
Coagulopathies after Vaccination against SARS-CoV-2 May Be Derived from a Combined Effect of SARS-CoV-2 Spike Protein and Adenovirus Vector-Triggered Signaling Pathways


{kind=link}
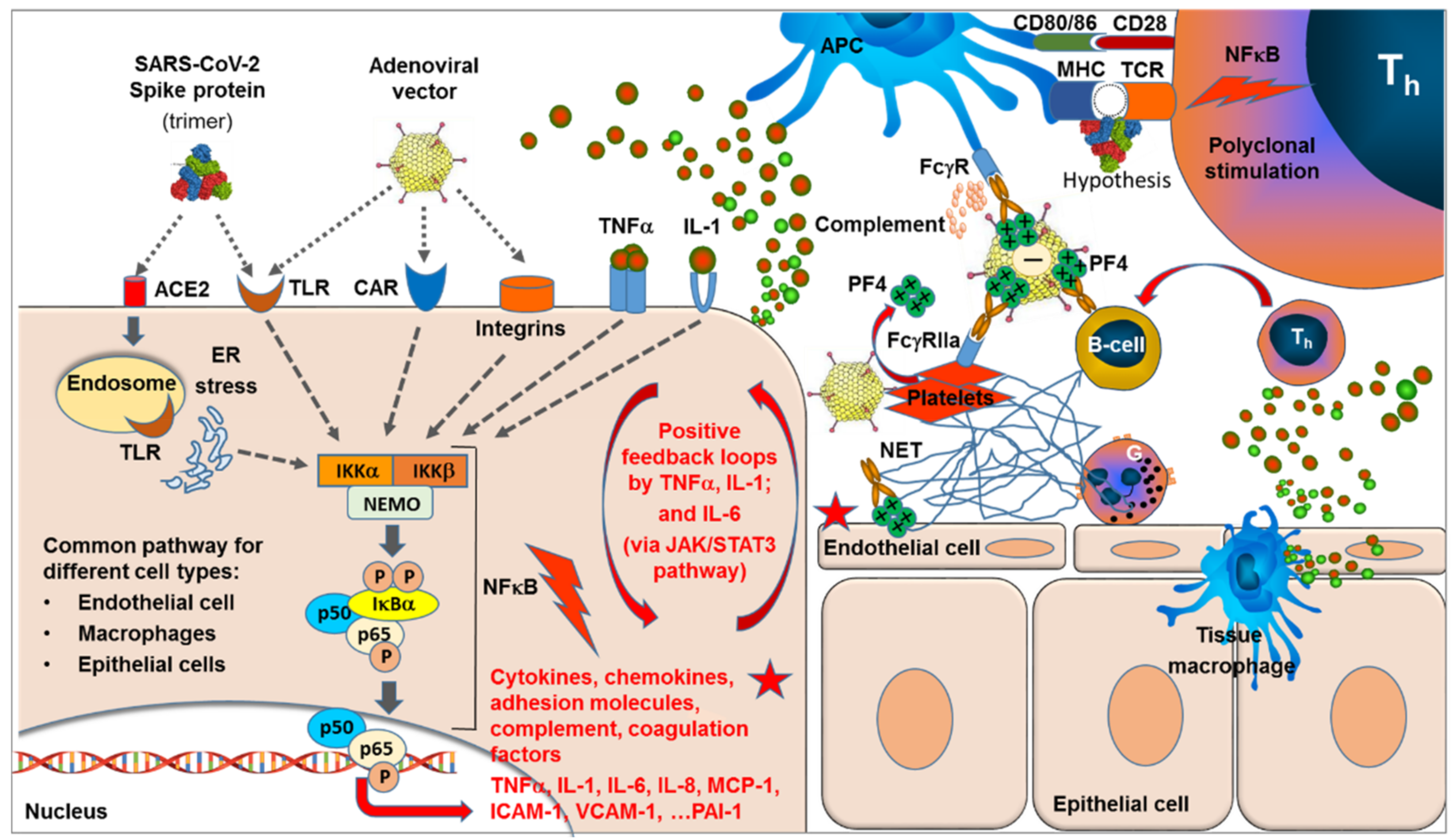
Abstract
:1. Introduction
2. SARS-CoV-2 and COVID-19—The Virus and the Disease
3. Frequently Described Side Effects after Vaccination against SARS-CoV-2
4. Rare Serious Side Effects after Vaccination Visible during Mass Vaccination after Market Approval
5. Superantigen Hypothesis
5.1. Superantigen Induce Procoagulant Activity
5.2. Superantigens and NF-kappaB
6. NF-κB Pathway
6.1. NF-κB Pathway Is Part of Normal T Cell Activation
6.2. SARS-CoV-2 Spike Protein Induces NF-κB
6.3. NF-κB Activation Is Central to Coagulation Events
7. SARS-CoV-2 Spike Protein Subunit S1 Induces JAK/STAT3 Activation
8. Platelet Factor 4
Additional Activities of PF4
9. The Impact of the Adenoviral Vector
10. Additive and Synergistic Effects of the S Protein and Adenoviral Vector
10.1. Antigen Mimickry
10.2. NF-κB Promotes Leaky Expression of Adenovirus Genes in a Replication Incompetent Adenovirus Vector
11. Adenoviral and Producer Cell Protein Impurities, and Additives in the ChAdOx1 nCov-19 Vaccine Formulation
12. Discussion of an Integrated Mechanistic Model for VITT Following Vaccination with Adenovirus Vector-Based Vaccines
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramasamy, M.N.; Minassian, A.M.; Ewer, K.J.; Flaxman, A.L.; Folegatti, P.M.; Owens, D.R.; Voysey, M.; Aley, P.K.; Angus, B.; Babbage, G.; et al. Safety and immunogenicity of ChAdOx1 nCoV-19 vaccine administered in a prime-boost regimen in young and old adults (COV002): A single-blind, random-ised, controlled, phase 2/3 trial. Lancet 2021, 396, 1979–1993. [Google Scholar] [CrossRef]
- Voysey, M.; Clemens, S.A.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2020, 384, 403–416. [Google Scholar] [CrossRef]
- Sadoff, J.; Gray, G.; Vandebosch, A.; Cárdenas, V.; Shukarev, G.; Grinsztejn, B.; Goepfert, P.A.; Truyers, C.; Fennema, H.; Spiessens, B.; et al. Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against Covid-19. N. Engl. J. Med. 2021, 384, 2187–2201. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.H.; Sørvoll, I.H.; Michelsen, A.E.; Munthe, L.A.; Lund-Johansen, F.; Ahlen, M.T.; Wiedmann, M.; Aamodt, A.-H.; Skattør, T.H.; Tjønnfjord, G.E.; et al. Thrombosis and Thrombocytopenia after ChAdOx1 nCoV-19 Vaccination. N. Engl. J. Med. 2021, 384, 2124–2130. [Google Scholar] [CrossRef]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef]
- Scully, M.; Singh, D.; Lown, R.; Poles, A.; Solomon, T.; Levi, M.; Goldblatt, D.; Kotoucek, P.; Thomas, W.; Lester, W. Pathologic Antibodies to Platelet Factor 4 after ChAdOx1 nCoV-19 Vaccination. N. Engl. J. Med. 2021, 384, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Berlit, P.; Diener, H.C.; Gerloff, C.; Greinacher, A.; Klein, C.; Petzold, G.; Poli, S.; Piccininni, M.; Kurth, T.; et al. COVID-19 vaccine-associated cerebral venous thrombosis in Germany: A descriptive study. medRxiv 2021. [Google Scholar] [CrossRef]
- Muir, K.-L.; Kallam, A.; Koepsell, S.A.; Gundabolu, K. Thrombotic Thrombocytopenia after Ad26.COV2.S Vaccination. N. Engl. J. Med. 2021, 384, 1964–1965. [Google Scholar] [CrossRef]
- See, I.; Su, J.R.; Lale, A.; Woo, E.J.; Guh, A.Y.; Shimabukuro, T.T.; Streiff, M.B.; Rao, A.K.; Wheeler, A.P.; Beavers, S.F.; et al. US Case Reports of Cerebral Venous Sinus Thrombosis With Thrombocytopenia After Ad26.COV2.S Vaccination, March 2 to April 21, 2021. JAMA 2021, 325, 2448. [Google Scholar] [CrossRef]
- Abdelrahman, Z.; Li, M.; Wang, X. Comparative Review of SARS-CoV-2, SARS-CoV, MERS-CoV, and Influenza A Respiratory Viruses. Front. Immunol. 2020, 11, 552909. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Pollock, A.M.; Lancaster, J. Asymptomatic transmission of covid-19. BMJ 2020, 371, m4851. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, M.; Lukasik, Z.M.; Agache, I.; Akdis, C.A.; Akdis, D.; Akdis, M.; Barcik, W.; Brough, H.A.; Eiwegger, T.; Eljaszewicz, A.; et al. Immunology of COVID-19: Mechanisms, clinical outcome, diagnostics, and perspectives—A report of the European Academy of Allergy and Clinical Immunology (EAACI). Allergy 2020, 75, 2445–2476. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Zheng, Z.; Peng, F.; Xu, B.; Zhao, J.; Liu, H.; Peng, J.; Li, Q.; Jiang, C.; Zhou, Y.; Liu, S.; et al. Risk factors of critical & mortal COVID-19 cases: A systematic literature review and meta-analysis. J. Infect. 2020, 81, e16–e25. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-J.; Jung, S.I. Age-Related Morbidity and Mortality among Patients with COVID-19. Infect. Chemother. 2020, 52, 154–164. [Google Scholar] [CrossRef]
- Biswas, M.; Rahaman, S.; Biswas, T.K.; Haque, Z.; Ibrahim, B. Association of Sex, Age, and Comorbidities with Mortality in COVID-19 Patients: A Systematic Review and Meta-Analysis. Intervirology 2021, 64, 36–47. [Google Scholar] [CrossRef]
- O’Driscoll, M.; Dos Santos, G.R.; Wang, L.; Cummings, D.A.T.; Azman, A.S.; Paireau, J.; Fontanet, A.; Cauchemez, S.; Salje, H. Age-specific mortality and immunity patterns of SARS-CoV-2. Nat. Cell Biol. 2021, 590, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P. Pulmonary most-mortem findings in a large series of COVID-19 cases from Northern Italy. Lancet 2020, 20, P1135–P1140. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; Macary, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Schett, G.; Sticherling, M.; Neurath, M.F. COVID-19: Risk for cytokine targeting in chronic inflammatory diseases? Nat. Rev. Immunol. 2020, 20, 271–272. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.B.; June, C.H. Cytokine release syndrome in severe COVID-19. Science 2020, 368, 473–474. [Google Scholar] [CrossRef] [Green Version]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Huang, J.; Hume, A.J.; Abo, K.M.; Werder, R.B.; Villacorta-Martin, C.; Alysandratos, K.-D.; Beermann, M.L.; Simone-Roach, C.; Lindstrom-Vautrin, J.; Olejnik, J.; et al. SARS-CoV-2 Infection of Pluripotent Stem Cell-Derived Human Lung Alveolar Type 2 Cells Elicits a Rapid Epithelial-Intrinsic Inflammatory Response. Cell Stem Cell 2020, 27, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, C.J.; Cerikan, B.; Cortese, M.; Frankish, J.; Lee, J.Y.; Plociennikowska, A.; Heigwer, F.; Joecks, S.; Burkart, S.S.; Zander, D.Y.; et al. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-κB. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 37. [Google Scholar] [CrossRef]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef]
- Hong, K.S.; Ahn, J.H.; Jang, J.G.; Lee, J.H.; Kim, H.N.; Kim, D.; Lee, W. GSK-LSD1, an LSD1 inhibitor, quashes SARS-CoV-2-triggered cytokine release syndrome in-vitro. Signal Transduct. Target. Ther. 2020, 5, 267. [Google Scholar] [CrossRef]
- McAleavy, M.; Zhang, Q.; Xu, J.; Pan, L.; Wakai, M.; Ehmann, P.J.; Wipperman, M.F.; Shavlakadze, T.; Hamon, S.C.; Boyapati, A.; et al. Activin A correlates with the worst outcomes in COVID-19 patients, and can be induced by cytokines via the IKK/NF-kappa B pathway. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hariharan, A.; Hakeem, A.R.; Radhakrishnan, S.; Reddy, M.S.; Rela, M. The Role and Therapeutic Potential of NF-kappa-B Pathway in Severe COVID-19 Patients. Inflammopharmacology 2021, 29, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Kircheis, R.; Haasbach, E.; Lueftenegger, D.; Heyken, W.T.; Ocker, M.; Planz, O. NF-κB Pathway as a Potential Target for Treatment of Critical Stage COVID-19 Patients. Front. Immunol. 2020, 11, 598444. [Google Scholar] [CrossRef]
- Lo, M.W.; Kemper, C.; Woodruff, T.M. COVID-19: Complement, Coagulation, and Collateral Damage. J. Immunol. 2020, 205, 1488–1495. [Google Scholar] [CrossRef]
- Holter, J.C.; Pischke, S.E.; de Boer, E.; Lind, A.; Jenum, S.; Holten, A.R.; Tonby, K.; Barratt-Due, A.; Sokolova, M.; Schjalm, C.; et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl. Acad. Sci. USA 2020, 117, 25018–25025. [Google Scholar] [CrossRef]
- Polycarpou, A.; Howard, M.; Farrar, C.A.; Greenlaw, R.; Fanelli, G.; Wallis, R.; Klavinskis, L.S.; Sacks, S. Rationale for targeting complement in COVID-19. EMBO Mol. Med. 2020, 12, e12642. [Google Scholar] [CrossRef] [PubMed]
- Fletcher-Sandersjöö, A.; Bellander, B.-M. Is COVID-19 associated thrombosis caused by overactivation of the complement cascade? A literature review. Thromb. Res. 2020, 194, 36–41. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, C.G.; Wilczynski, S.; Wenceslau, C.F.; Webb, R.C. A new storm on the horizon in COVID-19: Bradykinin-induced vascular complications. Vasc. Pharmacol. 2021, 137, 106826. [Google Scholar] [CrossRef]
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Stürzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine 2020, 58, 102925. [Google Scholar] [CrossRef]
- Kvietys, P.R.; Fakhoury, H.M.A.; Kadan, S.; Yaqinuddin, A.; Al-Mutairy, E.; Al-Kattan, K. COVID-19: Lung-Centric Immunothrombosis. Front. Cell. Infect. Microbiol. 2021, 11, 679878. [Google Scholar] [CrossRef]
- Gando, S.; Wada, T. Pathomechanisms Underlying Hypoxemia in Two COVID-19-Associated Acute Respiratory Distress Syndrome Phenotypes: Insights from Thrombosis and Hemostasis. Shock 2021. [Google Scholar] [CrossRef]
- De Buhr, N.; von Köckritz-Blickwede, M. The Balance of Neutrophil Extracellular Trap Formation and Nuclease Degradation: An Unknown Role of Bacterial Coinfections in COVID-19 Patients? mBio 2021, 12, e03304-20. [Google Scholar] [CrossRef] [PubMed]
- Thachil, J.; Tang, N.; Gando, S.; Falanga, A.; Cattaneo, M.; Levi, M.; Clark, C.; Iba, T. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 1023–1026. [Google Scholar] [CrossRef]
- Gu, S.X.; Tyagi, T.; Jain, K.; Gu, V.W.; Lee, S.H.; Hwa, J.M.; Kwan, J.M.; Krause, D.S.; Lee, A.I.; Halene, S.; et al. Thrombocytopathy and endotheliopathy: Crucial contributors to COVID-19 thromboinflammation. Nat. Rev. Cardiol. 2021, 18, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Estes, S.K.; Ali, R.A.; Gandhi, A.A.; Yalavarthi, S.; Shi, H.; Sule, G.; Gockman, K.; Madison, J.A.; Zuo, M.; et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl. Med. 2020, 12, eabd3876. [Google Scholar] [CrossRef]
- Oxley, T.J.; Mocco, J.; Majidi, S.; Kellner, C.P.; Shoirah, H.; Singh, I.P.; De Leacy, R.A.; Shigematsu, T.; Ladner, T.R.; Yaeger, K.A.; et al. Large-Vessel Stroke as a Presenting Feature of Covid-19 in the Young. N. Engl. J. Med. 2020, 382, e60. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 2020, 191, 148–150. [Google Scholar] [CrossRef]
- Diorio, C.; McNerney, K.O.; Lambert, M.; Paessler, M.; Anderson, E.M.; Henrickson, S.E.; Chase, J.; Liebling, E.J.; Burudpakdee, C.; Lee, J.H.; et al. Evidence of thrombotic microangiopathy in children with SARS-CoV-2 across the spectrum of clinical presentations. Blood Adv. 2020, 4, 6051–6063. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor–enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Yu, J.; Yuan, X.; Chen, H.; Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood 2020, 136, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- George, J.N.; Nester, C.M. Syndromes of Thrombotic Microangiopathy. N. Engl. J. Med. 2014, 371, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brocklebank, V.; Wood, K.M.; Kavanagh, D. Thrombotic Microangiopathy and the Kidney. Clin. J. Am. Soc. Nephrol. 2017, 13, 300–317. [Google Scholar] [CrossRef] [Green Version]
- Arnold, D.M.; Patriquin, C.J.; Nazy, I. Thrombotic microangiopathies: A general approach to diagnosis and management. Can. Med Assoc. J. 2017, 189, E153–E159. [Google Scholar] [CrossRef] [Green Version]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; et al. Post-mortem examination of COVID19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings of lungs and other organs suggesting vascular dysfunction. Histopathology 2020, 77, 198–209. [Google Scholar] [CrossRef]
- Nadkarni, G.N.; Lala, A.; Bagiella, E.; Chang, H.L.; Moreno, P.R.; Pujadas, E.; Arvind, V.; Bose, S.; Charney, A.W.; Chen, M.D.; et al. Anticoagulation, Bleeding, Mortality, and Pathology in Hospitalized Patients With COVID-19. J. Am. Coll. Cardiol. 2020, 76, 1815–1826. [Google Scholar] [CrossRef] [PubMed]
- Logunov, D.Y.; Dolzhikova, I.V.; Shcheblyakov, D.V.; Tukhvatulin, A.I.; Zubkova, O.V.; Dzharullaeva, A.S.; Kovyrshina, A.V.; Lubenets, N.L.; Grousova, D.M.; Erokhova, A.S.; et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: An interim analysis of a randomised controlled phase 3 trial in Russia. Lancet 2021, 397, 671–681. [Google Scholar] [CrossRef]
- Hunter, P.R. Thrombosis after covid-19 vaccination. BMJ 2021, 373, n958. [Google Scholar] [CrossRef]
- Rivas, M.N.; Porritt, R.A.; Cheng, M.H.; Bahar, I.; Arditi, M. COVID-19–associated multisystem inflammatory syndrome in children (MIS-C): A novel disease that mimics toxic shock syndrome—the superantigen hypothesis. J. Allergy Clin. Immunol. 2021, 147, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.H.; Zhang, S.; Porritt, R.A.; Rivas, M.N.; Paschold, L.; Willscher, E.; Binder, M.; Arditi, M.; Bahar, I. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation. Proc. Natl. Acad. Sci. USA 2020, 117, 25254–25262. [Google Scholar] [CrossRef]
- Kouo, T.; Chaisawangwong, W. SARS-CoV-2 as a superantigen in multisystem inflammatory syndrome in children (MIS-C). J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Scaglioni, V.; Soriano, E.R. Are superantigens the cause of cytokine storm and viral sepsis in severe COVID-19? Observations and hypothesis. Scand. J. Immunol. 2020, 92, e12944. [Google Scholar] [CrossRef] [PubMed]
- Rieper, K.; Sturm, A. Erste Fälle des Multisystem Inflammatory Syndrome nach SARS-CoV-2-Infektion bei jungen Erwachsenen in Deutschland [First Cases of Multisystem Inflammatory Syndrome following SARS-CoV-2 infection in Adults in Germany]. Dtsch. Med. Wochenschr. 2021, 146, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Umemura, Y.; Wada, H.; Levy, H. The Roles of Coagulation Disorder and Microthrombosis in Sepsis: Pathophysiology, Diagnosis, and Treatment. Arch. Med Res. 2021, 31, S0188-4409(21)00162-4. [Google Scholar] [CrossRef]
- Mattsson, E.; Herwald, H.; Egesten, A. Superantigens from Staphylococcus aureus induce procoagulant activity and monocyte tissue factor expression in whole blood and mononuclear cells via IL-1beta. J. Thromb. Haemost. 2003, 1, 2569–2576. [Google Scholar] [CrossRef] [Green Version]
- Kulhankova, K.; King, J.; Salgado-Pabón, W. Staphylococcal toxic shock syndrome: Superantigen-mediated enhancement of endotoxin shock and adaptive immune suppression. Immunol. Res. 2014, 59, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Trede, N.S.; Castigli, E.; Geha, R.S.; Chatila, T. Microbial superantigens induce NF-kappa B in the human monocytic cell line THP-1. J. Immunol. 1993, 150, 5604–5613. [Google Scholar] [PubMed]
- Zollner, T.M.; Podda, M.; Pien, C.; Elliott, P.J.; Kaufmann, R.; Boehncke, W.H. Proteasome inhibition reduces superantigen-mediated T cell activation and the severity of psoriasis in a SCID-hu model. J. Clin. Investig. 2002, 109, 671–679. [Google Scholar] [CrossRef]
- Krakauer, T.; Buckley, M. The Potency of Anti-Oxidants in Attenuating Superantigen-Induced Proinflammatory Cytokines Correlates with Inactivation of NF-κB. Immunopharmacol. Immunotoxicol. 2008, 30, 163–179. [Google Scholar] [CrossRef]
- Fu, X.; Xu, M.; Song, Y.; Li, Y.; Zhang, H.; Zhang, J.; Zhang, C. Enhanced interaction between SEC2 mutant and TCR Vβ induces MHC II–independent activation of T cells via PKCθ/NF-κB and IL-2R/STAT5 signaling pathways. J. Biol. Chem. 2018, 293, 19771–19784. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Schaefer, B.C. A new look at T cell receptor signaling to nuclear factor-κB. Trends Immunol. 2013, 34, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. bioRxiv 2021. [Google Scholar] [CrossRef]
- Patra, T.; Meyer, K.; Geerling, L.; Isbell, T.S.; Hoft, D.F.; Brien, J.; Pinto, A.K.; Ray, R.B.; Ray, R. SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells. PLoS Pathog. 2020, 16, e1009128. [Google Scholar] [CrossRef]
- Hsu, A.C.-Y.; Wang, G.; Reid, A.T.; Veerati, P.C.; Pathinayake, P.S.; Daly, K.; Mayall, J.R.; Hansbro, P.M.; Horvat, J.C.; Wang, F.; et al. SARS-CoV-2 Spike protein promotes hyper-inflammatory response that can be ameliorated by Spike-antagonistic peptide and FDA-approved ER stress and MAP kinase inhibitors in vitro. bioRxiv 2020. [Google Scholar] [CrossRef]
- Olajide, O.A.; Iwuanyanwu, V.U.; Lepiarz-Raba, I.; Al-Hindawi, A.A. Induction of Exaggerated Cytokine Production in Human Peripheral Blood Mononuclear Cells by a Recombinant SARS-CoV-2 Spike Glycoprotein S1 and Its Inhibition by Dexamethasone. Inflamm. 2021, 44, 1865–1877. [Google Scholar] [CrossRef]
- Petruk, G.; Puthia, M.; Petrlova, J.; Samsudin, F.; Strömdahl, A.-C.; Cerps, S.; Uller, L.; Kjellström, S.; Bond, P.J.; Schmidtchen, A.A. SARS-CoV-2 spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J. Mol. Cell Biol. 2021, 12, 916–932. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ye, L.; Ye, L.; Li, B.; Gao, B.; Zeng, Y.; Kong, L.; Fang, X.; Zheng, H.; Wu, Z.; et al. Up-regulation of IL-6 and TNF-α induced by SARS-coronavirus spike protein in murine macrophages via NF-κB pathway. Virus Res. 2007, 128, 1–8. [Google Scholar] [CrossRef]
- Biancatelli, R.M.L.C.; Solopov, P.A.; Sharlow, E.R.; Lazo, J.S.; Marik, P.E.; Catravas, J.D. The SARS-CoV-2 spike protein subunit S1 induces COVID-19-like acute lung injury in Κ18-hACE2 transgenic mice and barrier dysfunction in human endothelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2021, 321, L477–L484. [Google Scholar] [CrossRef]
- Hou, B.; Eren, M.; Painter, C.; Covington, J.W.; Dixon, J.D.; Schoenhard, J.A.; Vaughan, D.E. Tumor Necrosis Factor α Activates the Human Plasminogen Activator Inhibitor-1 Gene through a Distal Nuclear Factor κB Site. J. Biol. Chem. 2004, 279, 18127–18136. [Google Scholar] [CrossRef] [Green Version]
- Morgan, E.N.; Pohlman, T.H.; Vocelka, C.; Farr, A.; Lindley, G.; Chandler, W.; Griscavage-Ennis, J.M.; Verrier, E.D. Nuclear factor κB mediates a procoagulant response in monocytes during extracorporeal circulation. J. Thorac. Cardiovasc. Surg. 2003, 125, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Kastl, S.P.; Speidl, W.S.; Kaun, C.; Rega, G.; Assadian, A.; Weiss, T.W.; Valent, P.; Hagmueller, G.W.; Maurer, G.; Huber, K.; et al. The complement component C5a induces the expression of plasminogen activator inhibitor-1 in human macrophages via NF-?B activation. J. Thromb. Haemost. 2006, 4, 1790–1797. [Google Scholar] [CrossRef]
- Kojok, K.; El-Kadiry, A.E.-H.; Merhi, Y. Role of NF-κB in Platelet Function. Int. J. Mol. Sci. 2019, 20, 4185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, A.H.; Estrada, A.I.; Ayala-Marin, Y.M.; Alvidrez-Camacho, A.Y.; Rodriguez, G.; Robles-Escajeda, E.; Cadena-Medina, D.A.; Rodriguez, A.C.; Kirken, R.A. The Many Faces of JAKs and STATs Within the COVID-19 Storm. Front. Immunol. 2021, 12, 690477. [Google Scholar] [CrossRef]
- Jafarzadeh, A.; Nemati, M.; Jafarzadeh, S. Contribution of STAT3 to the pathogenesis of COVID-19. Microb. Pathog. 2021, 154, 104836. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020, 27, 3209–3225. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Thomas, T.; Dzieciatkowska, M.; Hill, R.C.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hod, E.A.; Spitalnik, S.L.; Hansen, K.C. Serum Proteomics in COVID-19 Patients: Altered Coagulation and Complement Status as a Function of IL-6 Level. J. Proteome Res. 2020, 19, 4417–4427. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, X.; Tu, Z.; Fu, J.; Xu, D.; Zhou, Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct. Target. Ther. 2021, 6, 255. [Google Scholar] [CrossRef]
- Li, J.; Jie, X.; Liang, X.; Chen, Z.; Xie, P.; Pan, X.; Zhou, B.; Li, J. Sinensetin suppresses influenza a virus-triggered inflammation through inhibition of NF-κB and MAPKs signalings. BMC Complement. Med. Ther. 2020, 20, 135. [Google Scholar] [CrossRef]
- Leitzke, M.; Stefanovic, D.; Meyer, J.-J.; Schimpf, S.; Schönknecht, P. Autonomic balance determines the severity of COVID-19 courses. Bioelectron. Med. 2020, 6, 22. [Google Scholar] [CrossRef]
- Cai, Z.; Greene, M.I.; Zhu, Z.; Zhang, H. Structural Features and PF4 Functions that Occur in Heparin-Induced Thrombocytopenia (HIT) Complicated by COVID-19. Antibodies 2020, 9, 52. [Google Scholar] [CrossRef]
- Nevzorova, T.A.; Mordakhanova, E.R.; Daminova, A.G.; Ponomareva, A.A.; Andrianova, I.A.; Le Minh, G.; Rauova, L.; Litvinov, R.I.; Weisel, J.W. Platelet factor 4-containing immune complexes induce platelet activation followed by calpain-dependent platelet death. Cell Death Discov. 2019, 5, 106. [Google Scholar] [CrossRef] [Green Version]
- Cines, D.B.; Rauova, L.; Arepally, G.; Reilly, M.P.; McKenzie, S.; Sachais, B.S.; Poncz, M.E. Heparin-induced thrombocytopenia: An autoimmune disorder regulated through dynamic autoantigen assembly/disassembly. J. Clin. Apher. 2007, 22, 31–36. [Google Scholar] [CrossRef]
- Krauel, K.; Pötschke, C.; Weber, C.; Kessler, W.; Fürll, B.; Ittermann, T.; Maier, S.; Hammerschmidt, S.; Bröker, B.; Greinacher, A. Platelet factor 4 binds to bacteria, inducing antibodies cross-reacting with the major antigen in heparin-induced thrombocytopenia. Blood 2011, 117, 1370–1378. [Google Scholar] [CrossRef] [Green Version]
- Karlin, S.; Brendel, V. Charge configurations in viral proteins. Proc. Natl. Acad. Sci. USA 1988, 85, 9396–9400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruščić, J.; Ambriović-Ristov, A.; Majhen, D.; Kolundžija, S.; Barut, M.; Benihoud, K.; Krajačić, M. Manipulating adenoviral vector ion-exchange chromatography: Hexon versus fiber. J. Sep. Sci. 2016, 39, 4299–4304. [Google Scholar] [CrossRef] [PubMed]
- Fasbender, A.; Zabner, J.; Chillon, M.; Moninger, T.O.; Puga, A.P.; Davidson, B.L.; Welsh, M.J. Complexes of Adenovirus with Polycationic Polymers and Cationic Lipids Increase the Efficiency of Gene Transfer in Vitro and in Vivo. J. Biol. Chem. 1997, 272, 6479–6489. [Google Scholar] [CrossRef] [Green Version]
- Arcasoy, S.M.; Latoche, J.; Gondor, M.; Pitt, B.R.; Pilewski, J.M. Polycations increase the efficiency of adenovirus-mediated gene transfer to epithelial and endothelial cells in vitro. Gene Ther. 1997, 4, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Baker, A.T.; Boyd, R.J.; Sarkar, D.; Vant, J.W.; Teijeira-Crespo, A.; Truong, C.D.; Bates, E.A.; Wilson, E.A.; Chan, C.K.; Lipka-Lloyd, M.; et al. The Structure of ChAdOx1/AZD-1222 Reveals Interactions with CAR and PF4 with Implications for Vaccine-induced Immune Thrombotic Thrombocytopenia. bioRxiv 2021. [Google Scholar] [CrossRef]
- Greinacher, A.; Selleng, K.; Mayerle, J.; Palankar, R.; Wesche, J.; Reiche, S.; Aebischer, A.; E Warkentin, T.; Muenchhoff, M.; Hellmuth, J.C.; et al. Anti-Platelet Factor 4 Antibodies Causing VITT do not Cross-React with SARS-CoV-2 Spike Protein. Blood 2021. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Selleng, K.; Wesche, J.; Handtke, S.; Palankar, R.; Aurich, K.; Lalk, M.; Methling, K.; Völker, U.; Hentsch-ker, C.; et al. Towards Understanding ChAdOx1 nCov-19 Vaccine-induced Immune Thrombotic Thrombocytopenia (VITT). Res. Sq. 2021. [Google Scholar] [CrossRef]
- Shi, C.; Yang, L.; Braun, A.; Anders, H.-J. Extracellular DNA—A Danger Signal Triggering Immunothrombosis. Front. Immunol. 2020, 11, 568513. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.M.; Kausar, F.; Day, A.; Osborne, M.; Hussain, K.; Mueller, A.; Lin, J.; Tsuchiya, T.; Kanegasaki, S.; Pease, J.E. CXCL4/Platelet Factor 4 is an agonist of CCR1 and drives human monocyte migration. Sci. Rep. 2018, 8, 9466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalska, M.A.; Rauova, L.; Poncz, M. Role of the platelet chemokine platelet factor 4 (PF4) in hemostasis and thrombosis. Thromb. Res. 2010, 125, 292–296. [Google Scholar] [CrossRef]
- Almuqrin, A.; Davidson, A.D.; Williamson, M.K.; Lewis, P.A.; Heesom, K.J.; Morris, S.; Gilbert, S.C.; Matthews, D.A. SARS-CoV-2 vaccine ChAdOx1 nCoV-19 infection of human cell lines reveals low levels of viral backbone gene transcription alongside very high levels of SARS-CoV-2 S glycoprotein gene transcription. Genome Med. 2021, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, L. Factors Which Contribute to the Immunogenicity of Non-replicating Adenoviral Vectored Vaccines. Front. Immunol. 2020, 11, 909. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.; Liu, Y.; Shayakhmetov, D.; Li, Z.-Y.; Ni, S.; Lieber, A. Adenovirus-Platelet Interaction in Blood Causes Virus Sequestration to the Reticuloendothelial System of the Liver. J. Virol. 2007, 81, 4866–4871. [Google Scholar] [CrossRef] [Green Version]
- Othman, M.; Labelle, A.; Mazzetti, I.; Elbatarny, H.S.; Lillicrap, D. Adenovirus-induced thrombocytopenia: The role of von Willebrand factor and P-selectin in mediating accelerated platelet clearance. Blood 2006, 109, 2832–2839. [Google Scholar] [CrossRef]
- Muruve, D.A.; Barnes, M.J.; Stillman, I.E.; Libermann, T.A. Adenoviral Gene Therapy Leads to Rapid Induction of Multiple Chemokines and Acute Neutrophil-Dependent Hepatic Injury in Vivo. Hum. Gene Ther. 1999, 10, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Melotti, P.; Nicolis, E.; Tamanini, A.; Rolfini, R.; Pavirani, A.; Cabrini, G. Activation of NF-kB mediates ICAM-1 induction in respiratory cells exposed to an adenovirus-derived vector. Gene Ther. 2001, 8, 1436–1442. [Google Scholar] [CrossRef] [Green Version]
- Tamanini, A.; Rolfini, R.; Nicolis, E.; Melotti, P.; Cabrini, G. MAP kinases and NF-κB collaborate to induce ICAM-1 gene expression in the early phase of adenovirus infection. Virology 2003, 307, 228–242. [Google Scholar] [CrossRef] [Green Version]
- Tamanini, A.; Nicolis, E.; Bonizzato, A.; Bezzerri, V.; Melotti, P.; Assael, B.M.; Cabrini, G. Interaction of Adenovirus Type 5 Fiber with the Coxsackievirus and Adenovirus Receptor Activates Inflammatory Response in Human Respiratory Cells. J. Virol. 2006, 80, 11241–11254. [Google Scholar] [CrossRef] [Green Version]
- Molinier-Frenkel, V.; Lengagne, R.; Gaden, F.; Hong, S.-S.; Choppin, J.; Gahery-Ségard, H.; Boulanger, P.; Guillet, J.-G. Adenovirus Hexon Protein Is a Potent Adjuvant for Activation of a Cellular Immune Response. J. Virol. 2002, 76, 127–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, A.E.; Larregina, A.T.; Ganster, R.W.; Zahorchak, A.F.; Plowey, J.M.; Takayama, T.; Logar, A.J.; Robbins, P.D.; Falo, L.D.; Thomson, A.W. Recombinant Adenovirus Induces Maturation of Dendritic Cells via an NF-κB-Dependent Pathway. J. Virol. 2000, 74, 9617–9628. [Google Scholar] [CrossRef] [Green Version]
- Talotta, R.; Robertson, E.S. Antiphospholipid antibodies and risk of post-COVID-19 vaccination thrombophilia: The straw that breaks the camel's back? Cytokine Growth Factor Rev. 2021, 60, 52–60. [Google Scholar] [CrossRef]
- Goldman, M.; Hermans, C. Thrombotic thrombocytopenia associated with COVID-19 infection or vaccination: Possible paths to platelet factor 4 autoimmunity. PLoS Med. 2021, 18, e1003648. [Google Scholar] [CrossRef]
- Mercier, S.; Gahéry-Segard, H.; Monteil, M.; Lengagne, R.; Guillet, J.-G.; Eloit, M.; Denesvre, C. Distinct Roles of Adenovirus Vector-Transduced Dendritic Cells, Myoblasts, and Endothelial Cells in Mediating an Immune Response against a Transgene Product. J. Virol. 2002, 76, 2899–2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef]
- Nuovo, G.J.; Magro, C.; Shaffer, T.; Awad, H.; Suster, D.; Mikhail, S.; He, B.; Michaille, J.-J.; Liechty, B.; Tili, E. Endothelial cell damage is the central part of COVID-19 and a mouse model induced by injection of the S1 subunit of the spike protein. Ann. Diagn. Pathol. 2021, 51, 151682. [Google Scholar] [CrossRef]
- Liu, L.; Chopra, P.; Li, X.; Wolfert, M.A.; Tompkins, S.M.; Boons, G.J. Heparan sulfate proteoglycans as attachment factor for SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Kowarz, E.; Krutzke, L.; Reis, J.; Bracharz, S.; Kochanek, S.; Marschalek, R. “Vaccine-Induced Covid-19 Mimicry” Syndrome: Splice reactions within the SARS-CoV-2 Spike open reading frame result in Spike protein variants that may cause thromboembolic events in patients immunized with vector-based vaccines. Preprint Res. Sq. 2021. [Google Scholar] [CrossRef]
- Machitani, M.; Sakurai, F.; Wakabayashi, K.; Nakatani, K.; Shimizu, K.; Tachibana, M.; Mizuguchi, H. NF-κB promotes leaky expression of adenovirus genes in a replication-incompetent adenovirus vector. Sci. Rep. 2016, 6, 19922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krutzke, L.; Roesler, R.; Wiese, S.; Kochanek, S. Process-related impurities in the ChAdOx1 nCov-19 vaccine. Preprint Res. Sq. 2021. [Google Scholar] [CrossRef]
- Appledorn, D.M.; Patial, S.; McBride, A.; Godbehere, S.; Van Rooijen, N.; Parameswaran, N.; Amalfitano, A. Adenovirus Vector-Induced Innate Inflammatory Mediators, MAPK Signaling, As Well As Adaptive Immune Responses Are Dependent upon Both TLR2 and TLR9 In Vivo. J. Immunol. 2008, 181, 2134–2144. [Google Scholar] [CrossRef]
- Appledorn, D.; Patial, S.; Godbehere, S.; Parameswaran, N.; Amalfitano, A. TRIF, and TRIF-Interacting TLRs Differentially Modulate Several Adenovirus Vector-Induced Immune Responses. J. Innate Immun. 2009, 1, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, S.; de Fougerolles, A.R.; Blaikie, P.; Khan, L.; Pepe, A.; Green, C.D.; Koteliansky, V.; Giancotti, F.G. α5β1 Integrin Activates an NF-κB-Dependent Program of Gene Expression Important for Angiogenesis and Inflammation. Mol. Cell. Biol. 2002, 22, 5912–5922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, A.; Sanders, J.M.; Bevard, M.; Coleman, E.; Sarembock, I.J.; Schwartz, M.A. The subendothelial extracellular matrix modulates NF-kappaB activation by flow: A potential role in atherosclerosis. J. Cell Biol. 2005, 169, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Ledford, H. COVID vaccines and blood clots: What researchers know so far. Nat. Cell Biol. 2021, 596, 479–481. [Google Scholar] [CrossRef]
- Clesham, G.; Adam, P.; Proudfoot, D.; Flynn, P.; Efstathiou, S.; Weissberg, P. High adenoviral loads stimulate NFκB-dependent gene expression in human vascular smooth muscle cells. Gene Ther. 1998, 5, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Tan, P.H.; Xue, S.-A.; Manunta, M.; Beutelspacher, S.C.; Fazekasova, H.; Alam, A.S.; McClure, M.O.; George, A.J. Effect of Vectors on Human Endothelial Cell Signal Transduction. Arter. Thromb. Vasc. Biol. 2006, 26, 462–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.F.; Yin, K.S.; Zhu, Z.L.; Zhu, Y.; Yao, X.; Mao, H.; Xie, W.P.; Huang, M. Adenovirus-mediated overexpression of novel mutated IkappaBalpha inhibits nuclear factor kappaB activation in endothelial cells. Chin. Med. J. 2005, 118, 1422–1428. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kircheis, R. Coagulopathies after Vaccination against SARS-CoV-2 May Be Derived from a Combined Effect of SARS-CoV-2 Spike Protein and Adenovirus Vector-Triggered Signaling Pathways. Int. J. Mol. Sci. 2021, 22, 10791. https://doi.org/10.3390/ijms221910791
Kircheis R. Coagulopathies after Vaccination against SARS-CoV-2 May Be Derived from a Combined Effect of SARS-CoV-2 Spike Protein and Adenovirus Vector-Triggered Signaling Pathways. International Journal of Molecular Sciences. 2021; 22(19):10791. https://doi.org/10.3390/ijms221910791
Chicago/Turabian StyleKircheis, Ralf. 2021. "Coagulopathies after Vaccination against SARS-CoV-2 May Be Derived from a Combined Effect of SARS-CoV-2 Spike Protein and Adenovirus Vector-Triggered Signaling Pathways" International Journal of Molecular Sciences 22, no. 19: 10791. https://doi.org/10.3390/ijms221910791
APA StyleKircheis, R. (2021). Coagulopathies after Vaccination against SARS-CoV-2 May Be Derived from a Combined Effect of SARS-CoV-2 Spike Protein and Adenovirus Vector-Triggered Signaling Pathways. International Journal of Molecular Sciences, 22(19), 10791. https://doi.org/10.3390/ijms221910791