (Parkin-Interacting Substrate)
PARIS is regulated by the ubiquitin proteasome system via the E3 ubiquitin ligase parkin [111]. PARIS is a transcriptional repressor containing four zinc finger domains and one KRAB domain to regulate DNA binding in the promoter region of target genes and play transcriptional repressor activity [112]. PARIS was SUMOylated by SUMO1 in K189 and K286 and was deSUMOylated by DJ-1 [75,76]. In cardiac hypertrophy, PARIS was SUMOylated and inhibited PGC1α transcription. As one of the major regulators of mitochondrial function and biogenesis, the PGC1α transcription decrease causes mitochondrial dysfunction and ROS accumulation [113]. The accumulation of ROS in cardiac hypertrophy induced the degradation of DJ-1 and augmented the SUMOylation of PARIS, and reduced the promoter activity of PGC1α [114]. These results indicated the critical roles of PARIS SUMOylation in the regulation of mitochondrial function, and inhibition of the SUMOylation of PARIS may be a potential treatment of cardiac hypertrophy.
3.3.4. PPARγ1 (Peroxisome Proliferation-Activated Receptor γ1)
The utilization of cardiac energy is partially determined by the PPAR transcription factors, including PPARα, PPARβ, and PPARγ, and the heterodimer-forming RXRα as well as coactivator PGC-1α and inhibitor NCoR (nuclear receptor corepressor). The PPAR family regulates the transcription of genes related to energy regulation and lipid metabolism [115]. PPARγ1 plays critical roles in the development of myocardial fibrosis and the protection of cardiac oxidative stress. PPARγ1 was covalently modified by SUMO1 in K107 and K365 with E3 ligase PIAS1 and PIASxβ, and SUMOylation inhibited the transcriptional activity of PPARγ1 [77,116]. The SUMOylation of PPARγ1 regulated NF-κB in response to hypertrophy and inflammatory stimuli, suggesting a protective role of PPARγ1 SUMOylation in cardiac hypertrophy [115]. These results indicated that the SUMOylation of PPARγ1 is a potential target for the treatment of cardiac hypertrophy.
3.3.5. SERCA2a (Sarcoplasmic/Endoplasmic Reticulum Ca2+-ATPase 2a)
SERCA2a is a key pump that regulates the calcium cycle in cardiomyocytes and is responsible for the reuptake of calcium during excitation-contraction coupling [117]. Reduced activity and expression of SERCA2a are markers of HF in the body [118,119]. It has been demonstrated that SERCA2a can be modified by SUMO1 at lysine 480 and 585, and SUMOylation is essential to maintain the stability and activity of SERCA2a [78]. The content of SUMO1 and SUMOylated SERCA2a was greatly reduced in heart failure. After the knockout of SUMO1, the activity of SERCA2a was also reduced, which accelerated the impairment of heart function and the death of animals [120]. After restoring SUMO1 levels, SERCA2a levels rose, and the heart function of mice and swine went into remission [121,122]. SUMOylation of SERCA2a increased the stability and activity of SERCA2a, and the contractility of the myocardium increased. The SUMOylation of SERCA2a is of great significance for the treatment of HF [123]. There is a small molecule, N106, that can activate E1 ligase and trigger the SUMOylation of SERCA2a and enhance the contractility of cardiomyocytes and improve ventricular function in heart failure mice [124]. These studies indicate that the SUMOylation of SERCA2a is a potential target and a promising treatment for HF.
3.4. Ischemic Cardiomyopathy
AMI (acute myocardial infarction) is ischemic heart disease with acute hypoxia in the myocardial tissue and is one of the leading causes of death [125]. Restoration of reperfusion is the main therapeutic method to limit the size of myocardial infarction, save the viable myocardium, maintain the systolic function of the heart, and delay the occurrence of HF [14,82]. Readjustment of blood flow restores oxygen and nutrient supply but may damage the cardiac muscle and induce myocardial I/R injury. Studies of animal models of AMI have shown that fatal reperfusion injury accounts for half of the myocardial infarction [126]. However, the underlying mechanisms of myocardial I/R injury have not been fully elucidated [127]. SUMOylation has been found to play a significant role in myocardial I/R injury, and several SUMOylated proteins interfere with multiple mechanisms of myocardial I/R injury (Figure 6).
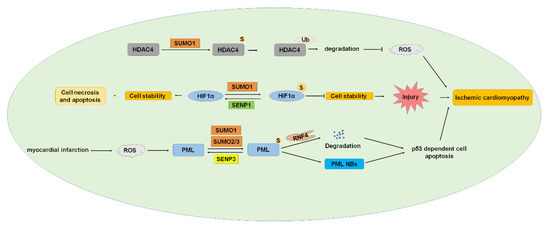
Figure 6.
SUMOylation and ischemic cardiomyopathy. HDAC4, HIF1α, and PML were SUMOylated, and SUMOylation of these proteins played essential roles in developing ischemic cardiomyopathy. HDAC4, histone deacetylase 4; HIF1α, hypoxia-inducible factor 1α; PML, promyelocytic leukemia. S: SUMO.
3.4.1. HDAC4 (Histone Deacetylase 4)
HDAC induces deacetylation, chromatin concentration, and transcription inhibition [128]. According to its function and sequence homology, HDAC can be divided into four types. HDAC4 belongs to class II HDACs and is distributed in the nucleus and cytoplasm. HDAC4 is highly expressed in skeletal muscle, brain, and heart [129,130,131]. HDAC4 contributes to myocardial I/R injury due to mitochondrial damage and reactive oxygen species (ROS) during reperfusion. Inhibiting HDAC4 expression promoted cardiac repair, improved cardiac function, and inhibited cardiac remodeling [132]. HDAC4 was modified by SUMO1, and SUMOylation of HDAC4 reduced the generation of ROS [80]. The SUMOylation of HDAC4 mediates ubiquitination and promotes the degradation of HDAC4 in I/R, which may be related to the decrease of ROS production after the degradation of HDAC4 [80]. ROS-mediated oxidative stress has been widely recognized as one of the important mechanisms of myocardial I/R injury [133]. These results indicate that the SUMOylation of HDAC4 plays important roles and is a potential therapeutic target for myocardial I/R injury.
3.4.2. HIF1α (Hypoxia-Inducible Factor 1α)
HIF1α is an important transcription factor that plays a critical role in protecting the I/R process by participating in the transcription response under hypoxia conditions. HIF1α was modified by SUMO1 to regulate the expression of heme oxygenase-1 in the rostral ventrolateral medulla (RVLM), playing a critical role in the regulation of brain death [8]. SENP1-mediated deSUMOylation of HIF1α leads to cell necrosis and apoptosis and plays an indispensable role in myocardial infarction after I/R in response to cell hypoxia. SUMOylation of HIF1α reduces its stability and leads to degradation, whereas SENP1 deSUMOylates HIF1α and improves its stability [134,135]. These studies showed that the decrease of SENP1 levels aggravated the I/R injury of cardiomyocytes by the HIF1α-dependent pathway, and the overexpression of HIF1α reversed the worsening effect of SENP1 downregulation on cell death [82]. These results suggest that the SUMOylation of HIF1α plays an important role in I/R injury and might be a potential target of AMI treatment.
3.4.3. PML (Promyelocytic Leukemia)
PML is the important organizer of nuclear bodies (NBs) and is expressed in several isoforms, including PML I to VII [136]. PML is SUMOylated by SUMO1 and SUMO2/3 and de-SUMOylated by SENP3 [42,137], and SUMOylation is critical in PML NBs formation [83]. Human ring-finger protein 4 (RNF4) is an E3 ubiquitin ligase and targets poly-SUMOylated proteins to degrade by the ubiquitin-proteasome pathway [138]. RNF4 is involved in controlling the degradation of PML and the transcriptional activity of several transcription factors [139,140]. PML is an ROS sensor, and myocardial infarction (MI) generated ROS, and ROS induced the increase of PML SUMOylation and PML-NB formation, leading to p53-dependent cell apoptosis. Overexpression of PML or knockdown of RNF4 enhances p53 recruitment and activation of PML SUMOylation and PML NBs accumulation, which aggravated ischemia-induced cardiomyocyte apoptosis in vivo and cell injury [84]. These results indicated the important roles of PML SUMOylation in ischemia and suggested that intervening in the dynamic pattern of PML SUMOylation under oxidative stress is a potential treatment of MI.
3.4.4. Target Protein Network
It has been reported that SUMOylation is enhanced to protect the brain during cardiac ischemia [141,142]. To identify the potential target proteins of SUMOylation in the mouse heart with cardiac I/R, an unbiased proteomic analysis was conducted. There were 102 proteins that significantly interacted with SUMO1, including the composition of the SUMO mechanism and the ubiquitin proteasome system [143,144], multiple subunits of 20S proteasome core particle, and metabolic enzymes involved in glycolytic and oxidative metabolic pathways [145]. High molecular weight SUMO1 conjugates were significantly reduced 24 h after reperfusion. In total, 15 proteins showed significant changes in SUMOylation at ischemia, and 13 and 12 proteins showed changes in SUMOylation at 1 and 24 h after reperfusion, respectively. Further, 55 proteins were found to be potential SUMO2/3 target proteins, including ubiquitin and proteasome subunits, gene expression regulators and transcription factors, RNA binding proteins and splicing regulators, heat shock proteins, and specific proteins that play important roles in cardiac function. Myocardial ischemia decreased the SUMO level of the whole protein and the number and catalytic activity of de-SUMO enzymes [145]. The heart SUMO2 target protein undergoes a dynamic adaptation process in cardiac I/R. Only one protein was SUMOylated differently at ischemia, and 8 and 11 target proteins were significantly altered 1 and 24 h after reperfusion, respectively. SENP3 is the major deSUMOylation enzyme in mouse hearts [145]. These results identified potential targets of SUMOylation during cardiac I/R and provided potential therapeutic targets for the treatment of cardiac ischemia.
4. Concluding Remarks and Perspective
Cardiovascular development and function are regulated by complex factors and signal pathways, and cardiovascular disorders lead to a series of stress responses [146,147]. SUMOylation is the PTM of proteins by a series of enzyme-linked reactions. Acute cardiac hypoxia induces elevated late sodium current and action potential prolongation in the heart and causes pro-arrhythmic change by SUMOylation of Nav1.5 channels [148]. In the decades since its discovery, studies related to SUMOylation in a variety of diseases have been extensively reported, including cardiovascular development and function [29,30].
Many factors regulated by SUMOylation and the imbalance of protein SUMOylation are involved in different types of CVDs, including atherosclerosis, CHD, HF, and ischemic cardiomyopathy. The previous studies indicated that SUMOylation is closely related to the occurrence and development of CVDs (Figure 7). The disruption of SUMOylation balance leads to severe CVDs. However, the molecular mechanism of SUMOylation in CVD has been preliminarily studied. It is possible to determine endogenous SUMOylated proteins under native conditions and identify endogenous SUMOylation sites on factors important in cardiac functions [149,150]. More SUMOylated proteins will be discovered during cardiovascular development and maintenance of cardiac function with further studies. Although the therapeutic strategies of CVDs have been greatly improved, there is still a lack of accurate, effective, and scientific unified treatment. Target therapy is one favorable way for future medical development and destination of CVD treatment. SUMOylation not only regulates the function of target proteins but also affects other post-translational modification processes. It is possible that SUMOylation co-regulates the same physiological process with other post-translational modifications. Therefore, the synergistic effects of SUMOylation and other post-translational modifications on the progression of CVD should be considered in the clinical treatment. Exploring the dynamic balance between the target of SUMOylation and also other PTM will help to better understand the biological functions of SUMOylation and the clinical treatment of CVDs.
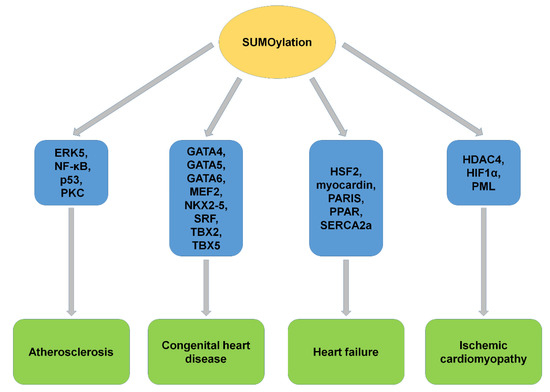
Figure 7.
The SUMOylation of substrates and CVD. SUMOylation of target proteins played critical roles and showed potential clinical implications in CVD, including atherosclerosis, congenital heart disease, heart failure, and ischemic cardiomyopathy. ERK5, extracellular signal-regulated kinase 5; PKC, protein kinase C; MEF2, myocyte enhancer factor 2; SRF, serum responsive factor; HSF2, heat shock transcription factor 2; PARIS, parkin-interacting substrate; PPARγ1, peroxisome proliferation activated receptor γ1; SERCA2a, sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 2a; HDAC4, histone deacetylase 4; HIF1α, hypoxia-inducible factor 1α; PML, promyelocytic leukemia.
Author Contributions
Y.Q. and H.W. conceived the idea for the review. C.D. performed the retrieval and collection of relevant literatures. X.C., Q.S., W.L., Q.W., H.Y., Z.Z., and X.W. provided suggestions. C.D., H.W., and Y.Q. wrote the paper. C.D. designed and prepared the figures. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (81671294 and 81870241 to Y.Q.) and the Fundamental Research Funds for the Central Universities (GK201903066 to H.W.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We are thankful for the financial support of the National Natural Science Foundation of China (81671294 and 81870241 to Y.Q.) and the Fundamental Research Funds for the Central Universities (GK201903066 to H.W.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Abe, J.I.; Sandhu, U.G.; Hoang, N.M.; Thangam, M.; Quintana-Quezada, R.A.; Fujiwara, K.; Le, N.T. Coordination of Cellular Localization-Dependent Effects of Sumoylation in Regulating Cardiovascular and Neurological Diseases. Adv. Exp. Med. Biol. 2017, 963, 337–358. [Google Scholar] [PubMed]
- Dehnavi, S.; Sadeghi, M.; Penson, P.E.; Banach, M.; Jamialahmadi, T.; Sahebkar, A. The Role of Protein SUMOylation in the Pathogenesis of Atherosclerosis. J. Clin. Med. 2019, 8, 1856. [Google Scholar] [CrossRef] [PubMed]
- Heo, K.S.; Chang, E.; Le, N.T.; Cushman, H.; Yeh, E.T.; Fujiwara, K.; Abe, J. De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow-induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ. Res. 2013, 112, 911–923. [Google Scholar] [CrossRef]
- Ahuja, P.; Sdek, P.; MacLellan, W.R. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 2007, 87, 521–544. [Google Scholar] [CrossRef] [PubMed]
- Henning, R.J. Diagnosis and treatment of adults with congenital heart disease. Future Cardiol. 2020, 16, 317–342. [Google Scholar] [CrossRef] [PubMed]
- Bui, A.L.; Horwich, T.B.; Fonarow, G.C. Epidemiology and risk profile of heart failure. Nat. Rev. Cardiol. 2011, 8, 30–41. [Google Scholar] [CrossRef]
- Mendler, L.; Braun, T.; Muller, S. The Ubiquitin-Like SUMO System and Heart Function: From Development to Disease. Circ. Res. 2016, 118, 132–144. [Google Scholar] [CrossRef]
- Chan, J.Y.; Tsai, C.Y.; Wu, C.H.; Li, F.C.; Dai, K.Y.; Sun, E.Y.; Chan, S.H.; Chang, A.Y. Sumoylation of hypoxia-inducible factor-1alpha ameliorates failure of brain stem cardiovascular regulation in experimental brain death. PLoS ONE 2011, 6, e17375. [Google Scholar] [CrossRef]
- Ulrich, H.D. Regulating post-translational modifications of the eukaryotic replication clamp PCNA. DNA Repair. 2009, 8, 461–469. [Google Scholar] [CrossRef]
- Yeh, E.T. SUMOylation and De-SUMOylation: Wrestling with life’s processes. J. Biol. Chem. 2009, 284, 8223–8227. [Google Scholar] [CrossRef]
- Matunis, M.J.; Coutavas, E.; Blobel, G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 1996, 135 Pt 1, 1457–1470. [Google Scholar] [CrossRef]
- Mahajan, R.; Delphin, C.; Guan, T.; Gerace, L.; Melchior, F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 1997, 88, 97–107. [Google Scholar] [CrossRef]
- Kim, E.Y.; Chen, L.; Ma, Y.; Yu, W.; Chang, J.; Moskowitz, I.P.; Wang, J. Enhanced desumoylation in murine hearts by overexpressed SENP2 leads to congenital heart defects and cardiac dysfunction. J. Mol. Cell. Cardiol. 2012, 52, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, X.; Sheng, H.; Liu, S.; Li, Y.; Zhao, J.Q.; Warner, D.S.; Paschen, W.; Yang, W. Neuron-specific SUMO knockdown suppresses global gene expression response and worsens functional outcome after transient forebrain ischemia in mice. Neuroscience 2017, 343, 190–212. [Google Scholar] [CrossRef] [PubMed]
- Treuter, E.; Gustafsson, J.A. Wrestling rules in transrepression: As easy as SUMO-1, -2, -3? Mol. Cell 2007, 25, 178–180. [Google Scholar] [CrossRef]
- Bohren, K.M.; Nadkarni, V.; Song, J.H.; Gabbay, K.H.; Owerbach, D. A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J. Biol. Chem. 2004, 279, 27233–27238. [Google Scholar] [CrossRef]
- Boulanger, M.; Paolillo, R.; Piechaczyk, M.; Bossis, G. The SUMO Pathway in Hematomalignancies and Their Response to Therapies. Int. J. Mol. Sci. 2019, 20, 3895. [Google Scholar] [CrossRef]
- Mukhopadhyay, D.; Dasso, M. Modification in reverse: The SUMO proteases. Trends Biochem. Sci. 2007, 32, 286–295. [Google Scholar] [CrossRef]
- Cheng, J.; Kang, X.L.; Zhang, S.; Yeh, E.T.H. SUMO-Specific protease 1 is essential for stabilization of HIF1 alpha during hypoxia. Cell 2007, 131, 584–595. [Google Scholar] [CrossRef]
- Kang, X.L.; Qi, Y.T.; Zuo, Y.; Wang, Q.; Zou, Y.Q.; Schwartz, R.J.; Cheng, J.K.; Yeh, E.T.H. SUMO-Specific Protease 2 Is Essential for Suppression of Polycomb Group Protein-Mediated Gene Silencing during Embryonic Development. Mol. Cell 2010, 38, 191–201. [Google Scholar] [CrossRef]
- Qi, Y.T.; Zuo, Y.; Yeh, E.T.H.; Cheng, J.K. An Essential Role of Small Ubiquitin-like Modifier (SUMO)-specific Protease 2 in Myostatin Expression and Myogenesis. J. Biol. Chem. 2014, 289, 3288–3293. [Google Scholar] [CrossRef]
- Qi, Y.T.; Wang, J.X.; Bomben, V.C.; Li, D.P.; Chen, S.R.; Sun, H.; Xi, Y.T.; Reed, J.G.; Cheng, J.K.; Pan, H.L.; et al. Hyper-SUMOylation of the Kv7 Potassium Channel Diminishes the M-Current Leading to Seizures and Sudden Death. Neuron 2014, 83, 1159–1171. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, S.; Huang, J.; Dong, W.; Xiao, H.; Shao, H.; Cheng, J.; Wu, H.; Qi, Y. Hyper-SUMOylation of K(+) Channels in Sudden Unexplained Death in Epilepsy: Isolation and Primary Culture of Dissociated Hippocampal Neurons from Newborn Mice for Subcellular Localization. Methods Mol. Biol. 2018, 1684, 63–71. [Google Scholar]
- Wu, H.; Chen, X.; Cheng, J.; Qi, Y. SUMOylation and Potassium Channels: Links to Epilepsy and Sudden Death. Adv. Protein Chem. Struct. Biol. 2016, 103, 295–321. [Google Scholar]
- Smits, V.A.; Freire, R. USP7/HAUSP: A SUMO deubiquitinase at the heart of DNA replication. Bioessays 2016, 38, 863–868. [Google Scholar] [CrossRef]
- Bertke, M.M.; Dubiak, K.M.; Cronin, L.; Zeng, E.; Huber, P.W. A deficiency in SUMOylation activity disrupts multiple pathways leading to neural tube and heart defects in Xenopus embryos. BMC Genom. 2019, 20, 386. [Google Scholar] [CrossRef]
- Hay, R.T. SUMO: A history of modification. Mol. Cell 2005, 18, 1–12. [Google Scholar] [CrossRef]
- Johnson, E.S. Protein modification by SUMO. Annu. Rev. Biochem. 2004, 73, 355–382. [Google Scholar] [CrossRef]
- Gao, J.; Shao, K.; Chen, X.; Li, Z.; Liu, Z.; Yu, Z.; Aung, L.H.H.; Wang, Y.; Li, P. The involvement of post-translational modifications in cardiovascular pathologies: Focus on SUMOylation, neddylation, succinylation, and prenylation. J. Mol. Cell. Cardiol. 2020, 138, 49–58. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, T.H.; Li, D.W. Roles of SUMOylation in Heart Development and Cardiovascular Diseases. Curr. Mol. Med. 2017, 16, 877–884. [Google Scholar] [CrossRef]
- McMurray, J.J.; Smith, G.L. Calcium handling in the failing heart and SUMO—Weighing the evidence. N. Engl. J. Med. 2011, 365, 1738–1739. [Google Scholar] [CrossRef]
- Abe, J.; Manabe, I.; Aikawa, M.; Aikawa, E. Cardiovascular Inflammation 2012: Reactive Oxygen Species, SUMOylation, and Biomarkers in Cardiovascular Inflammation. Int. J. Inflam. 2013, 2013, 953463. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; McLendon, P.M.; Gulick, J.; James, J.; Khalili, K.; Robbins, J. UBC9-Mediated Sumoylation Favorably Impacts Cardiac Function in Compromised Hearts. Circ. Res. 2016, 118, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Nacerddine, K.; Lehembre, F.; Bhaumik, M.; Artus, J.; Cohen-Tannoudji, M.; Babinet, C.; Pandolfi, P.P.; Dejean, A. The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev. Cell 2005, 9, 769–779. [Google Scholar] [CrossRef]
- Lee, Y.J.; Mou, Y.; Maric, D.; Klimanis, D.; Auh, S.; Hallenbeck, J.M. Elevated global SUMOylation in Ubc9 transgenic mice protects their brains against focal cerebral ischemic damage. PLoS ONE 2011, 6, e25852. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L.; Wen, S.; Zhu, H.; Yu, W.; Moskowitz, I.P.; Shaw, G.M.; Finnell, R.H.; Schwartz, R.J. Defective sumoylation pathway directs congenital heart disease. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 468–476. [Google Scholar] [CrossRef]
- Pai, P.; Shibu, M.A.; Chang, R.L.; Yang, J.J.; Su, C.C.; Lai, C.H.; Liao, H.E.; Viswanadha, V.P.; Kuo, W.W.; Huang, C.Y. ERbeta targets ZAK and attenuates cellular hypertrophy via SUMO-1 modification in H9c2 cells. J. Cell. Biochem. 2018, 119, 7855–7864. [Google Scholar] [CrossRef]
- Lee, A.; Jeong, D.; Mitsuyama, S.; Oh, J.G.; Liang, L.; Ikeda, Y.; Sadoshima, J.; Hajjar, R.J.; Kho, C. The role of SUMO-1 in cardiac oxidative stress and hypertrophy. Antioxid. Redox Signal. 2014, 21, 1986–2001. [Google Scholar] [CrossRef] [PubMed]
- Haindl, M.; Harasim, T.; Eick, D.; Muller, S. The nucleolar SUMO-specific protease SENP3 reverses SUMO modification of nucleophosmin and is required for rRNA processing. EMBO Rep. 2008, 9, 273–279. [Google Scholar] [CrossRef]
- Kim, Y.R.; Jacobs, J.S.; Li, Q.; Gaddam, R.R.; Vikram, A.; Liu, J.; Kassan, M.; Irani, K.; Kumar, S. SUMO2 regulates vascular endothelial function and oxidative stress in mice. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H1292–H1300. [Google Scholar] [CrossRef]
- Kim, E.Y.; Zhang, Y.; Ye, B.; Segura, A.M.; Beketaev, I.; Xi, Y.; Yu, W.; Chang, J.; Li, F.; Wang, J. Involvement of activated SUMO-2 conjugation in cardiomyopathy. Biochim. Biophys. Acta 2015, 1852, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Huang, C.; Sun, X.; Xiang, B.; Wang, M.; Yeh, E.T.; Chen, Y.; Li, H.; Shi, G.; Cang, H.; et al. SENP3-mediated de-conjugation of SUMO2/3 from promyelocytic leukemia is correlated with accelerated cell proliferation under mild oxidative stress. J. Biol. Chem. 2010, 285, 12906–12915. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Zhang, Y.; Beketaev, I.; Segura, A.M.; Yu, W.; Xi, Y.; Chang, J.; Wang, J. SENP5, a SUMO isopeptidase, induces apoptosis and cardiomyopathy. J. Mol. Cell. Cardiol. 2015, 78, 154–164. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, H.; Iyer, D.; Feng, X.H.; Schwartz, R.J. Regulation of cardiac specific nkx2.5 gene activity by small ubiquitin-like modifier. J. Biol. Chem. 2008, 283, 23235–23243. [Google Scholar] [CrossRef]
- Rawlings, N.; Lee, L.; Nakamura, Y.; Wilkinson, K.A.; Henley, J.M. Protective role of the deSUMOylating enzyme SENP3 in myocardial ischemia-reperfusion injury. PLoS ONE 2019, 14, e0213331. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Yeh, E.T. Characterization of a family of nucleolar SUMO-specific proteases with preference for SUMO-2 or SUMO-3. J. Biol. Chem. 2006, 281, 15869–15877. [Google Scholar] [CrossRef]
- Zunino, R.; Schauss, A.; Rippstein, P.; Andrade-Navarro, M.; McBride, H.M. The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. J. Cell Sci. 2007, 120 Pt 7, 1178–1188. [Google Scholar] [CrossRef]
- Zunino, R.; Braschi, E.; Xu, L.; McBride, H.M. Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J. Biol. Chem. 2009, 284, 17783–17795. [Google Scholar] [CrossRef]
- Bjorkbacka, H.; Nilsson, J. Innate immunity in atherosclerosis. J. Innate Immun. 2010, 2, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Heo, K.S.; Lee, H.; Nigro, P.; Thomas, T.; Le, N.T.; Chang, E.; McClain, C.; Reinhart-King, C.A.; King, M.R.; Berk, B.C.; et al. PKCzeta mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J. Cell Biol. 2011, 193, 867–884. [Google Scholar] [CrossRef]
- Nicorescu, I.; Dallinga, G.M.; de Winther, M.P.J.; Stroes, E.S.G.; Bahjat, M. Potential epigenetic therapeutics for atherosclerosis treatment. Atherosclerosis 2019, 281, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Woo, C.H.; Shishido, T.; McClain, C.; Lim, J.H.; Li, J.D.; Yang, J.; Yan, C.; Abe, J. Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ. Res. 2008, 102, 538–545. [Google Scholar] [CrossRef]
- Sato, M.; Kawai-Kowase, K.; Sato, H.; Oyama, Y.; Kanai, H.; Ohyama, Y.; Suga, T.; Maeno, T.; Aoki, Y.; Tamura, J.; et al. c-Src and hydrogen peroxide mediate transforming growth factor-beta1-induced smooth muscle cell-gene expression in 10T1/2 cells. Arter. Thromb. Vasc. Biol. 2005, 25, 341–347. [Google Scholar] [CrossRef]
- Bornfeldt, K.E.; Tabas, I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011, 14, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Gareus, R.; Kotsaki, E.; Xanthoulea, S.; van der Made, I.; Gijbels, M.J.; Kardakaris, R.; Polykratis, A.; Kollias, G.; de Winther, M.P.; Pasparakis, M. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008, 8, 372–383. [Google Scholar] [CrossRef]
- Chang, E.; Abe, J.I. Kinase-SUMO networks in diabetes-mediated cardiovascular disease. Metabolism 2016, 65, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Desterro, J.M.; Rodriguez, M.S.; Hay, R.T. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol. Cell 1998, 2, 233–239. [Google Scholar] [CrossRef]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Teodoro, J.G.; Parker, A.E.; Zhu, X.; Green, M.R. p53-mediated inhibition of angiogenesis through up-regulation of a collagen prolyl hydroxylase. Science 2006, 313, 968–971. [Google Scholar] [CrossRef]
- Heo, K.S.; Le, N.T.; Cushman, H.J.; Giancursio, C.J.; Chang, E.; Woo, C.H.; Sullivan, M.A.; Taunton, J.; Yeh, E.T.; Fujiwara, K.; et al. Disturbed flow-activated p90RSK kinase accelerates atherosclerosis by inhibiting SENP2 function. J. Clin. Investig. 2015, 125, 1299–1310. [Google Scholar] [CrossRef]
- Tabit, C.E.; Shenouda, S.M.; Holbrook, M.; Fetterman, J.L.; Kiani, S.; Frame, A.A.; Kluge, M.A.; Held, A.; Dohadwala, M.M.; Gokce, N.; et al. Protein kinase C-beta contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation 2013, 127, 86–95. [Google Scholar] [CrossRef]
- Dentice, M.; Cordeddu, V.; Rosica, A.; Ferrara, A.M.; Santarpia, L.; Salvatore, D.; Chiovato, L.; Perri, A.; Moschini, L.; Fazzini, C.; et al. Missense mutation in the transcription factor NKX2-5: A novel molecular event in the pathogenesis of thyroid dysgenesis. J. Clin. Endocrinol. Metab. 2006, 91, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Yuan, H.; Liu, X.; Wang, H.; Chen, S.; Chen, Z.; de The, H.; Zhou, J.; Zhu, J. GATA5 SUMOylation is indispensable for zebrafish cardiac development. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sun, W.; Zhu, J.; Yuan, H.; Chu, M.; Wen, B. Modification of cardiac transcription factor Gata6 by SUMO. Biochimie 2020, 170, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Liu, B.; You, S.; Chen, L.; Dongmei, Q.; Gu, M.; Lu, Y.; Chen, Y.; Zhang, F.; Yu, B. SENP2 regulates MEF2A de-SUMOylation in an activity dependent manner. Mol. Biol. Rep. 2013, 40, 2485–2490. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Chen, Z.; Bartunkova, S.; Yamasaki, N.; Izumo, S. The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development 1999, 126, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Hikichi, T.; Miura, H.; Shibata, H.; Mitsunaga, K.; Yamada, Y.; Woltjen, K.; Miyamoto, K.; Hiratani, I.; Yamada, Y.; et al. Srf destabilizes cellular identity by suppressing cell-type-specific gene expression programs. Nat. Commun. 2018, 9, 1387. [Google Scholar] [CrossRef]
- Iwanicka-Pronicka, K.; Socha, M.; Jedrzejowska, M.; Krajewska-Walasek, M.; Jamsheer, A. Life-threatening cardiac episode in a Polish patient carrying contiguous gene microdeletion of the TBX5 and the TBX3 genes. Springerplus 2016, 5, 1638. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Z.; Zhang, W.; Nam, Y.J. Stoichiometric optimization of Gata4, Hand2, Mef2c, and Tbx5 expression for contractile cardiomyocyte reprogramming. Sci. Rep. 2019, 9, 14970. [Google Scholar] [CrossRef]
- Shalizi, A.; Bilimoria, P.M.; Stegmuller, J.; Gaudilliere, B.; Yang, Y.; Shuai, K.; Bonni, A. PIASx is a MEF2 SUMO E3 ligase that promotes postsynaptic dendritic morphogenesis. J. Neurosci. 2007, 27, 10037–10046. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Kuo, C.H.; Pai, P.Y.; Ho, T.J.; Lin, Y.M.; Chen, R.J.; Tsai, F.J.; Vijaya Padma, V.; Kuo, W.W.; Huang, C.Y. Inhibition of HSF2 SUMOylation via MEL18 upregulates IGF-IIR and leads to hypertension-induced cardiac hypertrophy. Int. J. Cardiol. 2018, 257, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Jiang, Y.; Yin, Y.; Li, Q.; He, J.; Jing, Y.; Qi, Y.T.; Xu, Q.; Li, W.; Lu, B.; et al. A regulatory circuit of miR-148a/152 and DNMT1 in modulating cell transformation and tumor angiogenesis through IGF-IR and IRS1. J. Mol. Cell Biol. 2013, 5, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Zhang, T.C.; Cao, D.; Wang, Z.; Antos, C.L.; Li, S.; Wang, Y.; Olson, E.N.; Wang, D.Z. Myocardin induces cardiomyocyte hypertrophy. Circ. Res. 2006, 98, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; Squitieri, F.; Krieger, E.; Vanacore, N.; van Swieten, J.C.; Brice, A.; van Duijn, C.M.; Oostra, B.; Meco, G.; et al. DJ-1 (PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol. Sci. 2003, 24, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Ohshima, T.; Koga, H.; Shimotohno, K. Transcriptional activity of peroxisome proliferator-activated receptor gamma is modulated by SUMO-1 modification. J. Biol. Chem. 2004, 279, 29551–29557. [Google Scholar] [CrossRef]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Chaanine, A.H.; Kizana, E.; Park, W.J.; Hajjar, R.J. SUMO1-dependent modulation of SERCA2a in heart failure. Nature 2011, 477, 601–605. [Google Scholar] [CrossRef]
- Oh, J.G.; Watanabe, S.; Lee, A.; Gorski, P.A.; Lee, P.; Jeong, D.; Liang, L.; Liang, Y.; Baccarini, A.; Sahoo, S.; et al. miR-146a Suppresses SUMO1 Expression and Induces Cardiac Dysfunction in Maladaptive Hypertrophy. Circ. Res. 2018, 123, 673–685. [Google Scholar] [CrossRef]
- Du, J.; Zhang, L.; Zhuang, S.; Qin, G.J.; Zhao, T.C. HDAC4 degradation mediates HDAC inhibition-induced protective effects against hypoxia/reoxygenation injury. J. Cell. Physiol. 2015, 230, 1321–1331. [Google Scholar] [CrossRef]
- Ghosh, T.K.; Aparicio-Sanchez, J.J.; Buxton, S.; Brook, J.D. HDAC4 and 5 repression of TBX5 is relieved by protein kinase D1. Sci. Rep. 2019, 9, 17992. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Fan, Y.; Liu, X.; Zhou, L.; Cheng, J.; Cai, R.; Xue, S. SENP1 protects against myocardial ischaemia/reperfusion injury via a HIF1alpha-dependent pathway. Cardiovasc. Res. 2014, 104, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Ishov, A.M.; Sotnikov, A.G.; Negorev, D.; Vladimirova, O.V.; Neff, N.; Kamitani, T.; Yeh, E.T.; Strauss, J.F., 3rd; Maul, G.G. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 1999, 147, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Han, Y.; Shao, X.; Paulo, P.; Li, W.; Zhu, M.; Tang, N.; Guo, S.; Chen, Y.; Wu, H.; et al. Knockdown of endogenous RNF4 exacerbates ischaemia-induced cardiomyocyte apoptosis in mice. J. Cell. Mol. Med. 2020, 24, 9545–9559. [Google Scholar] [CrossRef] [PubMed]
- Evans, T. Regulation of Cardiac Gene Expression by GATA-4/5/6. Trends Cardiovasc. Med. 1997, 7, 75–83. [Google Scholar] [CrossRef]
- Winick, J.; Abel, T.; Leonard, M.W.; Michelson, A.M.; Chardon-Loriaux, I.; Holmgren, R.A.; Maniatis, T.; Engel, J.D. A GATA family transcription factor is expressed along the embryonic dorsoventral axis in Drosophila melanogaster. Development 1993, 119, 1055–1065. [Google Scholar] [CrossRef]
- Qiu, C.; Wang, Y.; Zhao, H.; Qin, L.; Shi, Y.; Zhu, X.; Song, L.; Zhou, X.; Chen, J.; Zhou, H.; et al. The critical role of SENP1-mediated GATA2 deSUMOylation in promoting endothelial activation in graft arteriosclerosis. Nat. Commun. 2017, 8, 15426. [Google Scholar] [CrossRef]
- Liu, X.Y.; Yang, Y.Q.; Ma, J.; Lin, X.P.; Zheng, J.H.; Bai, K.; Chen, Y.H. Novel GATA4 mutations identified in patients with congenital atrial septal defects. Zhonghua Xin Xue Guan Bing Za Zhi 2010, 38, 724–727. [Google Scholar]
- Riquelme, C.; Barthel, K.K.; Liu, X. SUMO-1 modification of MEF2A regulates its transcriptional activity. J. Cell. Mol. Med. 2006, 10, 132–144. [Google Scholar] [CrossRef]
- Clark, C.D.; Lee, K.H. Second heart field-specific expression of Nkx2-5 requires promoter proximal interaction with Srf. Mech. Dev. 2020, 162, 103615. [Google Scholar] [CrossRef]
- Lyons, I.; Parsons, L.M.; Hartley, L.; Li, R.; Andrews, J.E.; Robb, L.; Harvey, R.P. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 1995, 9, 1654–1666. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Chen, L.; Ma, Y.; Yu, W.; Chang, J.; Moskowitz, I.P.; Wang, J. Expression of sumoylation deficient Nkx2.5 mutant in Nkx2.5 haploinsufficient mice leads to congenital heart defects. PLoS ONE 2011, 6, e20803. [Google Scholar] [CrossRef]
- Norman, C.; Runswick, M.; Pollock, R.; Treisman, R. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell 1988, 55, 989–1003. [Google Scholar] [CrossRef]
- Johansen, F.E.; Prywes, R. Serum response factor: Transcriptional regulation of genes induced by growth factors and differentiation. Biochim. Biophys. Acta 1995, 1242, 1–10. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Minami, T.; Tojo, M.; Honda, Y.; Uchimura, Y.; Saitoh, H.; Yasuda, H.; Nagahiro, S.; Saya, H.; Nakao, M. Serum response factor is modulated by the SUMO-1 conjugation system. Biochem. Biophys. Res. Commun. 2003, 306, 32–38. [Google Scholar] [CrossRef]
- Niu, Z.; Yu, W.; Zhang, S.X.; Barron, M.; Belaguli, N.S.; Schneider, M.D.; Parmacek, M.; Nordheim, A.; Schwartz, R.J. Conditional mutagenesis of the murine serum response factor gene blocks cardiogenesis and the transcription of downstream gene targets. J. Biol. Chem. 2005, 280, 32531–32538. [Google Scholar] [CrossRef]
- Di Gregorio, A. T-Box Genes and Developmental Gene Regulatory Networks in Ascidians. Curr. Top. Dev. Biol. 2017, 122, 55–91. [Google Scholar] [PubMed]
- Pang, S.; Liu, Y.; Zhao, Z.; Huang, W.; Chen, D.; Yan, B. Novel and functional sequence variants within the TBX2 gene promoter in ventricular septal defects. Biochimie 2013, 95, 1807–1809. [Google Scholar] [CrossRef]
- Zhang, R.R.; Cai, K.; Liu, L.; Yang, Q.; Zhang, P.; Gui, Y.H.; Wang, F. A regulatory variant in TBX2 promoter is related to the decreased susceptibility of congenital heart disease in the Han Chinese population. Mol. Genet. Genom. Med. 2019, 7, e00530. [Google Scholar] [CrossRef] [PubMed]
- Huber, P.; Crum, T.; Clary, L.M.; Ronan, T.; Packard, A.V.; Okkema, P.G. Function of the C. elegans T-box factor TBX-2 depends on SUMOylation. Cell. Mol. Life Sci. 2013, 70, 4157–4168. [Google Scholar] [CrossRef]
- Steimle, J.D.; Moskowitz, I.P. TBX5: A Key Regulator of Heart Development. Curr. Top. Dev. Biol. 2017, 122, 195–221. [Google Scholar]
- Jia, Y.; Chang, Y.; Guo, Z.; Li, H. Transcription factor Tbx5 promotes cardiomyogenic differentiation of cardiac fibroblasts treated with 5-azacytidine. J. Cell. Biochem. 2019, 120, 16503–16515. [Google Scholar] [CrossRef] [PubMed]
- Kathiriya, I.S.; Rao, K.S.; Iacono, G.; Devine, W.P.; Blair, A.P.; Hota, S.K.; Lai, M.H.; Garay, B.I.; Thomas, R.; Gong, H.Z.; et al. Modeling Human TBX5 Haploinsufficiency Predicts Regulatory Networks for Congenital Heart Disease. Dev. Cell 2021, 56, 292–309.e9. [Google Scholar] [CrossRef]
- Beketaev, I.; Kim, E.Y.; Zhang, Y.; Yu, W.; Qian, L.; Wang, J. Potentiation of Tbx5-mediated transactivation by SUMO conjugation and protein inhibitor of activated STAT 1 (PIAS1). Int. J. Biochem. Cell Biol. 2014, 50, 82–92. [Google Scholar] [CrossRef]
- Frey, N.; Olson, E.N. Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef]
- Booz, G.W. Putting the brakes on cardiac hypertrophy: Exploiting the NO-cGMP counter-regulatory system. Hypertension 2005, 45, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Miano, J.M. Myocardin in biology and disease. J. Biomed. Res. 2015, 29, 3–19. [Google Scholar]
- Trembley, M.A.; Quijada, P.; Agullo-Pascual, E.; Tylock, K.M.; Colpan, M.; Dirkx, R.A., Jr.; Myers, J.R.; Mickelsen, D.M.; de Mesy Bentley, K.; Rothenberg, E.; et al. Mechanosensitive Gene Regulation by Myocardin-Related Transcription Factors Is Required for Cardiomyocyte Integrity in Load-Induced Ventricular Hypertrophy. Circulation 2018, 138, 1864–1878. [Google Scholar] [CrossRef] [PubMed]
- Cen, B.; Selvaraj, A.; Prywes, R. Myocardin/MKL family of SRF coactivators: Key regulators of immediate early and muscle specific gene expression. J. Cell. Biochem. 2004, 93, 74–82. [Google Scholar] [CrossRef]
- Wang, D.Z.; Olson, E.N. Control of smooth muscle development by the myocardin family of transcriptional coactivators. Curr. Opin. Genet. Dev. 2004, 14, 558–566. [Google Scholar] [CrossRef]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef]
- Nishida, T.; Yamada, Y. SUMOylation of the KRAB zinc-finger transcription factor PARIS/ZNF746 regulates its transcriptional activity. Biochem. Biophys. Res. Commun. 2016, 473, 1261–1267. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-induced production of mitochondrial reactive oxygen species: Potential mechanisms and relevance for cardiovascular disease. Antioxid. Redox Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef]
- Mukherjee, D.; Chander, V.; Bandyopadhyay, A. PARIS-DJ-1 Interaction Regulates Mitochondrial Functions in Cardiomyocytes, Which Is Critically Important in Cardiac Hypertrophy. Mol. Cell. Biol. 2020, 41, e00106-20. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Willis, M.S. The story so far: Post-translational regulation of peroxisome proliferator-activated receptors by ubiquitination and SUMOylation. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H515–H526. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 2005, 437, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Gwathmey, J.K.; Copelas, L.; MacKinnon, R.; Schoen, F.J.; Feldman, M.D.; Grossman, W.; Morgan, J.P. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ. Res. 1987, 61, 70–76. [Google Scholar] [CrossRef] [PubMed]
- del Monte, F.; O’Gara, P.; Poole-Wilson, P.A.; Yacoub, M.; Harding, S.E. Cell geometry and contractile abnormalities of myocytes from failing human left ventricle. Cardiovasc. Res. 1995, 30, 281–290. [Google Scholar] [CrossRef]
- Hasenfuss, G. Alterations of calcium-regulatory proteins in heart failure. Cardiovasc. Res. 1998, 37, 279–289. [Google Scholar] [CrossRef]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Gorski, P.A.; Fish, K.; Sanchez, R.; DeVita, R.J.; Christensen, G.; Dahl, R.; et al. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Xu, J.; Zhao, H.; Zheng, Q.; Xiao, X.; Ma, X.; Li, Y.; Du, X.; Liu, X. Zinc-Induced SUMOylation of Dynamin-Related Protein 1 Protects the Heart against Ischemia-Reperfusion Injury. Oxid. Med. Cell. Longev. 2019, 2019, 1232146. [Google Scholar] [CrossRef]
- Tilemann, L.; Lee, A.; Ishikawa, K.; Aguero, J.; Rapti, K.; Santos-Gallego, C.; Kohlbrenner, E.; Fish, K.M.; Kho, C.; Hajjar, R.J. SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci. Transl. Med. 2013, 5, 211ra159. [Google Scholar] [CrossRef]
- Shah, S.J.; Wasserstrom, J.A. SERCA2a gene therapy for the prevention of sudden cardiac death: A future theranostic for heart failure? Circulation 2012, 126, 2047–2050. [Google Scholar] [CrossRef]
- Samuel, T.J.; Rosenberry, R.P.; Lee, S.; Pan, Z. Correcting Calcium Dysregulation in Chronic Heart Failure Using SERCA2a Gene Therapy. Int. J. Mol. Sci. 2018, 19, 1086. [Google Scholar] [CrossRef]
- Lowenstein, C.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar]
- Frank, A.; Bonney, M.; Bonney, S.; Weitzel, L.; Koeppen, M.; Eckle, T. Myocardial ischemia reperfusion injury: From basic science to clinical bedside. Semin. Cardiothorac. Vasc. Anesth. 2012, 16, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Luo, Y.; Wang, S.; Zhu, H.; Li, D. Roles and mechanisms of SUMOylation on key proteins in myocardial ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2019, 134, 154–164. [Google Scholar] [CrossRef]
- Kuo, M.H.; Allis, C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 1998, 20, 615–626. [Google Scholar] [CrossRef]
- Fischle, W.; Emiliani, S.; Hendzel, M.J.; Nagase, T.; Nomura, N.; Voelter, W.; Verdin, E. A new family of human histone deacetylases related to Saccharomyces cerevisiae HDA1p. J. Biol. Chem. 1999, 274, 11713–11720. [Google Scholar] [CrossRef] [PubMed]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.H.; Bertos, N.R.; Vezmar, M.; Pelletier, N.; Crosato, M.; Heng, H.H.; Th’ng, J.; Han, J.; Yang, X.J. HDAC4, a human histone deacetylase related to yeast HDA1, is a transcriptional corepressor. Mol. Cell. Biol. 1999, 19, 7816–7827. [Google Scholar] [CrossRef]
- Zhang, L.X.; DeNicola, M.; Qin, X.; Du, J.; Ma, J.; Tina Zhao, Y.; Zhuang, S.; Liu, P.Y.; Wei, L.; Qin, G.; et al. Specific inhibition of HDAC4 in cardiac progenitor cells enhances myocardial repairs. Am. J. Physiol. Cell Physiol. 2014, 307, C358–C372. [Google Scholar] [CrossRef]
- Rodrigo, R.; Fernandez-Gajardo, R.; Gutierrez, R.; Matamala, J.M.; Carrasco, R.; Miranda-Merchak, A.; Feuerhake, W. Oxidative stress and pathophysiology of ischemic stroke: Novel therapeutic opportunities. CNS Neurol. Disord. Drug Targets 2013, 12, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Han, Y.; Wang, Y.; Sun, X.; Yan, S.; Yeh, E.T.; Chen, Y.; Cang, H.; Li, H.; Shi, G.; et al. SENP3 is responsible for HIF-1 transactivation under mild oxidative stress via p300 de-SUMOylation. EMBO J. 2009, 28, 2748–2762. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, J.; Yang, K.; Cang, H.; Huang, X.Z.; Li, H.; Yi, J. The biphasic redox sensing of SENP3 accounts for the HIF-1 transcriptional activity shift by oxidative stress. Acta Pharmacol. Sin. 2012, 33, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Maroui, M.A.; Kheddache-Atmane, S.; El Asmi, F.; Dianoux, L.; Aubry, M.; Chelbi-Alix, M.K. Requirement of PML SUMO interacting motif for RNF4- or arsenic trioxide-induced degradation of nuclear PML isoforms. PLoS ONE 2012, 7, e44949. [Google Scholar] [CrossRef]
- Niwa-Kawakita, M.; Ferhi, O.; Soilihi, H.; Le Bras, M.; Lallemand-Breitenbach, V.; de The, H. PML is a ROS sensor activating p53 upon oxidative stress. J. Exp. Med. 2017, 214, 3197–3206. [Google Scholar] [CrossRef]
- Plechanovova, A.; Jaffray, E.G.; McMahon, S.A.; Johnson, K.A.; Navratilova, I.; Naismith, J.H.; Hay, R.T. Mechanism of ubiquitylation by dimeric RING ligase RNF4. Nat. Struct. Mol. Biol. 2011, 18, 1052–1059. [Google Scholar] [CrossRef]
- Guo, B.; Sharrocks, A.D. Extracellular signal-regulated kinase mitogen-activated protein kinase signaling initiates a dynamic interplay between sumoylation and ubiquitination to regulate the activity of the transcriptional activator PEA3. Mol. Cell. Biol. 2009, 29, 3204–3218. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef]
- Yang, W.; Sheng, H.; Thompson, J.W.; Zhao, S.; Wang, L.; Miao, P.; Liu, X.; Moseley, M.A.; Paschen, W. Small ubiquitin-like modifier 3-modified proteome regulated by brain ischemia in novel small ubiquitin-like modifier transgenic mice: Putative protective proteins/pathways. Stroke 2014, 45, 1115–1122. [Google Scholar] [CrossRef]
- Gao, L.; Zhao, Y.; He, J.; Yan, Y.; Xu, L.; Lin, N.; Ji, Q.; Tong, R.; Fu, Y.; Gao, Y.; et al. The desumoylating enzyme sentrin-specific protease 3 contributes to myocardial ischemia reperfusion injury. J. Genet. Genom. 2018, 45, 125–135. [Google Scholar] [CrossRef]
- Gartner, A.; Muller, S. PML, SUMO, and RNF4: Guardians of nuclear protein quality. Mol. Cell 2014, 55, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Keiten-Schmitz, J.; Wagner, K.; Piller, T.; Kaulich, M.; Alberti, S.; Muller, S. The Nuclear SUMO-Targeted Ubiquitin Quality Control Network Regulates the Dynamics of Cytoplasmic Stress Granules. Mol. Cell 2020, 79, 54–67.e7. [Google Scholar] [CrossRef] [PubMed]
- Hotz, P.W.; Wiesnet, M.; Tascher, G.; Braun, T.; Muller, S.; Mendler, L. Profiling the Murine SUMO Proteome in Response to Cardiac Ischemia and Reperfusion Injury. Molecules 2020, 25, 5571. [Google Scholar] [CrossRef] [PubMed]
- Gruber, P.J. Cardiac development: New concepts. Clin. Perinatol. 2005, 32, 845–855, vii. [Google Scholar] [CrossRef] [PubMed]
- Glynne-Jones, P.; Boltryk, R.J.; Hill, M.; Zhang, F.; Dong, L.; Wilkinson, J.S.; Melvin, T.; Harris, N.R.; Brown, T. Flexible acoustic particle manipulation device with integrated optical waveguide for enhanced microbead assays. Anal. Sci. 2009, 25, 285–291. [Google Scholar] [CrossRef]
- Plant, L.D.; Xiong, D.; Romero, J.; Dai, H.; Goldstein, S.A.N. Hypoxia Produces Pro-arrhythmic Late Sodium Current in Cardiac Myocytes by SUMOylation of NaV1.5 Channels. Cell Rep. 2020, 30, 2225–2236.e4. [Google Scholar] [CrossRef]
- Lumpkin, R.J.; Gu, H.; Zhu, Y.; Leonard, M.; Ahmad, A.S.; Clauser, K.R.; Meyer, J.G.; Bennett, E.J.; Komives, E.A. Site-specific identification and quantitation of endogenous SUMO modifications under native conditions. Nat. Commun. 2017, 8, 1171. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Lyon, D.; Su, D.; Skotte, N.H.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Site-specific characterization of endogenous SUMOylation across species and organs. Nat. Commun. 2018, 9, 2456. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).