
The NLRP3 Inflammasome: Relevance in Solid Organ Transplantation


{kind=link}
Abstract
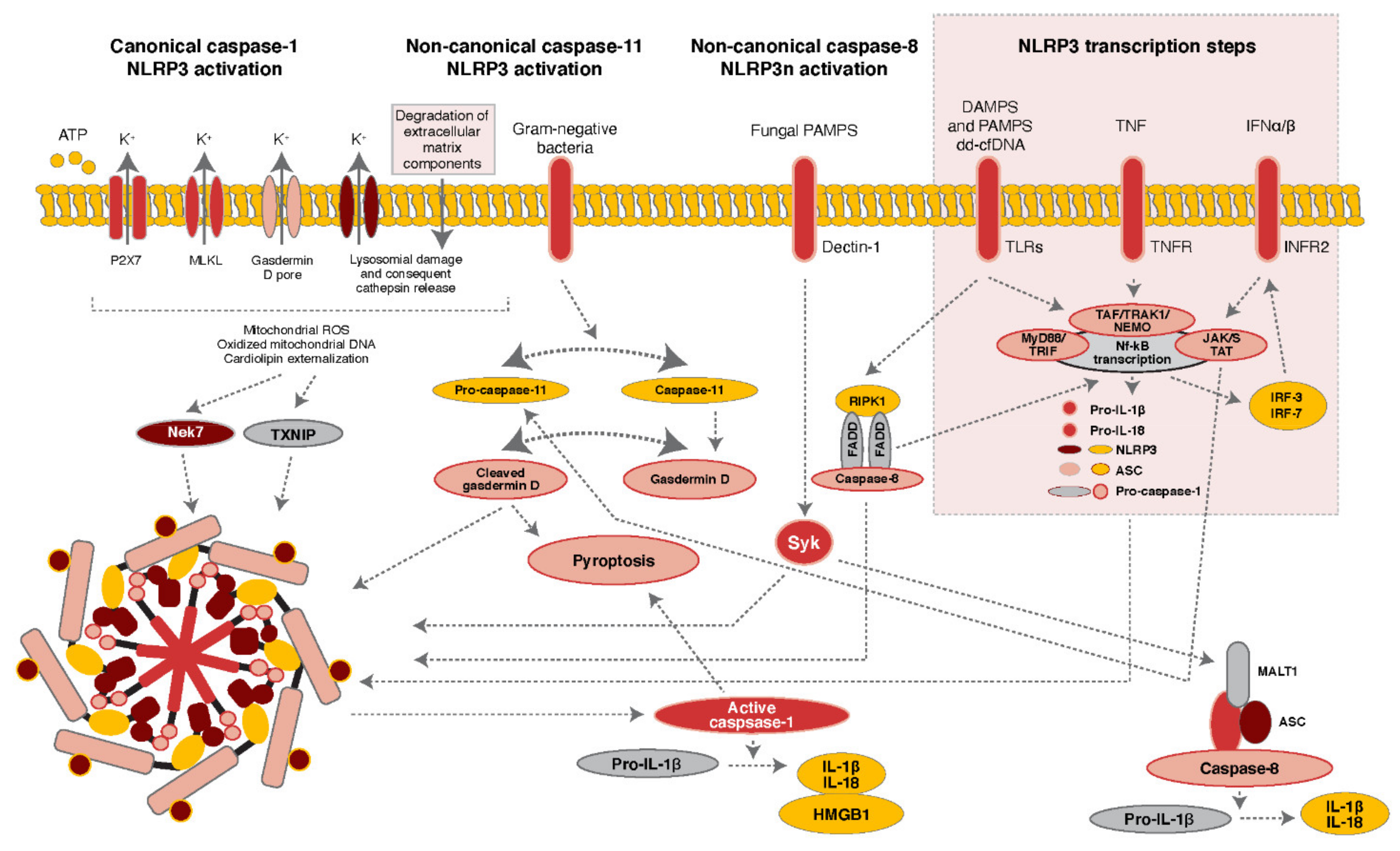
:1. Composition and Assembly of the NLRP3 Inflammasome
2. Regulation of NLRP3 Inflammasome Activity
2.1. Role of Post-Translational Modifications in NLRP3 Inflammasome Activation
2.1.1. Phosphorylation of NLRP3 and Kinases Associated with Inflammasome Activation
2.1.2. Ubiquitination of NLRP3 and Regulatory Molecules Associated with Inflammasome Activation
2.2. Direct and Indirect Pharmacological Inhibition of NLRP3 Inflammasome Activation
3. The NLRP3 Inflammasome and DNA Sensing
3.1. The Role of DNA in Autoimmune Pathology
3.2. Neutrophils and the NLRP3 Inflammasome—Casting a Wide NET
4. NLRP3 Inflammasome Activation in End-Stage Organ Failure and Transplant
4.1. Liver
4.2. Lung
4.3. Kidney
4.4. Heart
4.5. Skin
5. Summary and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchi, L.; Warner, N.; Viani, K.; Nunez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef] [Green Version]
- Oroz, J.; Barrera-Vilarmau, S.; Alfonso, C.; Rivas, G.; de Alba, E. ASC Pyrin Domain Self-associates and Binds NLRP3 Protein Using Equivalent Binding Interfaces. J. Biol. Chem. 2016, 291, 19487–19501. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F. Detection of immune danger signals by NALP3. J. Leukoc. Biol. 2008, 83, 507–511. [Google Scholar] [CrossRef]
- Schmidt, F.I.; Lu, A.; Chen, J.W.; Ruan, J.; Tang, C.; Wu, H.; Ploegh, H.L. A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assembly. J. Exp. Med. 2016, 213, 771–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, B.S.; Bossaller, L.; De Nardo, D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 2014, 15, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.R.; Timmer, A.M.; Bilgrami, S.; Park, E.J.; Eckmann, L.; Nizet, V.; Karin, M. Anthrax toxin induces macrophage death by p38 MAPK inhibition but leads to inflammasome activation via ATP leakage. Immunity 2011, 35, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutterwala, F.S.; Ogura, Y.; Szczepanik, M.; Lara-Tejero, M.; Lichtenberger, G.S.; Grant, E.P.; Bertin, J.; Coyle, A.J.; Galan, J.E.; Askenase, P.W.; et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 2006, 24, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Inohara; Chamaillard; McDonald, C.; Nunez, G. NOD-LRR proteins: Role in host-microbial interactions and inflammatory disease. Annu. Rev. Biochem. 2005, 74, 355–383. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Celhar, T.; Magalhaes, R.; Fairhurst, A.M. TLR7 and TLR9 in SLE: When sensing self goes wrong. Immunol. Res. 2012, 53, 58–77. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Greenwald, D.; Hulmes, J.D.; Chang, M.; Pan, Y.C.; Mathison, J.; Ulevitch, R.; Cerami, A. Identity of tumour necrosis factor and the macrophage-secreted factor cachectin. Nature 1985, 316, 552–554. [Google Scholar] [CrossRef]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukoc. Biol. 2013, 93, 847–863. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef]
- Hennessy, E.J.; Parker, A.E.; O’Neill, L.A. Targeting Toll-like receptors: Emerging therapeutics? Nat. Rev. Drug Discov. 2010, 9, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Nunez, G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J. Immunol. 2009, 183, 792–796. [Google Scholar] [CrossRef]
- Song, N.; Liu, Z.S.; Xue, W.; Bai, Z.F.; Wang, Q.Y.; Dai, J.; Liu, X.; Huang, Y.J.; Cai, H.; Zhan, X.Y.; et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol. Cell 2017, 68, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Stutz, A.; Kolbe, C.C.; Stahl, R.; Horvath, G.L.; Franklin, B.S.; van Ray, O.; Brinkschulte, R.; Geyer, M.; Meissner, F.; Latz, E. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 2017, 214, 1725–1736. [Google Scholar] [CrossRef]
- Mortimer, L.; Moreau, F.; MacDonald, J.A.; Chadee, K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat. Immunol. 2016, 17, 1176–1186. [Google Scholar] [CrossRef]
- Kuemmerle-Deschner, J.B. CAPS—pathogenesis, presentation and treatment of an autoinflammatory disease. Semin. Immunopathol. 2015, 37, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Spalinger, M.R.; Kasper, S.; Gottier, C.; Lang, S.; Atrott, K.; Vavricka, S.R.; Scharl, S.; Raselli, T.; Frey-Wagner, I.; Gutte, P.M.; et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J. Clin. Investig. 2016, 126, 1783–1800. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.N.; Wang, X.; Wang, J.; Yang, Z.; Li, S.; Yang, J.; Liu, L.; Lei, X.; Shao, F. Chemical probing reveals insights into the signaling mechanism of inflammasome activation. Cell Res. 2010, 20, 1289–1305. [Google Scholar] [CrossRef]
- Li, L.; Chen, Y.; Li, J.; Yin, H.; Guo, X.; Doan, J.; Molkentin, J.D.; Liu, Q. TAK1 Regulates Myocardial Response to Pathological Stress via NFAT, NFkappaB, and Bnip3 Pathways. Sci. Rep. 2015, 5, 16626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Fan, Z.; Sun, G.; Qi, G. Sacubitril/valsartan (LCZ696) reduces myocardial injury following myocardial infarction by inhibiting NLRP3induced pyroptosis via the TAK1/JNK signaling pathway. Mol. Med. Rep. 2021, 24. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Gurung, P.; Mavuluri, J.; Dasari, T.K.; Klco, J.M.; Chi, H.; Kanneganti, T.D. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med. 2018, 215, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Malireddi, R.K.S.; Gurung, P.; Kesavardhana, S.; Samir, P.; Burton, A.; Mummareddy, H.; Vogel, P.; Pelletier, S.; Burgula, S.; Kanneganti, T.D. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Jiang, Z.; Zamanian-Daryoush, M.; Nie, H.; Silva, A.M.; Williams, B.R.; Li, X. Poly(I-C)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFkappa B and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J. Biol. Chem. 2003, 278, 16713–16719. [Google Scholar] [CrossRef] [Green Version]
- Byrd-Leifer, C.A.; Block, E.F.; Takeda, K.; Akira, S.; Ding, A. The role of MyD88 and TLR4 in the LPS-mimetic activity of Taxol. Eur. J. Immunol. 2001, 31, 2448–2457. [Google Scholar] [CrossRef]
- Song, N.; Li, T. Regulation of NLRP3 Inflammasome by Phosphorylation. Front. Immunol. 2018, 9, 2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, R.; Xue, Z.; Fan, G.; Zhang, N.; Wang, C.; Li, G.; Da, Y. Pin1 Promotes NLRP3 Inflammasome Activation by Phosphorylation of p38 MAPK Pathway in Septic Shock. Front. Immunol. 2021, 12, 620238. [Google Scholar] [CrossRef] [PubMed]
- Henriksbo, B.D.; Tamrakar, A.K.; Phulka, J.S.; Barra, N.G.; Schertzer, J.D. Statins activate the NLRP3 inflammasome and impair insulin signaling via p38 and mTOR. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E110–E116. [Google Scholar] [CrossRef] [PubMed]
- Boza, P.; Ayala, P.; Vivar, R.; Humeres, C.; Caceres, F.T.; Munoz, C.; Garcia, L.; Hermoso, M.A.; Diaz-Araya, G. Expression and function of toll-like receptor 4 and inflammasomes in cardiac fibroblasts and myofibroblasts: IL-1beta synthesis, secretion, and degradation. Mol. Immunol. 2016, 74, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Chei, S.; Oh, H.J.; Song, J.H.; Seo, Y.J.; Lee, K.; Kim, K.J.; Lee, B.Y. Spirulina maxima extract prevents activation of the NLRP3 inflammasome by inhibiting ERK signaling. Sci. Rep. 2020, 10, 2075. [Google Scholar] [CrossRef]
- Yin, R.; Zhu, X.; Wang, J.; Yang, S.; Ma, A.; Xiao, Q.; Song, J.; Pan, X. MicroRNA-155 promotes the ox-LDL-induced activation of NLRP3 inflammasomes via the ERK1/2 pathway in THP-1 macrophages and aggravates atherosclerosis in ApoE-/- mice. Ann. Palliat. Med. 2019, 8, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Ghonime, M.G.; Shamaa, O.R.; Das, S.; Eldomany, R.A.; Fernandes-Alnemri, T.; Alnemri, E.S.; Gavrilin, M.A.; Wewers, M.D. Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J. Immunol. 2014, 192, 3881–3888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Tu, S.; Lin, G.; Guo, H.; Yan, C.; Liu, Q.; Huang, L.; Tang, N.; Xiao, Y.; Pope, R.M.; et al. Sequential ubiquitination of NLRP3 by RNF125 and Cbl-b limits inflammasome activation and endotoxemia. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Lear, T.B.; Jerome, J.A.; Rajbhandari, S.; Snavely, C.A.; Gulick, D.L.; Gibson, K.F.; Zou, C.; Chen, B.B.; Mallampalli, R.K. Lipopolysaccharide Primes the NALP3 Inflammasome by Inhibiting Its Ubiquitination and Degradation Mediated by the SCFFBXL2 E3 Ligase. J. Biol. Chem. 2015, 290, 18124–18133. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Damianou, A.; Di Daniel, E.; Kessler, B.M. Inflammasome activation controlled by the interplay between post-translational modifications: Emerging drug target opportunities. Cell Commun. Signal. 2021, 19, 23. [Google Scholar] [CrossRef]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef]
- Bartoloni, E.; Ludovini, V.; Alunno, A.; Pistola, L.; Bistoni, O.; Crino, L.; Gerli, R. Increased levels of circulating DNA in patients with systemic autoimmune diseases: A possible marker of disease activity in Sjogren’s syndrome. Lupus 2011, 20, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Atamaniuk, J.; Kopecky, C.; Skoupy, S.; Saemann, M.D.; Weichhart, T. Apoptotic cell-free DNA promotes inflammation in haemodialysis patients. Nephrol. Dial. Transplant. 2012, 27, 902–905. [Google Scholar] [CrossRef] [Green Version]
- Holzhauser, L.; Clerkin, K.J.; Fujino, T.; Alenghat, F.J.; Raikhelkar, J.; Kim, G.; Sayer, G.; Uriel, N. Donor-derived cell-free DNA is associated with cardiac allograft vasculopathy. Clin. Transplant. 2021, 35, e14206. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.; Gillespie, M.; Ammerman, N.; Vo, A.; Lim, K.; Peng, A.; Najjar, R.; Sethi, S.; Jordan, S.C.; Mirocha, J.; et al. Donor-derived Cell-free DNA Combined with Histology Improves Prediction of Estimated Glomerular Filtration Rate Over Time in Kidney Transplant Recipients Compared With Histology Alone. Transplant. Direct 2020, 6, e580. [Google Scholar] [CrossRef]
- Kanwar, M.K.; Khush, K.K.; Pinney, S.; Sherman, C.; Hall, S.; Teuteberg, J.; Uriel, N.; Kobashigawa, J. Impact of cytomegalovirus infection on gene expression profile in heart transplant recipients. J. Heart Lung Transplant. 2021, 40, 101–107. [Google Scholar] [CrossRef]
- Levine, D.J.; Ross, D.J.; Sako, E. Single Center “Snapshot” Experience With Donor-Derived Cell-Free DNA After Lung Transplantation. Biomark. Insights 2020, 15, 1177271920958704. [Google Scholar] [CrossRef]
- Sayah, D.; Weigt, S.S.; Ramsey, A.; Ardehali, A.; Golden, J.; Ross, D.J. Plasma Donor-derived Cell-free DNA Levels Are Increased During Acute Cellular Rejection After Lung Transplant: Pilot Data. Transplant. Direct 2020, 6, e608. [Google Scholar] [CrossRef] [PubMed]
- Bloom, R.D.; Bromberg, J.S.; Poggio, E.D.; Bunnapradist, S.; Langone, A.J.; Sood, P.; Matas, A.J.; Mehta, S.; Mannon, R.B.; Sharfuddin, A.; et al. Cell-Free DNA and Active Rejection in Kidney Allografts. J. Am. Soc. Nephrol. 2017, 28, 2221–2232. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.S.; Brennan, D.C.; Poggio, E.; Bunnapradist, S.; Langone, A.; Sood, P.; Matas, A.J.; Mannon, R.B.; Mehta, S.; Sharfuddin, A.; et al. Biological Variation of Donor-Derived Cell-Free DNA in Renal Transplant Recipients: Clinical Implications. J. Appl. Lab. Med. 2017, 2, 309–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stites, E.; Kumar, D.; Olaitan, O.; John Swanson, S.; Leca, N.; Weir, M.; Bromberg, J.; Melancon, J.; Agha, I.; Fattah, H.; et al. High levels of dd-cfDNA identify patients with TCMR 1A and borderline allograft rejection at elevated risk of graft injury. Am. J. Transplant. 2020, 20, 2491–2498. [Google Scholar] [CrossRef] [Green Version]
- Knight, S.R.; Thorne, A.; Lo Faro, M.L. Donor-specific Cell-free DNA as a Biomarker in Solid Organ Transplantation. A Systematic Review. Transplantation 2019, 103, 273–283. [Google Scholar] [CrossRef]
- Garcia Moreira, V.; Prieto Garcia, B.; Baltar Martin, J.M.; Ortega Suarez, F.; Alvarez, F.V. Cell-free DNA as a noninvasive acute rejection marker in renal transplantation. Clin. Chem. 2009, 55, 1958–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, T.; Yoshida, K.; Hashiramoto, A.; Matsui, K. Cell-Free DNA in Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 8941. [Google Scholar] [CrossRef]
- Busani, S.; De Biasi, S.; Nasi, M.; Paolini, A.; Venturelli, S.; Tosi, M.; Girardis, M.; Cossarizza, A. Increased Plasma Levels of Mitochondrial DNA and Normal Inflammasome Gene Expression in Monocytes Characterize Patients With Septic Shock Due to Multidrug Resistant Bacteria. Front. Immunol. 2020, 11, 768. [Google Scholar] [CrossRef]
- Bohata, J.; Horvathova, V.; Pavlikova, M.; Stiburkova, B. Circulating microRNA alternations in primary hyperuricemia and gout. Arthritis Res. Ther. 2021, 23, 186. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Ren, Y.; He, X. IFN-I Mediates Lupus Nephritis From the Beginning to Renal Fibrosis. Front. Immunol. 2021, 12, 676082. [Google Scholar] [CrossRef]
- Filev, A.D.; Shmarina, G.V.; Ershova, E.S.; Veiko, N.N.; Martynov, A.V.; Borzikova, M.A.; Poletkina, A.A.; Dolgikh, O.A.; Veiko, V.P.; Bekker, A.A.; et al. Oxidized Cell-Free DNA Role in the Antioxidant Defense Mechanisms under Stress. Oxid. Med. Cell. Longev. 2019, 2019, 1245749. [Google Scholar] [CrossRef] [PubMed]
- Halverson, T.W.; Wilton, M.; Poon, K.K.; Petri, B.; Lewenza, S. DNA is an antimicrobial component of neutrophil extracellular traps. PLoS Pathog. 2015, 11, e1004593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, J.S.; Carmona-Rivera, C.; Kaplan, M.J. Proteins derived from neutrophil extracellular traps may serve as self-antigens and mediate organ damage in autoimmune diseases. Front. Immunol. 2012, 3, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiam, H.R.; Wong, S.L.; Qiu, R.; Kittisopikul, M.; Vahabikashi, A.; Goldman, A.E.; Goldman, R.D.; Wagner, D.D.; Waterman, C.M. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc. Natl. Acad. Sci. USA 2020, 117, 7326–7337. [Google Scholar] [CrossRef] [Green Version]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef] [PubMed]
- Sorvillo, N.; Mizurini, D.M.; Coxon, C.; Martinod, K.; Tilvawala, R.; Cherpokova, D.; Salinger, A.J.; Seward, R.J.; Staudinger, C.; Weerapana, E.; et al. Plasma Peptidylarginine Deiminase IV Promotes VWF-Platelet String Formation and Accelerates Thrombosis After Vessel Injury. Circ. Res. 2019, 125, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Savchenko, A.S.; Borissoff, J.I.; Martinod, K.; De Meyer, S.F.; Gallant, M.; Erpenbeck, L.; Brill, A.; Wang, Y.; Wagner, D.D. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood 2014, 123, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Martinod, K.; Witsch, T.; Erpenbeck, L.; Savchenko, A.; Hayashi, H.; Cherpokova, D.; Gallant, M.; Mauler, M.; Cifuni, S.M.; Wagner, D.D. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J. Exp. Med. 2017, 214, 439–458. [Google Scholar] [CrossRef]
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Sturzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine 2020, 58, 102925. [Google Scholar] [CrossRef] [PubMed]
- Lintzmaier Petiz, L.; Glaser, T.; Scharfstein, J.; Ratajczak, M.Z.; Ulrich, H. P2Y14 Receptor as a Target for Neutrophilia Attenuation in Severe COVID-19 Cases: From Hematopoietic Stem Cell Recruitment and Chemotaxis to Thrombo-inflammation. Stem Cell Rev. Rep. 2021, 17, 241–252. [Google Scholar] [CrossRef]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Nazir, S.; Gadi, I.; Al-Dabet, M.M.; Elwakiel, A.; Kohli, S.; Ghosh, S.; Manoharan, J.; Ranjan, S.; Bock, F.; Braun-Dullaeus, R.C.; et al. Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition. Blood 2017, 130, 2664–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.; Sahu, A.; Prabhakar, A.; Chatterjee, T.; Tyagi, T.; Kumari, B.; Khan, N.; Nair, V.; Bajaj, N.; Sharma, M.; et al. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc. Natl. Acad. Sci. USA 2017, 114, 4763–4768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Demers, M.; Fuchs, T.A.; Wong, S.L.; Brill, A.; Gallant, M.; Hu, J.; Wang, Y.; Wagner, D.D. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8674–8679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.L.; Wagner, D.D. Peptidylarginine deiminase 4: A nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J. 2018. [Google Scholar] [CrossRef]
- Munzer, P.; Negro, R.; Fukui, S.; di Meglio, L.; Aymonnier, K.; Chu, L.; Cherpokova, D.; Gutch, S.; Sorvillo, N.; Shi, L.; et al. NLRP3 Inflammasome Assembly in Neutrophils Is Supported by PAD4 and Promotes NETosis Under Sterile Conditions. Front. Immunol. 2021, 12, 683803. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroja-Mazo, A.; Martin-Sanchez, F.; Gomez, A.I.; Martinez, C.M.; Amores-Iniesta, J.; Compan, V.; Barbera-Cremades, M.; Yague, J.; Ruiz-Ortiz, E.; Anton, J.; et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Menegazzo, L.; Rigato, M.; Scattolini, V.; Poncina, N.; Bruttocao, A.; Ciciliot, S.; Mammano, F.; Ciubotaru, C.D.; Brocco, E.; et al. NETosis Delays Diabetic Wound Healing in Mice and Humans. Diabetes 2016, 65, 1061–1071. [Google Scholar] [CrossRef] [Green Version]
- Mirza, R.E.; Fang, M.M.; Weinheimer-Haus, E.M.; Ennis, W.J.; Koh, T.J. Sustained inflammasome activity in macrophages impairs wound healing in type 2 diabetic humans and mice. Diabetes 2014, 63, 1103–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Jiao, J.; Liu, J.; Huang, M.; Hu, Y.; Ran, W.; Yan, L.; Xiong, Y.; Li, M.; Quan, Z.; et al. MFG-E8 accelerates wound healing in diabetes by regulating “NLRP3 inflammasome-neutrophil extracellular traps” axis. Cell Death Discov. 2020, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Hanayama, R.; Tanaka, M.; Miwa, K.; Shinohara, A.; Iwamatsu, A.; Nagata, S. Identification of a factor that links apoptotic cells to phagocytes. Nature 2002, 417, 182–187. [Google Scholar] [CrossRef]
- Motegi, S.I.; Ishikawa, O. Mesenchymal stem cells: The roles and functions in cutaneous wound healing and tumor growth. J. Dermatol. Sci. 2017, 86, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Brissette, M.J.; Laplante, P.; Qi, S.; Latour, M.; Cailhier, J.F. Milk fat globule epidermal growth factor-8 limits tissue damage through inflammasome modulation during renal injury. J. Leukoc. Biol. 2016, 100, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Deroide, N.; Li, X.; Lerouet, D.; Van Vre, E.; Baker, L.; Harrison, J.; Poittevin, M.; Masters, L.; Nih, L.; Margaill, I.; et al. MFGE8 inhibits inflammasome-induced IL-1beta production and limits postischemic cerebral injury. J. Clin. Investig. 2013, 123, 1176–1181. [Google Scholar] [CrossRef] [Green Version]
- Ratajczak, M.Z.; Bujko, K.; Cymer, M.; Thapa, A.; Adamiak, M.; Ratajczak, J.; Abdel-Latif, A.K.; Kucia, M. The Nlrp3 inflammasome as a “rising star” in studies of normal and malignant hematopoiesis. Leukemia 2020, 34, 1512–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratajczak, M.Z.; Kucia, M. Extracellular Adenosine Triphosphate (eATP) and Its Metabolite, Extracellular Adenosine (eAdo), as Opposing “Yin-Yang” Regulators of Nlrp3 Inflammasome in the Trafficking of Hematopoietic Stem/Progenitor Cells. Front. Immunol. 2020, 11, 603942. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Adamiak, M.; Ratajczak, J.; Kucia, M. Heme Oxygenase 1 (HO-1) as an Inhibitor of Trafficking of Normal and Malignant Hematopoietic Stem Cells—Clinical and Translational Implications. Stem Cell Rev. Rep. 2021, 17, 821–828. [Google Scholar] [CrossRef]
- Chen, C.; Rong, P.; Yang, M.; Ma, X.; Feng, Z.; Wang, W. The Role of Interleukin-1beta in Destruction of Transplanted Islets. Cell Transplant. 2020, 29, 963689720934413. [Google Scholar] [CrossRef] [PubMed]
- Zeiser, R.; Penack, O.; Holler, E.; Idzko, M. Danger signals activating innate immunity in graft-versus-host disease. J. Mol. Med. 2011, 89, 833–845. [Google Scholar] [CrossRef]
- Martinez-Mier, G.; Toledo-Pereyra, L.H.; McDuffie, J.E.; Warner, R.L.; Ward, P.A. Neutrophil depletion and chemokine response after liver ischemia and reperfusion. J. Investig. Surg. 2001, 14, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Shirasuna, K.; Kimura, H.; Usui, F.; Kawashima, A.; Karasawa, T.; Tago, K.; Dezaki, K.; Nishimura, S.; Sagara, J.; et al. NLRP3 regulates neutrophil functions and contributes to hepatic ischemia-reperfusion injury independently of inflammasomes. J. Immunol. 2014, 192, 4342–4351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, Y.; Hines, I.N.; Zibari, G.; Pavlick, K.; Gray, L.; Kitagawa, Y.; Grisham, M.B. Mouse model of liver ischemia and reperfusion injury: Method for studying reactive oxygen and nitrogen metabolites in vivo. Free Radic. Biol. Med. 2009, 46, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kuboki, S.; Shin, T.; Huber, N.; Eismann, T.; Galloway, E.; Schuster, R.; Blanchard, J.; Edwards, M.J.; Lentsch, A.B. Hepatocyte signaling through CXC chemokine receptor-2 is detrimental to liver recovery after ischemia/reperfusion in mice. Hepatology 2008, 48, 1213–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, A.; Gabay, C.; Okaya, T.; Lentsch, A.B. Specific role of interleukin-1 in hepatic neutrophil recruitment after ischemia/reperfusion. Am. J. Pathol. 2002, 161, 1797–1803. [Google Scholar] [CrossRef]
- Kamo, N.; Ke, B.; Ghaffari, A.A.; Shen, X.D.; Busuttil, R.W.; Cheng, G.; Kupiec-Weglinski, J.W. ASC/caspase-1/IL-1beta signaling triggers inflammatory responses by promoting HMGB1 induction in liver ischemia/reperfusion injury. Hepatology 2013, 58, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Amores-Iniesta, J.; Barbera-Cremades, M.; Martinez, C.M.; Pons, J.A.; Revilla-Nuin, B.; Martinez-Alarcon, L.; Di Virgilio, F.; Parrilla, P.; Baroja-Mazo, A.; Pelegrin, P. Extracellular ATP Activates the NLRP3 Inflammasome and Is an Early Danger Signal of Skin Allograft Rejection. Cell Rep. 2017, 21, 3414–3426. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Cheng, Y.; Pan, Q.; Zhang, Y.J.; Jia, D.G.; Liu, Y.F. Effect of the Selective NLRP3 Inflammasome Inhibitor mcc950 on Transplantation Outcome in a Pig Liver Transplantation Model With Organs From Donors After Circulatory Death Preserved by Hypothermic Machine Perfusion. Transplantation 2019, 103, 353–362. [Google Scholar] [CrossRef]
- Liu, H.; Lo, C.M.; Yeung, O.W.H.; Li, C.X.; Liu, X.B.; Qi, X.; Ng, K.T.P.; Liu, J.; Ma, Y.Y.; Lam, Y.F.; et al. NLRP3 inflammasome induced liver graft injury through activation of telomere-independent RAP1/KC axis. J. Pathol. 2017, 242, 284–296. [Google Scholar] [CrossRef]
- Knorr, J.; Wree, A.; Tacke, F.; Feldstein, A.E. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin. Liver Dis. 2020, 40, 298–306. [Google Scholar] [CrossRef]
- Wan, X.; Xu, C.; Yu, C.; Li, Y. Role of NLRP3 Inflammasome in the Progression of NAFLD to NASH. Can. J. Gastroenterol. Hepatol. 2016, 2016, 6489012. [Google Scholar] [CrossRef] [Green Version]
- Banday, M.M.; Kumar, A.; Vestal, G.; Sethi, J.; Patel, K.N.; O’Neill, E.B.; Finan, J.; Cheng, F.; Lin, M.; Davis, N.M.; et al. N-myc-interactor mediates microbiome induced epithelial to mesenchymal transition and is associated with chronic lung allograft dysfunction. J. Heart Lung Transplant. 2021, 40, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Al-Hattab, D.S.; Safi, H.A.; Nagalingam, R.S.; Bagchi, R.A.; Stecy, M.T.; Czubryt, M.P. Scleraxis regulates Twist1 and Snai1 expression in the epithelial-to-mesenchymal transition. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H658–H668. [Google Scholar] [CrossRef] [PubMed]
- Bugg, D.; Bretherton, R.; Kim, P.; Olszewski, E.; Nagle, A.; Schumacher, A.E.; Chu, N.; Gunaje, J.; DeForest, C.A.; Stevens, K.; et al. Infarct Collagen Topography Regulates Fibroblast Fate via p38-Yes-Associated Protein Transcriptional Enhanced Associate Domain Signals. Circ. Res. 2020, 127, 1306–1322. [Google Scholar] [CrossRef]
- Huo, J.L.; Jiao, L.; An, Q.; Chen, X.; Qi, Y.; Wei, B.; Zheng, Y.; Shi, X.; Gao, E.; Liu, H.M.; et al. Myofibroblast Deficiency of LSD1 Alleviates TAC-Induced Heart Failure. Circ. Res. 2021. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 2017, 136, 549–561. [Google Scholar] [CrossRef]
- Yariswamy, M.; Yoshida, T.; Valente, A.J.; Kandikattu, H.K.; Sakamuri, S.S.; Siddesha, J.M.; Sukhanov, S.; Saifudeen, Z.; Ma, L.; Siebenlist, U.; et al. Cardiac-restricted Overexpression of TRAF3 Interacting Protein 2 (TRAF3IP2) Results in Spontaneous Development of Myocardial Hypertrophy, Fibrosis, and Dysfunction. J. Biol. Chem. 2016, 291, 19425–19436. [Google Scholar] [CrossRef] [Green Version]
- Burke, R.M.; Dirkx, R.A., Jr.; Quijada, P.; Lighthouse, J.K.; Mohan, A.; O’Brien, M.; Wojciechowski, W.; Woeller, C.F.; Phipps, R.P.; Alexis, J.D.; et al. Prevention of Fibrosis and Pathological Cardiac Remodeling by Salinomycin. Circ. Res. 2021, 128, 1663–1678. [Google Scholar] [CrossRef]
- Cantu, E.; Lederer, D.J.; Meyer, K.; Milewski, K.; Suzuki, Y.; Shah, R.J.; Diamond, J.M.; Meyer, N.J.; Tobias, J.W.; Baldwin, D.A.; et al. Gene set enrichment analysis identifies key innate immune pathways in primary graft dysfunction after lung transplantation. Am. J. Transplant. 2013, 13, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.Y.; Tong, S.; Wu, C.Y.; Ding, X.C.; Chen, J.L.; Ming, Y.; Wang, S.H. Nlrp3 Inflammasome Inhibitor MCC950 Ameliorates Obliterative Bronchiolitis by Inhibiting Th1/Th17 Response and Promoting Treg Response After Orthotopic Tracheal Transplantation in Mice. Transplantation 2020, 104, e151–e163. [Google Scholar] [CrossRef]
- D’Amico, R.; Fusco, R.; Cordaro, M.; Siracusa, R.; Peritore, A.F.; Gugliandolo, E.; Crupi, R.; Scuto, M.; Cuzzocrea, S.; Di Paola, R.; et al. Modulation of NLRP3 Inflammasome through Formyl Peptide Receptor 1 (Fpr-1) Pathway as a New Therapeutic Target in Bronchiolitis Obliterans Syndrome. Int. J. Mol. Sci. 2020, 21, 2144. [Google Scholar] [CrossRef] [Green Version]
- McElvaney, O.J.; Zaslona, Z.; Becker-Flegler, K.; Palsson-McDermott, E.M.; Boland, F.; Gunaratnam, C.; Gulbins, E.; O’Neill, L.A.; Reeves, E.P.; McElvaney, N.G. Specific Inhibition of the NLRP3 Inflammasome as an Antiinflammatory Strategy in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhou, Y.; Li, P.; Duan, J.X.; Liu, Y.P.; Sun, G.Y.; Wan, L.; Dong, L.; Fang, X.; Jiang, J.X.; et al. Blocking triggering receptor expressed on myeloid cells-1 attenuates lipopolysaccharide-induced acute lung injury via inhibiting NLRP3 inflammasome activation. Sci. Rep. 2016, 6, 39473. [Google Scholar] [CrossRef] [Green Version]
- Andrade-Oliveira, V.; Foresto-Neto, O.; Watanabe, I.K.M.; Zatz, R.; Camara, N.O.S. Inflammation in Renal Diseases: New and Old Players. Front. Pharmacol. 2019, 10, 1192. [Google Scholar] [CrossRef]
- Turner, C.M.; Arulkumaran, N.; Singer, M.; Unwin, R.J.; Tam, F.W. Is the inflammasome a potential therapeutic target in renal disease? BMC Nephrol. 2014, 15, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knauf, F.; Asplin, J.R.; Granja, I.; Schmidt, I.M.; Moeckel, G.W.; David, R.J.; Flavell, R.A.; Aronson, P.S. NALP3-mediated inflammation is a principal cause of progressive renal failure in oxalate nephropathy. Kidney Int. 2013, 84, 895–901. [Google Scholar] [CrossRef] [Green Version]
- Dessing, M.C.; Kers, J.; Damman, J.; Navis, G.J.; Florquin, S.; Leemans, J.C. Donor and recipient genetic variants in NLRP3 associate with early acute rejection following kidney transplantation. Sci. Rep. 2016, 6, 36315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigt, S.S.; Palchevskiy, V.; Belperio, J.A. Inflammasomes and IL-1 biology in the pathogenesis of allograft dysfunction. J. Clin. Investig. 2017, 127, 2022–2029. [Google Scholar] [CrossRef]
- Mezzaroma, E.; Abbate, A.; Toldo, S. NLRP3 Inflammasome Inhibitors in Cardiovascular Diseases. Molecules 2021, 26, 976. [Google Scholar] [CrossRef] [PubMed]
- Quader, M.; Mezzaroma, E.; Kenning, K.; Toldo, S. Targeting the NLRP3 inflammasome to reduce warm ischemic injury in donation after circulatory death heart. Clin. Transplant. 2020, 34, e14044. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burke, R.M.; Dale, B.L.; Dholakia, S. The NLRP3 Inflammasome: Relevance in Solid Organ Transplantation. Int. J. Mol. Sci. 2021, 22, 10721. https://doi.org/10.3390/ijms221910721
Burke RM, Dale BL, Dholakia S. The NLRP3 Inflammasome: Relevance in Solid Organ Transplantation. International Journal of Molecular Sciences. 2021; 22(19):10721. https://doi.org/10.3390/ijms221910721
Chicago/Turabian StyleBurke, Ryan M., Bethany L. Dale, and Shamik Dholakia. 2021. "The NLRP3 Inflammasome: Relevance in Solid Organ Transplantation" International Journal of Molecular Sciences 22, no. 19: 10721. https://doi.org/10.3390/ijms221910721
APA StyleBurke, R. M., Dale, B. L., & Dholakia, S. (2021). The NLRP3 Inflammasome: Relevance in Solid Organ Transplantation. International Journal of Molecular Sciences, 22(19), 10721. https://doi.org/10.3390/ijms221910721