
Selenium and the 15kDa Selenoprotein Impact Colorectal Tumorigenesis by Modulating Intestinal Barrier Integrity

Abstract
:1. Introduction
2. Results
2.1. Growth Metrics
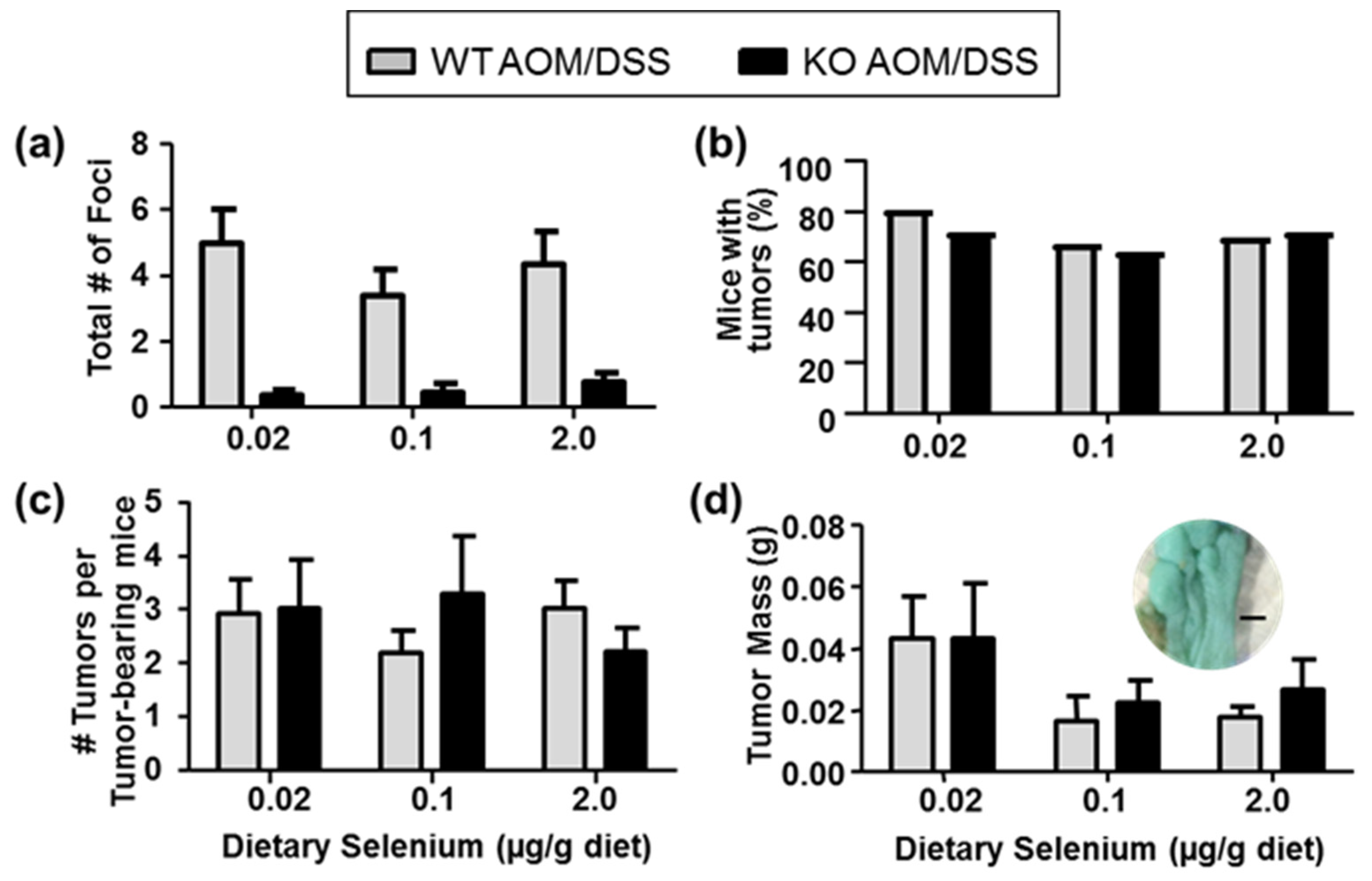
2.2. Aberrant Crypt Foci Formation and Tumorigenesis
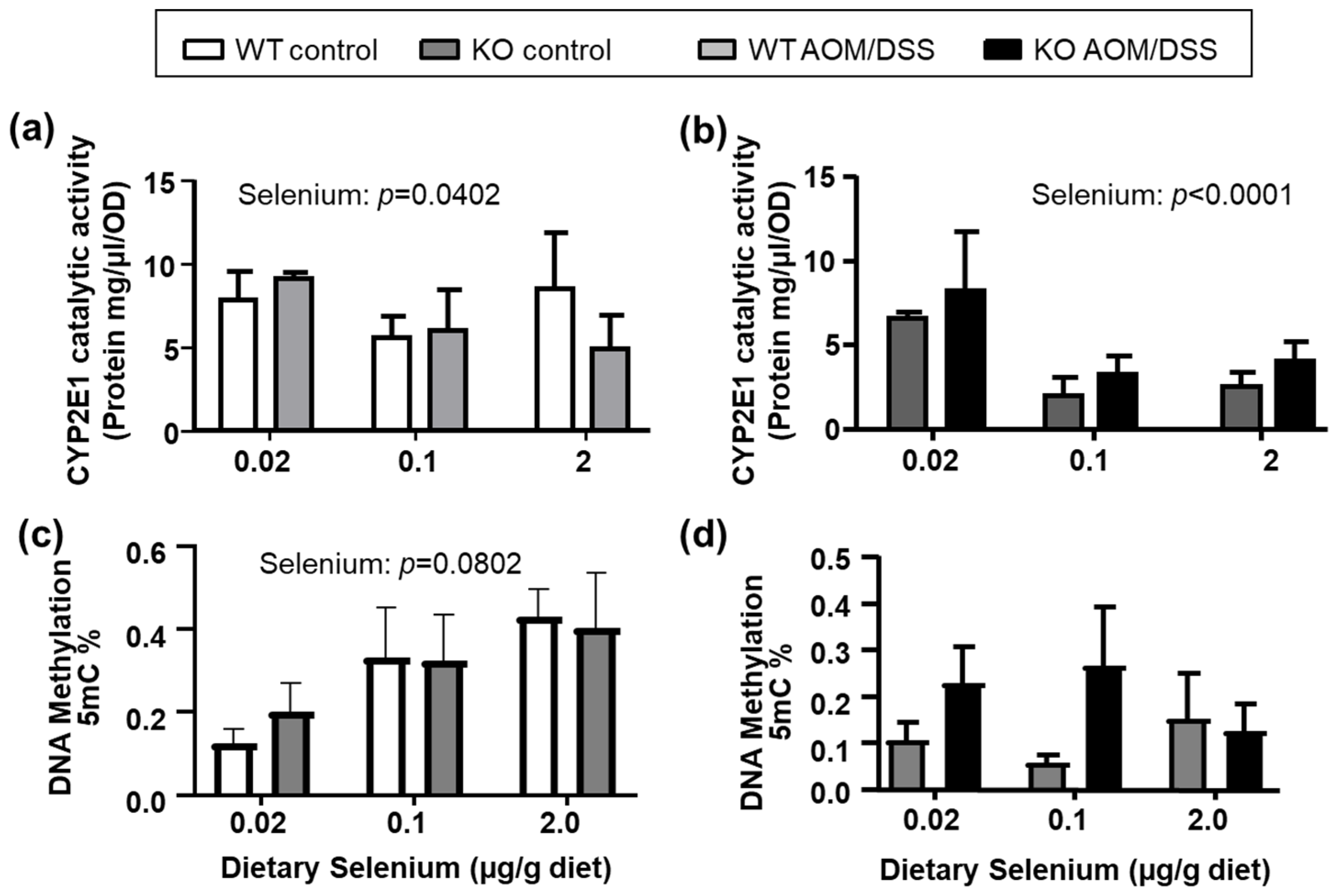
2.3. Expression and Catalytic Activity of Carcinogen-Activating Enzymes
2.4. Serum Inflammatory Markers
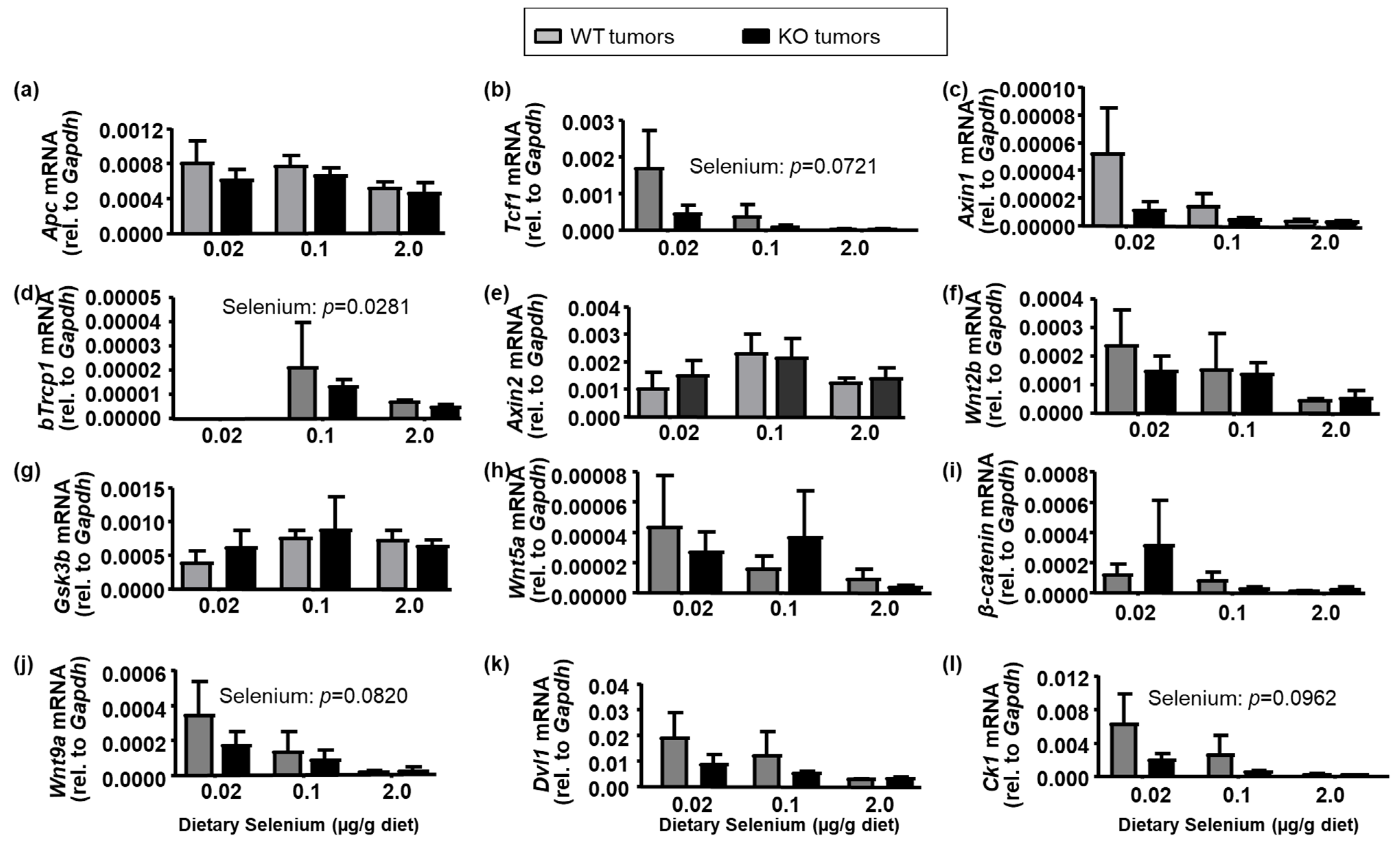
2.5. Colorectal Cancer Cell Signaling Pathways
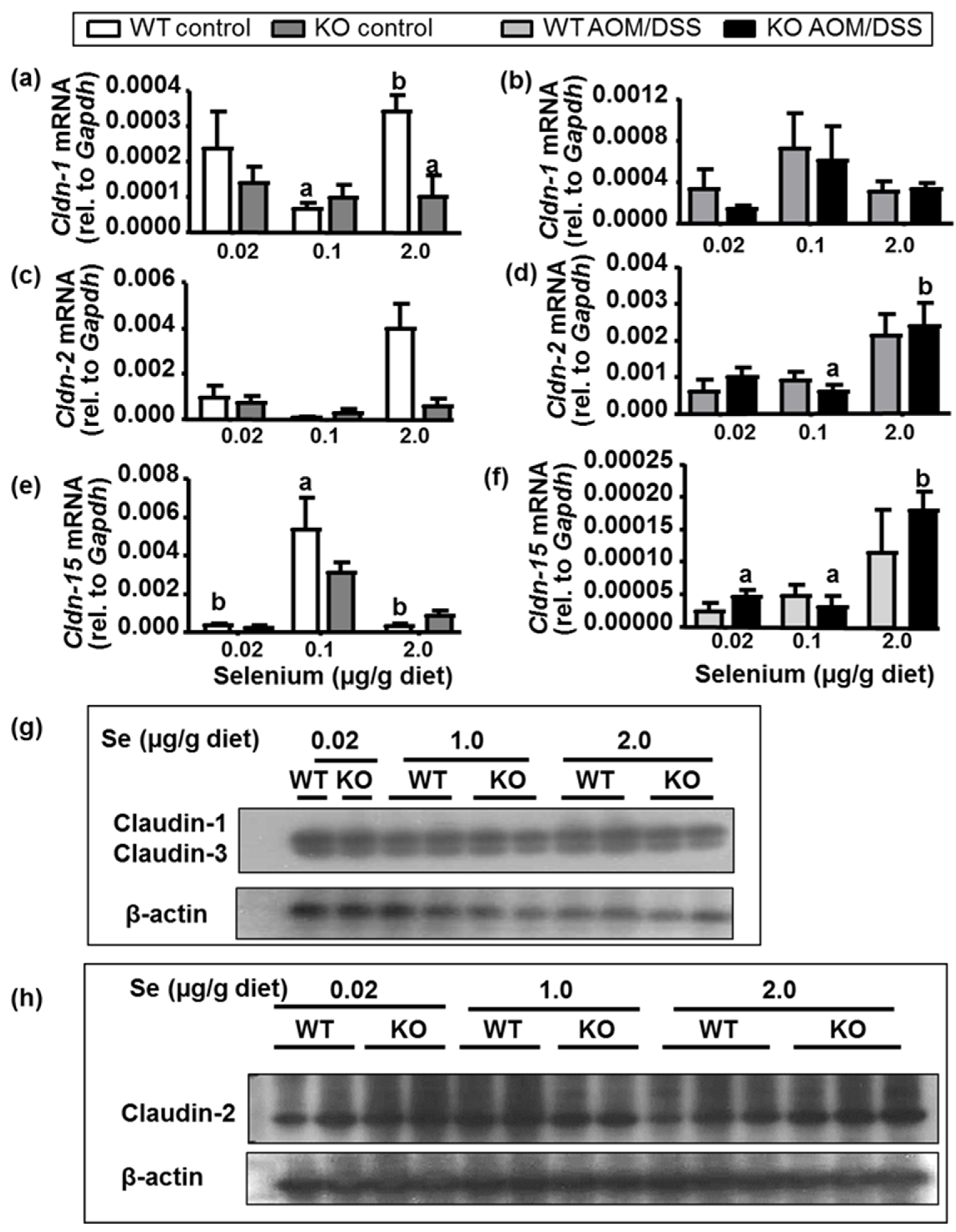
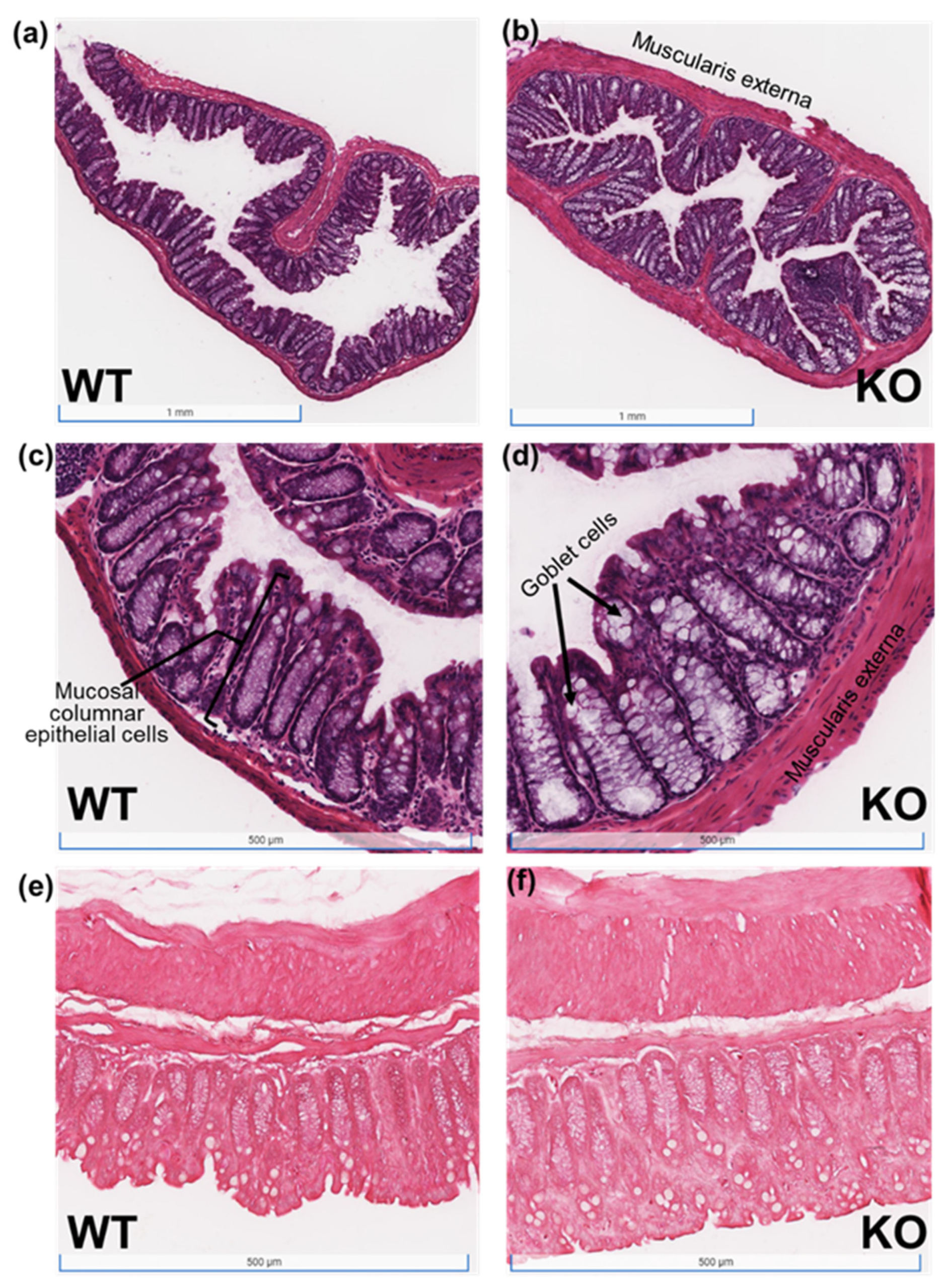
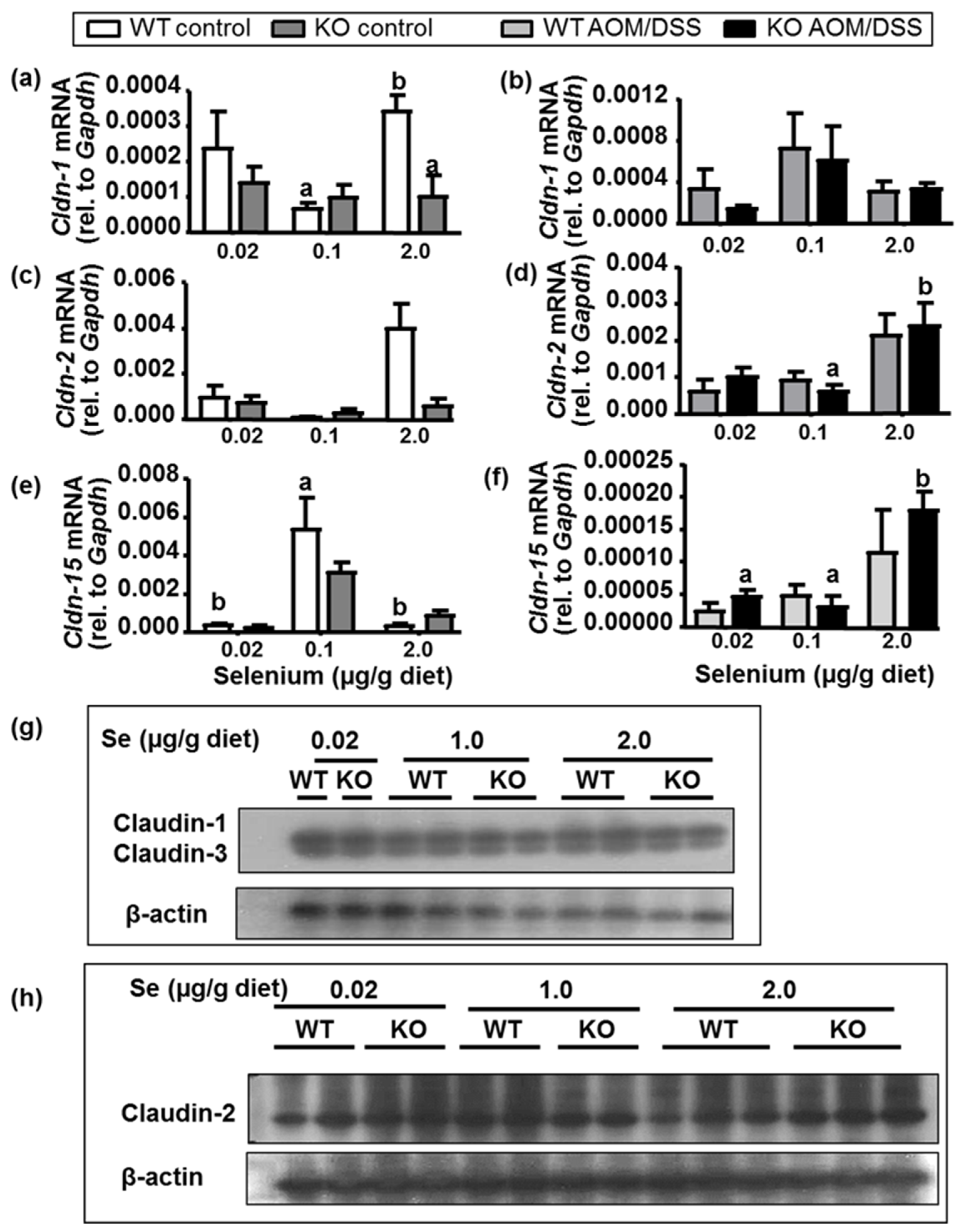
2.6. Intestinal Barrier Integrity
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Care Disclosure and Study Organization
4.3. Colorectal Tumor and ACF Analyses
4.4. Tissue Staining
4.5. Gene Expression Analysis of Mouse Liver and Colon Tissues
4.6. Cyp2e1 Catalytic Activity Assay
4.7. Serum Cytokine Analysis
4.8. Epigenetic Analyses
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2021. [Google Scholar]
- Takayama, T.; Katsuki, S.; Takahashi, Y.; Ohi, M.; Nojiri, S.; Sakamaki, S.; Kato, J.; Kogawa, K.; Miyake, H.; Niitsu, Y. Ab-errant crypt foci of the colon as precursors of adenoma and cancer. N. Engl. J. Med. 1998, 339, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Fichtner-Feigl, S.; Kesselring, R.; Strober, W. Chronic inflammation and the development of malignancy in the GI tract. Trends Immunol. 2015, 36, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I. Inflammation and colorectal cancer: Colitis-associated neoplasia. Semin. Immunopathol. 2013, 35, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Tuomisto, A.E.; Mäkinen, M.J.; Väyrynen, J. Systemic inflammation in colorectal cancer: Underlying factors, effects, and prognostic significance. World J. Gastroenterol. 2019, 25, 4383–4404. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Barrett, C.W.; Short, S.P.; Williams, C.S. Selenoproteins and oxidative stress-induced inflammatory tumorigenesis in the gut. Cell. Mol. Life Sci. 2016, 74, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Barrett, C.W.; Singh, K.; Motley, A.K.; Lintel, M.K.; Matafonova, E.; Bradley, A.M.; Ning, W.; Poindexter, S.V.; Parang, B.; Reddy, V.K.; et al. Dietary selenium deficiency exacerbates DSS-induced epithelial injury and AOM/DSS-induced tumor-igenesis. PLoS ONE 2013, 8, e67845. [Google Scholar] [CrossRef] [Green Version]
- Duntas, L.H. Selenium and Inflammation: Underlying Anti-inflammatory Mechanisms. Horm. Metab. Res. 2009, 41, 443–447. [Google Scholar] [CrossRef]
- Carlson, B.A.; Lee, B.C.; Tsuji, P.A.; Tobe, R.; Park, J.M.; Schweizer, U.; Gladyshev, V.N.; Hatfield, D.L. Selenocysteine tRNA[Ser]Sec: From nonsense suppressor tRNA to the quintessential consituent in selenoprotein biosynthesis. In Selenium—Its Molecular Biology and Role in Human Health; Hatfield, D.L., Schweizer, U., Tsuji, P.A., Gladyshev, V.N., Eds.; Springer Science Business Media, LLC: New York, NY, USA, 2016. [Google Scholar]
- Tsuji, P.A.; Davis, C.D.; Milner, J.A. Selenium: Dietary sources and human requirements. In Selenium—Its Molecular Biology and Role in Human Health, 3rd ed.; Hatfield, D.L., Berry, M.J., Gladyshev, V.N., Eds.; Springer Science Business Media, LLC: New York, NY, USA, 2012. [Google Scholar]
- Turanov, A.A.; Lobanov, A.V.; Fomenko, D.E.; Morrison, H.; Sogin, M.L.; Klobutcher, L.; Hatfield, D.L.; Gladyshev, V.N. Genetic Code Supports Targeted Insertion of Two Amino Acids by One Codon. Science 2009, 323, 259–261. [Google Scholar] [CrossRef] [Green Version]
- Hatfield, D.L.; Carlson, B.A.; Tsuji, P.A.; Tobe, R.; Gladyshev, V.N. Selenium and Cancer. In Molecular, Genetic, and Nutritional Aspects of Major and Trace Minerals; Collins, J.F., Ed.; Elsevier: New York, NY, USA, 2016; pp. 463–473. [Google Scholar]
- Hatfield, D.L.; Tsuji, P.A.; Carlson, B.A.; Gladyshev, V.N. Selenium and selenocysteine: Roles in cancer, health, and development. Trends Biochem. Sci. 2014, 39, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, P.A.; Carlson, B.A.; Lee, B.J.; Gladyshev, V.N.; Hatfield, D.L. Interplay of Selenoproteins and Different Antioxidant Systems in Various Cancers. In Selenium; Springer Science and Business Media, LLC: New York, NY, USA, 2016; pp. 441–449. [Google Scholar]
- Peters, K.M.; Carlson, B.A.; Gladyshev, V.N.; Tsuji, P.A. Selenoproteins in colon cancer. Free Radic. Biol. Med. 2018, 127, 14–25. [Google Scholar] [CrossRef]
- Irons, R.; Tsuji, P.A.; Carlson, B.A.; Ouyang, P.; Yoo, M.-H.; Xu, X.-M.; Hatfield, D.L.; Gladyshev, V.N.; Davis, C.D. Deficiency in the 15-kDa Selenoprotein Inhibits Tumorigenicity and Metastasis of Colon Cancer Cells. Cancer Prev. Res. 2010, 3, 630–639. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, P.A.; Naranjo-Suarez, S.; Carlson, B.A.; Tobe, R.; Yoo, M.-H.; Davis, C.D. Deficiency in the 15 kDa Selenoprotein Inhibits Human Colon Cancer Cell Growth. Nutrients 2011, 3, 805–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladyshev, V.N.; Jeang, K.-T.; Wootton, J.C.; Hatfield, D.L. A New Human Selenium-containing Protein. J. Biol. Chem. 1998, 273, 8910–8915. [Google Scholar] [CrossRef] [Green Version]
- Kasaikina, M.V.; Fomenko, D.E.; Labunskyy, V.M.; Lachke, S.A.; Qiu, W.; Moncaster, J.A.; Zhang, J.; Wojnarowicz, M.W.; Natarajan, S.K.; Malinouski, M.; et al. Roles of the 15-kDa selenoprotein (Sep15) in redox homeostasis and cataract devel-opment revealed by the analysis of Sep15 knockout mice. J. Biol. Chem. 2011, 286, 33203–33212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labunskyy, V.M.; Ferguson, A.D.; Fomenko, D.E.; Chelliah, Y.; Hatfield, D.L.; Gladyshev, V.N. A novel cysteine-rich do-main of Sep15 mediates the interaction with UDP-glucose:glycoprotein glucosyltransferase. J. Biol. Chem. 2005, 280, 37839–37845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labunskyy, V.M.; Yoo, M.-H.; Hatfield, D.L.; Gladyshev, V.N. Sep15, a Thioredoxin-like Selenoprotein, Is Involved in the Unfolded Protein Response and Differentially Regulated by Adaptive and Acute ER Stresses. Biochemistry 2009, 48, 8458–8465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, S.H.; Everley, R.A.; Schildberg, F.A.; Lee, S.G.; Orsi, A.; Barbati, Z.R.; Karatepe, K.; Fomenko, D.E.; Tsuji, P.A.; Luo, H.R.; et al. Role of Selenof as a gatekeeper of decreted disulfide-tich glycoproteins. Cell Rep. 2018, 23, 1387–1398. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, P.A.; Carlson, B.A.; Yoo, M.-H.; Naranjo-Suarez, S.; Xu, X.-M.; Esther, A.; Asaki, E.; Seifried, H.E.; Reinhold, W.; Davis, C.D.; et al. The 15 kDa Selenoprotein and Thioredoxin Reductase 1 Promote Colon Cancer by Different Pathways. PLoS ONE 2015, 10, e0124487. [Google Scholar] [CrossRef] [Green Version]
- Kasaikina, M.V.; Hatfield, D.L.; Gladyshev, V.N. Understanding selenoprotein function and regulation through the use of rodent models. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2012, 1823, 1633–1642. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, P.A.; Carlson, B.A.; Naranjo-Suarez, S.; Yoo, M.-H.; Xu, X.-M.; Fomenko, D.E.; Gladyshev, V.N.; Hatfield, D.L.; Davis, C.D. Knockout of the 15 kDa Selenoprotein Protects against Chemically-Induced Aberrant Crypt Formation in Mice. PLoS ONE 2012, 7, e50574. [Google Scholar] [CrossRef]
- Hu, Y.J.; Korotkov, K.; Mehta, R.; Hatfield, D.L.; Rotimi, C.N.; Luke, A.; Prewitt, T.E.; Cooper, R.S.; Stock, W.; Vokes, E.E.; et al. Distribution and functional consequences of nucleotide polymorphisms in the 3′-untranslated region of the human Sep15 gene. Cancer Res. 2001, 61, 2307–2310. [Google Scholar]
- Méplan, C. Association of Single Nucleotide Polymorphisms in Selenoprotein Genes with Cancer Risk. Adv. Struct. Saf. Stud. 2017, 1661, 313–324. [Google Scholar] [CrossRef]
- Mohammaddoust, S.; Salehi, Z.; Saedi, H.S. SEPP1 and SEP15 gene polymorphisms and susceptibility to breast cancer. Br. J. Biomed. Sci. 2017, 75, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Orlando, F.A.; Tan, D.; Baltodano, J.D.; Khoury, T.; Gibbs, J.F.; Hassid, V.J.; Ahmed, B.H.; Alrawi, S.J. Aberrant crypt foci as precursors in colorectal cancer progression. J. Surg. Oncol. 2008, 98, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Bedenne, L.; Faivre, J.; Boutron, M.C.; Piard, F.; Cauvin, J.M.; Hillon, P. Adenoma—Carcinoma sequence or “de novo” car-cinogenesis? A study of adenomatous remnants in a population-based series of large bowel cancers. Cancer 1992, 69, 883–888. [Google Scholar] [CrossRef]
- Gonzalez, F.J. The 2006 Bernard B. Brodie Award lecture. Cyp2e1. Drug Metab. Dispos. 2007, 35, 1–8. [Google Scholar] [CrossRef]
- Sohn, O.S.; Fiala, E.S.; Requeijo, S.P.; Weisburger, J.H.; Gonzalez, F.J. Differential effects of CYP2E1 status on the metabolic activation of the colon carcinogens azoxymethane and methylazoxymethanol. Cancer Res. 2001, 61, 8435–8440. [Google Scholar] [PubMed]
- Feinberg, A.; Zedeck, M.S. Production of a highly reactive alkylating agent from the organospecific carcinogen methyla-zoxymethanol by alcohol dehydrogenase. Cancer Res. 1980, 40, 4446–4450. [Google Scholar]
- Davis, C.D.; Uthus, E.O.; Finley, J.W. Dietary Selenium and Arsenic Affect DNA Methylation In Vitro in Caco-2 Cells and In Vivo in Rat Liver and Colon. J. Nutr. 2000, 130, 2903–2909. [Google Scholar] [CrossRef]
- Davis, C.D.; Uthus, E.O. Dietary Folate and Selenium Affect Dimethylhydrazine-Induced Aberrant Crypt Formation, Global DNA Methylation and One-Carbon Metabolism in Rats. J. Nutr. 2003, 133, 2907–2914. [Google Scholar] [CrossRef]
- Speckman, B.; Schulz, S.; Hiller, F.; Hesse, D.; Schumacher, F.; Kleuser, B.; Geisel, J.; Obeid, R.; Grune, T.; Kipp, A.P. Sele-nium increases hepatic DNA methylation and modulates one-carbon metabolism in the liver of mice. J. Nutr. Biochem. 2017, 48, 112–119. [Google Scholar] [CrossRef]
- Jablonska, E.; Reszka, E. Selenium and Epigenetics in Cancer: Focus on DNA Methylation. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2017; Volume 136, pp. 193–234. [Google Scholar]
- Kajla, S.; Mondol, A.S.; Nagasawa, A.; Zhang, Y.; Kato, M.; Matsuno, K.; Yabe-Nishimura, C.; Kamata, T. A crucial role for Nox 1 in redox-dependent regulation of Wnt-β-catenin signaling. FASEB J. 2012, 26, 2049–2059. [Google Scholar] [CrossRef]
- Liu, F.; Zuo, Z.; Liu, Y.; Deguchi, Y.; Moussali, M.; Chen, W.; Yang, P.; Wei, B.; Tan, L.; Lorenzi, P.L.; et al. Suppression of membranous LRP5 recycling, Wnt/β-catenin signaling, and colon carcinogenesis by 15-LOX-1 oeroxidation of linoleic acid in PI3P. Cell Rep. 2020, 32, 108049. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Zhang, J.; Zhong, J.; Yang, R. COL1A1 promotes metastasis in colorectal cancer by regulating the WNT/PCP pathway. Mol. Med. Rep. 2018, 17, 5037–5042. [Google Scholar] [CrossRef] [Green Version]
- Clapper, M.L.; Chang, W.-C.L.; Cooper, H.S. Dysplastic Aberrant Crypt Foci: Biomarkers of Early Colorectal Neoplasia and Response to Preventive Intervention. Cancer Prev. Res. 2020, 13, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Hamada, F.; Ishidate, T.; Anai, K.; Kawahara, K.; Toyoshima, K.; Akiyama, T. Axin, an inhibitor of the Wnt signalling pathway, interacts with beta-catenin, GSK-3beta and APC and reduces the beta-catenin level. Genes Cells 1998, 3, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Spink, K.E.; Fridman, S.G.; Weis, W.I. Molecular mechanisms of β-catenin recognition by adenomatous polyposis coli re-vealed by the structure of an APC–β-catenin complex. EMBO J. 2001, 20, 6203–6212. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinzler, K.W.; Vogelstein, B. Lessons from Hereditary Colorectal Cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Segditsas, S.; Tomlinson, I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006, 25, 7531–7537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fodde, R. The APC gene in colorectal cancer. Eur. J. Cancer 2002, 38, 867–871. [Google Scholar] [CrossRef]
- Najdi, R.; Holcombe, R.F.; Waterman, M.L. Wnt signaling and colon carcinogenesis: Beyond APC. J. Carcinog. 2011, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienz, M. β-Catenin: A Pivot between Cell Adhesion and Wnt Signalling. Curr. Biol. 2005, 15, R64–R67. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.H.; Zheng, G. E-Cadherin/β-Catenin Complex and the Epithelial Barrier. J. Biomed. Biotechnol. 2011, 2011, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Park, P.-G.; Jo, S.J.; Kim, M.J.; Kim, H.J.; Lee, J.H.; Park, C.K.; Kim, H.; Lee, K.Y.; Kim, H.; Park, J.H.; et al. Role of LOXL2 in the epithelial-mesenchymal transition and colorectal cancer metastasis. Oncotarget 2017, 8, 80325–80335. [Google Scholar] [CrossRef] [Green Version]
- Kipp, A.; Banning, A.; van Schothorst, E.M.; Méplan, C.; Schomburg, L.; Evelo, C.; Coort, S.; Gaj, S.; Keijer, J.; Hesketh, J.; et al. Four selenoproteins, protein synthesis, and Wnt signaling are particularly sensitive to limited selenium intake in mouse colon. Mol. Nutr. Food Res. 2009, 53, 1561–1572. [Google Scholar] [CrossRef]
- Adams, J.M.; Jafar-Nejad, H. The Roles of Notch Signaling in Liver Development and Disease. Biomolecules 2019, 9, 608. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Rosenberg, D.W. Role of Notch signaling in colon homeostasis and carcinogenesis. Cancer Sci. 2011, 102, 1938–1942. [Google Scholar] [CrossRef]
- Fazio, C.; Ricciardiello, L. Inflammation and Notch signaling: A crosstalk with opposite effects on tumorigenesis. Cell Death Dis. 2016, 7, e2515. [Google Scholar] [CrossRef]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel Integral Membrane Proteins Localizing at Tight Junctions with No Sequence Similarity to Occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Fanning, A.S.; Holmes, J.; Anderson, J. Occludin is required for cytokine-induced regulation of tight junction barriers. J. Cell Sci. 2010, 123, 2844–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, R.; Milatz, S.; Krug, S.M.; Oelrich, B.; Schulzke, J.-D.; Amasheh, S.; Günzel, D.; Fromm, M. Claudin-2, a compo-nent of the tight junction, forms a paracellular water channel. J. Cell Sci. 2010, 123, 1913–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Hernandez, V.; Quiros, M.; Nusrat, A. Intestinal epithelial claudins: Expression and regulation in homeostasis and inflammation. Ann. N. Y. Acad. Sci. 2017, 1397, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.R.; Nalle, S.C.; Tretiakova, M.; Rubin, D.T.; Turner, J.R. Claudin-1 and claudin-2 expression is elevated in in-flammatory bowel disease and may contribute to early neoplastic transformation. Lab. Investig. 2008, 88, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Yang, Y.; Yang, F.; Liu, S.; Zhu, Z.; Lei, Z.; Guo, J. Functions of EpCAM in physiological processes and diseases (Review). Int. J. Mol. Med. 2018, 42, 1771–1785. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.-S.; Hsu, H.-P.; Lai, M.-D.; Yen, M.-C.; Fang, J.-H.; Weng, T.-Y.; Chen, Y.-L. Suppression of mucin 2 promotes interleukin-6 secretion and tumor growth in an orthotopic immune-competent colon cancer animal model. Oncol. Rep. 2014, 32, 2335–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuan, S.F.; Byrd, J.C.; Basbaum, C.B.; Kim, Y.S. Characterization of quantitative mucin variants from a human colon cancer cell line. Cancer Res. 1987, 47, 5715–5724. [Google Scholar] [PubMed]
- Cho, M.; Dahiya, R.; Choi, S.; Siddiki, B.; Yeh, M.; Sleisenger, M.; Kirn, Y. Mucins secreted by cell lines derived from colorectal mucinous carcinoma and adenocarcinoma. Eur. J. Cancer 1997, 33, 931–941. [Google Scholar] [CrossRef]
- Schenkman, J.B.; Cinti, D.L. [6] Preparation of microsomes with calcium. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1978; Volume 52, pp. 83–89. [Google Scholar] [CrossRef]
- Cederbaum, A.I. Methodology to assay CYP2E1 mixed function oxidase catalytic activity and its induction. Redox Biol. 2014, 2, 1048–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swamynathan, S.K.; Wells, A. Conjunctival goblet cells: Ocular surface functions, disorders that affect them, and the potential for their regeneration. Ocul. Surf. 2020, 18, 19–26. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selenium (µg/g Diet) | WT Control 1 | WT AOM/DSS 1 | Selenof-KO Control 1 | Selenof-KO AOM/DSS 1 |
|---|---|---|---|---|
| 0.02 | 0/8 (0%) | 13/15 (86.7%) | 1/9 (11.1%) | 4/14 (28.6%) |
| 0.1 | 2/8 (25%) | 13/18 (72.2%) | 0/9 (0%) | 3/11 (27.3%) |
| 2.0 | 2/9 (22%) | 10/13 (76.9%) | 0/12 (0%) | 6/14 (42.9%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canter, J.A.; Ernst, S.E.; Peters, K.M.; Carlson, B.A.; Thielman, N.R.J.; Grysczyk, L.; Udofe, P.; Yu, Y.; Cao, L.; Davis, C.D.; et al. Selenium and the 15kDa Selenoprotein Impact Colorectal Tumorigenesis by Modulating Intestinal Barrier Integrity. Int. J. Mol. Sci. 2021, 22, 10651. https://doi.org/10.3390/ijms221910651
Canter JA, Ernst SE, Peters KM, Carlson BA, Thielman NRJ, Grysczyk L, Udofe P, Yu Y, Cao L, Davis CD, et al. Selenium and the 15kDa Selenoprotein Impact Colorectal Tumorigenesis by Modulating Intestinal Barrier Integrity. International Journal of Molecular Sciences. 2021; 22(19):10651. https://doi.org/10.3390/ijms221910651
Chicago/Turabian StyleCanter, Jessica A., Sarah E. Ernst, Kristin M. Peters, Bradley A. Carlson, Noelle R. J. Thielman, Lara Grysczyk, Precious Udofe, Yunkai Yu, Liang Cao, Cindy D. Davis, and et al. 2021. "Selenium and the 15kDa Selenoprotein Impact Colorectal Tumorigenesis by Modulating Intestinal Barrier Integrity" International Journal of Molecular Sciences 22, no. 19: 10651. https://doi.org/10.3390/ijms221910651
APA StyleCanter, J. A., Ernst, S. E., Peters, K. M., Carlson, B. A., Thielman, N. R. J., Grysczyk, L., Udofe, P., Yu, Y., Cao, L., Davis, C. D., Gladyshev, V. N., Hatfield, D. L., & Tsuji, P. A. (2021). Selenium and the 15kDa Selenoprotein Impact Colorectal Tumorigenesis by Modulating Intestinal Barrier Integrity. International Journal of Molecular Sciences, 22(19), 10651. https://doi.org/10.3390/ijms221910651