
Atrophy of White Adipose Tissue Accompanied with Decreased Insulin-Stimulated Glucose Uptake in Mice Lacking the Small GTPase Rac1 Specifically in Adipocytes

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
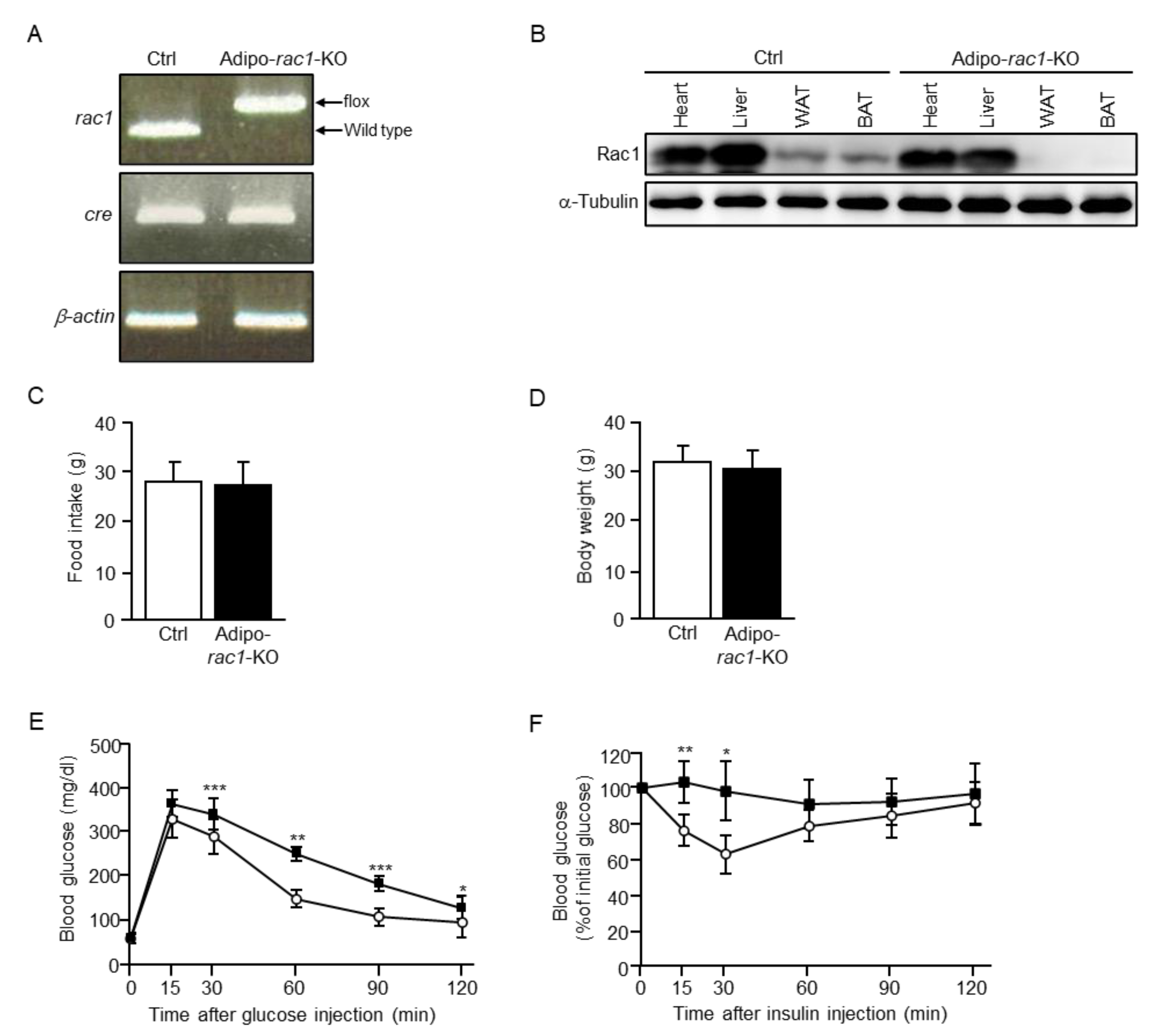
2.1. Atrophy of WAT in Adipocyte-Specific Rac1 Knockout (Adipo-Rac1-KO) Mice
2.2. Effect of Rac1 KO on Glucose Uptake and GLUT4 Translocation Induced by Insulin or a Constitutively Activated Mutant of PI3K, Akt2, or FLJ00068
2.3. The Activation of Rac1 by Insulin or a Constitutively Activated Mutant of PI3K, Akt2, or FLJ00068 and the Effect of an Akt2-Specific Inhibitor on Rac1 Activation
2.4. Effect of a Dominant-Negative RalA Mutant on GLUT4 Translocation Induced by Insulin or a Constitutively Activated Mutant of PI3K, Akt2, FLJ00068, or Rac1
2.5. Effect of Rac1 KO on the Activation of RalA Induced by Insulin or a Constitutively Activated Mutant of PI3K, Akt2, FLJ00068, or Rac1
2.6. Effect of Akt2- and Rac1-Specific Inhibitors on the Activation of RalA Induced by Insulin or a Constitutively Activated Mutant of PI3K
2.7. Effect of Rac1 KO on mRNA Levels of Various Lipogenic Enzymes
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Experiments
4.3. Genotyping
4.4. Immunoblot Analysis
4.5. Glucose and Insulin Tolerance Tests
4.6. Histological Analysis
4.7. Gene Transfer into White Adipocytes by Electroporation
4.8. Preparation of Mature White Adipocytes from sWAT and Their Primary Cultures
4.9. Measurement of the Uptake of 2-DG
4.10. Detection of GLUT4 Translocation to the Plasma Membrane by a Reporter Assay
4.11. Detection of the Activation of Rac1 and RalA
4.12. Quantitative RT-PCR Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-DG | 2-deoxy-D-glucose |
| ACC | acetyl-CoA carboxylase |
| ACLY | ATP citrate lyase |
| adipo-rac1-KO | adipocyte-specific rac1 knockout |
| BAT | brown adipose tissue |
| ctrl | control |
| DMEM | Dulbecco’s modified Eagle’s medium |
| eWAT | epididymal white adipose tissue |
| FASN | fatty acid synthase |
| GAP | GTPase-activating protein |
| GEF | guanine nucleotide exchange factor |
| GFP | green fluorescent protein |
| GPAT | glycerol-3-phosphate acyltransferase |
| GST | glutathione S-transferase |
| HA | hemagglutinin |
| Myr-Akt2 | an N-terminally myristoylated form of Akt2 |
| Myr-p110α | an N-terminally myristoylated form of the phosphoinositide 3-kinase catalytic subunit p110α |
| PBS | phosphate-buffered saline |
| PCR | polymerase chain reaction |
| PI3K | phosphoinositide 3-kinase |
| PPARγ | peroxisome proliferator-activated receptor γ |
| RT | reverse transcriptase |
| SCD1 | stearoyl-CoA desaturase 1 |
| sWAT | subcutaneous white adipose tissue |
| WAT | white adipose tissue |
References
- Rosen, E.D.; Spiegelman, B.M. What We Talk about When We Talk about Fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Czech, M.P. The GLUT4 Glucose Transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T. Molecular Mechanisms for the Regulation of Insulin-Stimulated Glucose Uptake by Small Guanosine Triphosphatases in Skeletal Muscle and Adipocytes. Int. J. Mol. Sci. 2014, 15, 18677–18692. [Google Scholar] [CrossRef] [Green Version]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Chiu, T.T.; Jensen, T.; Sylow, L.; Richter, E.A.; Klip, A. Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle. Cell. Signal. 2011, 23, 1546–1554. [Google Scholar] [CrossRef]
- Jaldin-Fincati, J.R.; Pavarotti, M.; Frendo-Cumbo, S.; Bilan, P.J.; Klip, A. Update on GLUT4 Vesicle Traffic: A Cornerstone of Insulin Action. Trends Endocrinol. Metab. 2017, 28, 597–611. [Google Scholar] [CrossRef]
- Kane, S.; Sano, H.; Liu, S.C.H.; Asara, J.M.; Lane, W.S.; Garner, C.C.; Lienhard, G.E. A Method to Identify Serine Kinase Substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J. Biol. Chem. 2002, 277, 22115–22118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Bilan, P.J.; Liu, Z.; Klip, A. Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc. Natl. Acad. Sci. USA 2010, 107, 19909–19914. [Google Scholar] [CrossRef] [Green Version]
- Sano, H.; Eguez, L.; Teruel, M.; Fukuda, M.; Chuang, T.D.; Chavez, J.A.; Lienhard, G.E.; McGraw, T.E. Rab10, a Target of the AS160 Rab GAP, Is Required for Insulin-Stimulated Translocation of GLUT4 to the Adipocyte Plasma Membrane. Cell Metab. 2007, 5, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, Y.; Zhang, J.; Deng, Y.; Jiang, L.; Song, E.; Wu, X.S.; Hammer, J.A.; Xu, T.; Lippincott-Schwartz, J. Rab10 and myosin-Va mediate insulin-stimulated GLUT4 storage vesicle translocation in adipocytes. J. Cell Biol. 2012, 198, 545–560. [Google Scholar] [CrossRef] [Green Version]
- Chiang, S.-H.; Baumann, C.A.; Kanzaki, M.; Thurmond, D.C.; Watson, R.T.; Neudauer, C.L.; Macara, I.G.; Pessin, J.E.; Saltiel, A. Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10. Nature 2001, 410, 944–948. [Google Scholar] [CrossRef] [Green Version]
- JeBailey, L.; Rudich, A.; Huang, X.; Di Ciano-Oliveira, C.; Kapus, A.; Klip, A. Skeletal Muscle Cells and Adipocytes Differ in Their Reliance on TC10 and Rac for Insulin-Induced Actin Remodeling. Mol. Endocrinol. 2004, 18, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Khayat, Z.; Tong, P.; Yaworsky, K.; Bloch, R.; Klip, A. Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J. Cell Sci. 2000, 113, 279–290. [Google Scholar] [CrossRef]
- Randhawa, V.K.; Ishikura, S.; Talior-Volodarsky, I.; Cheng, A.W.; Patel, N.; Hartwig, J.H.; Klip, A. GLUT4 Vesicle Recruitment and Fusion Are Differentially Regulated by Rac, AS160, and Rab8A in Muscle Cells. J. Biol. Chem. 2008, 283, 27208–27219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sylow, L.; Jensen, T.E.; Kleinert, M.; Højlund, K.; Kiens, B.; Wojtaszewski, J.; Prats, C.; Schjerling, P.; Richter, E.A. Rac1 Signaling Is Required for Insulin-Stimulated Glucose Uptake and Is Dysregulated in Insulin-Resistant Murine and Human Skeletal Muscle. Diabetes 2013, 62, 1865–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, S.; Kataoka, T.; Satoh, T. Activation of the small GTPase Rac1 by a specific guanine-nucleotide-exchange factor suffices to induce glucose uptake into skeletal-muscle cells. Biol. Cell 2008, 100, 645–661. [Google Scholar] [CrossRef]
- Ueda, S.; Kitazawa, S.; Ishida, K.; Nishikawa, Y.; Matsui, M.; Matsumoto, H.; Aoki, T.; Nozaki, S.; Takeda, T.; Tamori, Y.; et al. Crucial role of the small GTPase Rac1 in insulin-stimulated translocation of glucose transporter 4 to the mouse skeletal muscle sarcolemma. FASEB J. 2010, 24, 2254–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozaki, S.; Takeda, T.; Kitaura, T.; Takenaka, N.; Kataoka, T.; Satoh, T. Akt2 regulates Rac1 activity in the insulin-dependent signaling pathway leading to GLUT4 translocation to the plasma membrane in skeletal muscle cells. Cell. Signal. 2013, 25, 1361–1371. [Google Scholar] [CrossRef]
- Takenaka, N.; Izawa, R.; Wu, J.; Kitagawa, K.; Nihata, Y.; Hosooka, T.; Noguchi, T.; Ogawa, W.; Aiba, A.; Satoh, T. A critical role of the small GTPase Rac1 in Akt2-mediated GLUT4 translocation in mouse skeletal muscle. FEBS J. 2014, 281, 1493–1504. [Google Scholar] [CrossRef]
- Takenaka, N.; Nihata, Y.; Satoh, T. Immunofluorescent detection of the activation of the small GTPase Rac1 in mouse skeletal muscle fibers. Anal. Biochem. 2015, 476, 5–7. [Google Scholar] [CrossRef]
- Chiu, T.T.; Sun, Y.; Koshkina, A.; Klip, A. Rac-1 Superactivation Triggers Insulin-independent Glucose Transporter 4 (GLUT4) Translocation That Bypasses Signaling Defects Exerted by c-Jun N-terminal kinase (JNK)- and Ceramide-induced Insulin Resistance. J. Biol. Chem. 2013, 288, 17520–17531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sylow, L.; Kleinert, M.; Pehmøller, C.; Prats, C.; Chiu, T.T.; Klip, A.; Richter, E.A.; Jensen, T.E. Akt and Rac1 signaling are jointly required for insulin-stimulated glucose uptake in skeletal muscle and downregulated in insulin resistance. Cell. Signal. 2013, 26, 323–331. [Google Scholar] [CrossRef]
- Takenaka, N.; Nihata, Y.; Ueda, S.; Satoh, T. In situ detection of the activation of Rac1 and RalA small GTPases in mouse adipocytes by immunofluorescent microscopy following in vivo and ex vivo insulin stimulation. Cell. Signal. 2017, 39, 108–117. [Google Scholar] [CrossRef]
- Takenaka, N.; Nakao, M.; Matsui, S.; Satoh, T. A Crucial Role for the Small GTPase Rac1 Downstream of the Protein Kinase Akt2 in Insulin Signaling that Regulates Glucose Uptake in Mouse Adipocytes. Int. J. Mol. Sci. 2019, 20, 5443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takenaka, N.; Nihata, Y.; Satoh, T. Rac1 Activation Caused by Membrane Translocation of a Guanine Nucleotide Exchange Factor in Akt2-Mediated Insulin Signaling in Mouse Skeletal Muscle. PLoS ONE 2016, 11, e0155292. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, N.; Nakao, M.; Hasegawa, K.; Chan, M.P.; Satoh, T. The guanine nucleotide exchange factor FLJ00068 activates Rac1 in adipocyte insulin signaling. FEBS Lett. 2020, 594, 4370–4380. [Google Scholar] [CrossRef] [PubMed]
- Marcusohn, J.; Isakoff, S.; Rose, E.; Symons, M.; Skolnik, E. The GTP-binding protein Rac does not couple PI 3-kinase to insulin-stimulated glucose transport in adipocytes. Curr. Biol. 1995, 5, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Kassai, H.; Terashima, T.; Fukaya, M.; Nakao, K.; Sakahara, M.; Watanabe, M.; Aiba, A. Rac1 in cortical projection neurons is selectively required for midline crossing of commissural axonal formation. Eur. J. Neurosci. 2008, 28, 257–267. [Google Scholar] [CrossRef]
- Eguchi, J.; Wang, X.; Yu, S.; Kershaw, E.E.; Chiu, P.C.; Dushay, J.; Estall, J.; Klein, U.; Maratos-Flier, E.; Rosen, E.D. Transcriptional Control of Adipose Lipid Handling by IRF4. Cell Metab. 2011, 13, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Bogan, J.S.; McKee, A.E.; Lodish, H.F. Insulin-Responsive Compartments Containing GLUT4 in 3T3-L1 and CHO Cells: Regulation by Amino Acid Concentrations. Mol. Cell. Biol. 2001, 21, 4785–4806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.-W.; Leto, D.; Chiang, S.-H.; Wang, Q.; Saltiel, A.R. Activation of RalA Is Required for Insulin-Stimulated Glut4 Trafficking to the Plasma Membrane via the Exocyst and the Motor Protein Myo1c. Dev. Cell 2007, 13, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, S.; Ueda, S.; Takenaka, N.; Kataoka, T.; Satoh, T. Role of RalA downstream of Rac1 in insulin-dependent glucose uptake in muscle cells. Cell. Signal. 2012, 24, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, N.; Sumi, Y.; Matsuda, K.; Fujita, J.; Hosooka, T.; Noguchi, T.; Aiba, A.; Satoh, T. Role for RalA downstream of Rac1 in skeletal muscle insulin signalling. Biochem. J. 2015, 469, 445–454. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morigny, P.; Boucher, J.; Arner, P.; Langin, D. Lipid and glucose metabolism in white adipocytes: Pathways, dysfunction and therapeutics. Nat. Rev. Endocrinol. 2021, 17, 276–295. [Google Scholar] [CrossRef]
- Fiorenza, C.G.; Chou, S.H.; Mantzoros, C.S. Lipodystrophy: Pathophysiology and advances in treatment. Nat. Rev. Endocrinol. 2010, 7, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Magré, J.; Delépine, M.; Khallouf, E.; Gedde-Dahl, T.; Van Maldergem, L.; Sobel, E.; Papp, J.; Meier, M.; Mégarbané, A.; BSCL Working Group; et al. Identification of the gene altered in Berardinelli–Seip congenital lipodystrophy on chromosome 11q13. Nat. Genet. 2001, 28, 365–370. [Google Scholar] [CrossRef]
- Chudasama, K.K.; Winnay, J.; Johansson, S.; Claudi, T.; König, R.; Haldorsen, I.S.; Johansson, B.B.; Woo, J.R.; Aarskog, D.; Sagen, J.V.; et al. SHORT Syndrome with Partial Lipodystrophy Due to Impaired Phosphatidylinositol 3 Kinase Signaling. Am. J. Hum. Genet. 2013, 93, 150–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, S.; Rochford, J.; Wolfrum, C.; Gray, S.L.; Schinner, S.; Wilson, J.C.; Soos, M.A.; Murgatroyd, P.R.; Williams, R.M.; Acerini, C.; et al. A Family with Severe Insulin Resistance and Diabetes Due to a Mutation in AKT2. Science 2004, 304, 1325–1328. [Google Scholar] [CrossRef] [Green Version]
- Hussain, K.; Challis, B.; Rocha, N.; Payne, F.; Minic, M.; Thompson, A.; Daly, A.; Scott, C.; Harris, J.; Smillie, B.J.L.; et al. An Activating Mutation of AKT2 and Human Hypoglycemia. Science 2011, 334, 474. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.; Kimber, W.A.; Luan, J.; Soos, M.A.; Semple, R.K.; Wareham, N.J.; O’Rahilly, S.; Barroso, I. Analysis of Genetic Variation in Akt2/PKB- in Severe Insulin Resistance, Lipodystrophy, Type 2 Diabetes, and Related Metabolic Phenotypes. Diabetes 2007, 56, 714–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, R.S.; Orena, S.J.; Rafidi, K.; Torchia, A.J.; Stock, J.L.; Hildebrandt, A.L.; Coskran, T.; Black, S.C.; Brees, D.J.; Wicks, J.R.; et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKBβ. J. Clin. Investig. 2003, 112, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, N.; Yasuda, N.; Nihata, Y.; Hosooka, T.; Noguchi, T.; Aiba, A.; Satoh, T. Role of the guanine nucleotide exchange factor in Akt2-mediated plasma membrane translocation of GLUT4 in insulin-stimulated skeletal muscle. Cell. Signal. 2014, 26, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Stahl, A.; Evans, J.G.; Pattel, S.; Hirsch, D.; Lodish, H.F. Insulin Causes Fatty Acid Transport Protein Translocation and Enhanced Fatty Acid Uptake in Adipocytes. Dev. Cell 2002, 2, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Raun, S.H.; Ali, M.S.; Kjøbsted, R.; Møller, L.L.V.; Federspiel, M.A.; Richter, E.A.; Jensen, T.; Sylow, L. Rac1 muscle knockout exacerbates the detrimental effect of high-fat diet on insulin-stimulated muscle glucose uptake independently of Akt. J. Physiol. 2018, 596, 2283–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasegawa, K.; Takenaka, N.; Tanida, K.; Chan, M.P.; Sakata, M.; Aiba, A.; Satoh, T. Atrophy of White Adipose Tissue Accompanied with Decreased Insulin-Stimulated Glucose Uptake in Mice Lacking the Small GTPase Rac1 Specifically in Adipocytes. Int. J. Mol. Sci. 2021, 22, 10753. https://doi.org/10.3390/ijms221910753
Hasegawa K, Takenaka N, Tanida K, Chan MP, Sakata M, Aiba A, Satoh T. Atrophy of White Adipose Tissue Accompanied with Decreased Insulin-Stimulated Glucose Uptake in Mice Lacking the Small GTPase Rac1 Specifically in Adipocytes. International Journal of Molecular Sciences. 2021; 22(19):10753. https://doi.org/10.3390/ijms221910753
Chicago/Turabian StyleHasegawa, Kiko, Nobuyuki Takenaka, Kenya Tanida, Man Piu Chan, Mizuki Sakata, Atsu Aiba, and Takaya Satoh. 2021. "Atrophy of White Adipose Tissue Accompanied with Decreased Insulin-Stimulated Glucose Uptake in Mice Lacking the Small GTPase Rac1 Specifically in Adipocytes" International Journal of Molecular Sciences 22, no. 19: 10753. https://doi.org/10.3390/ijms221910753