
Exosite Binding in Thrombin: A Global Structural/Dynamic Overview of Complexes with Aptamers and Other Ligands

 ,
,  ,
,  , and
, and
Abstract
:
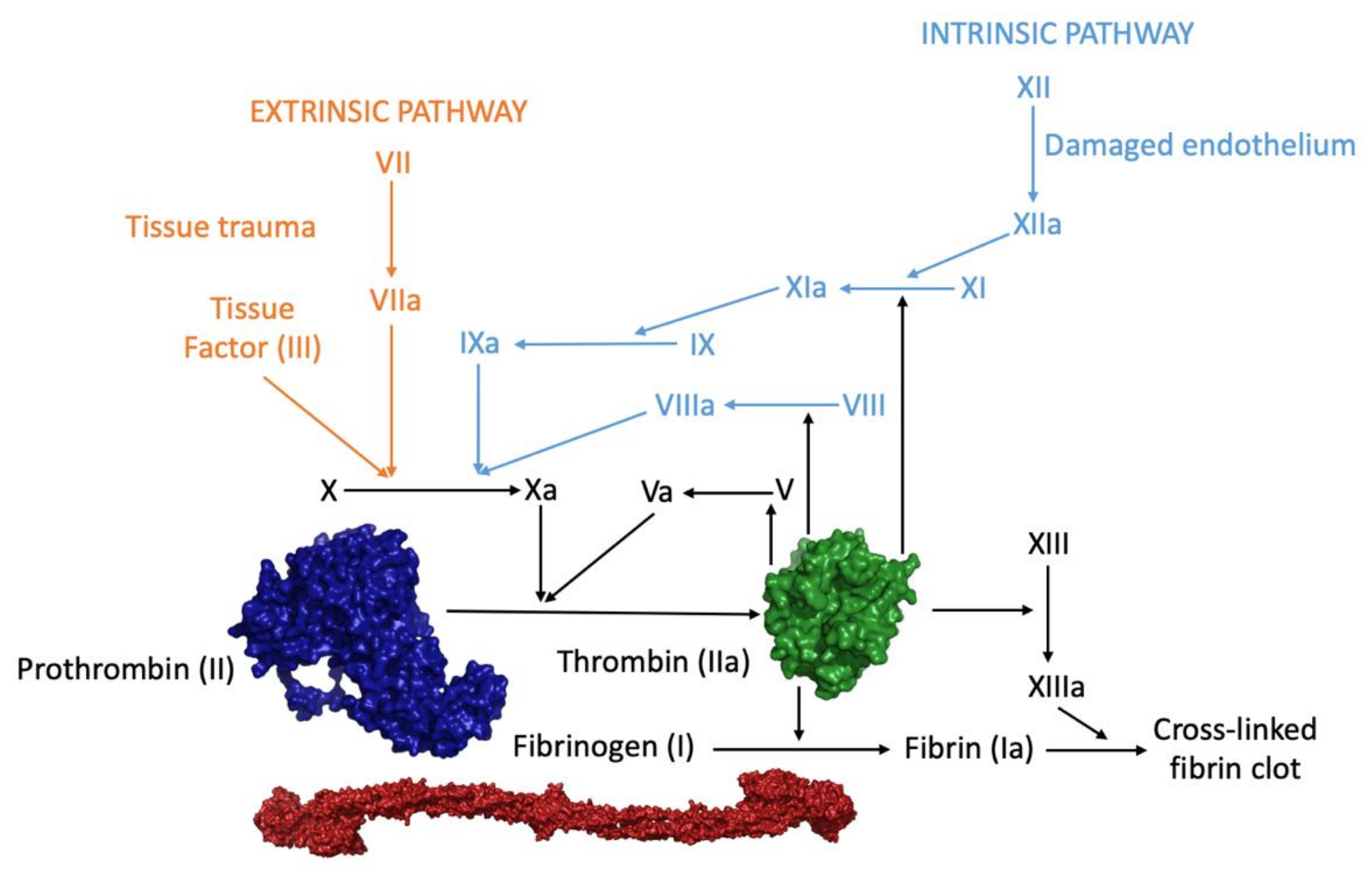
1. Introduction—Overview of Thrombin: A Multi-Partner Serine Protease Involved in Blood Coagulation
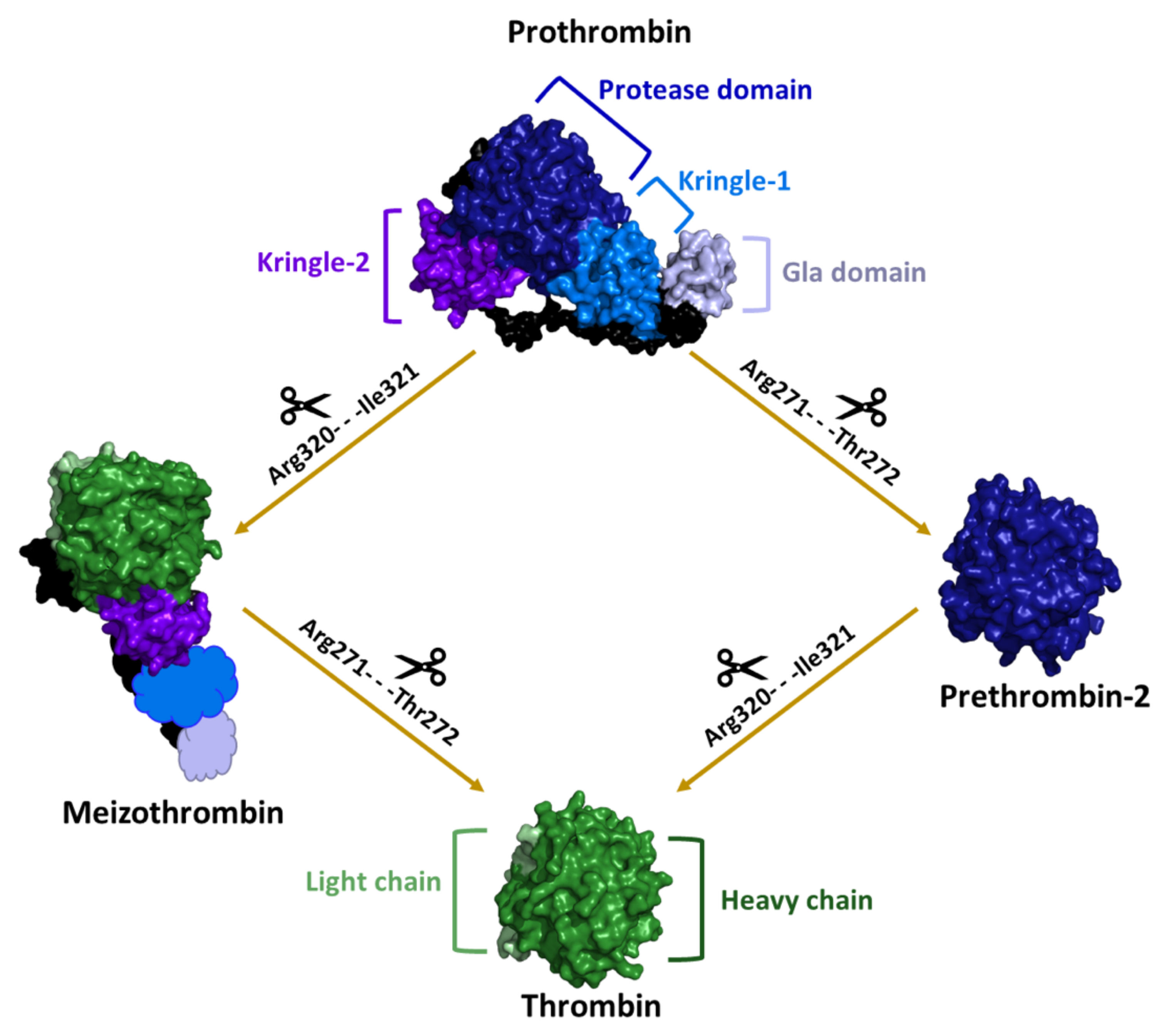
2. Structural Characterization of Thrombin: A Chronological Perspective
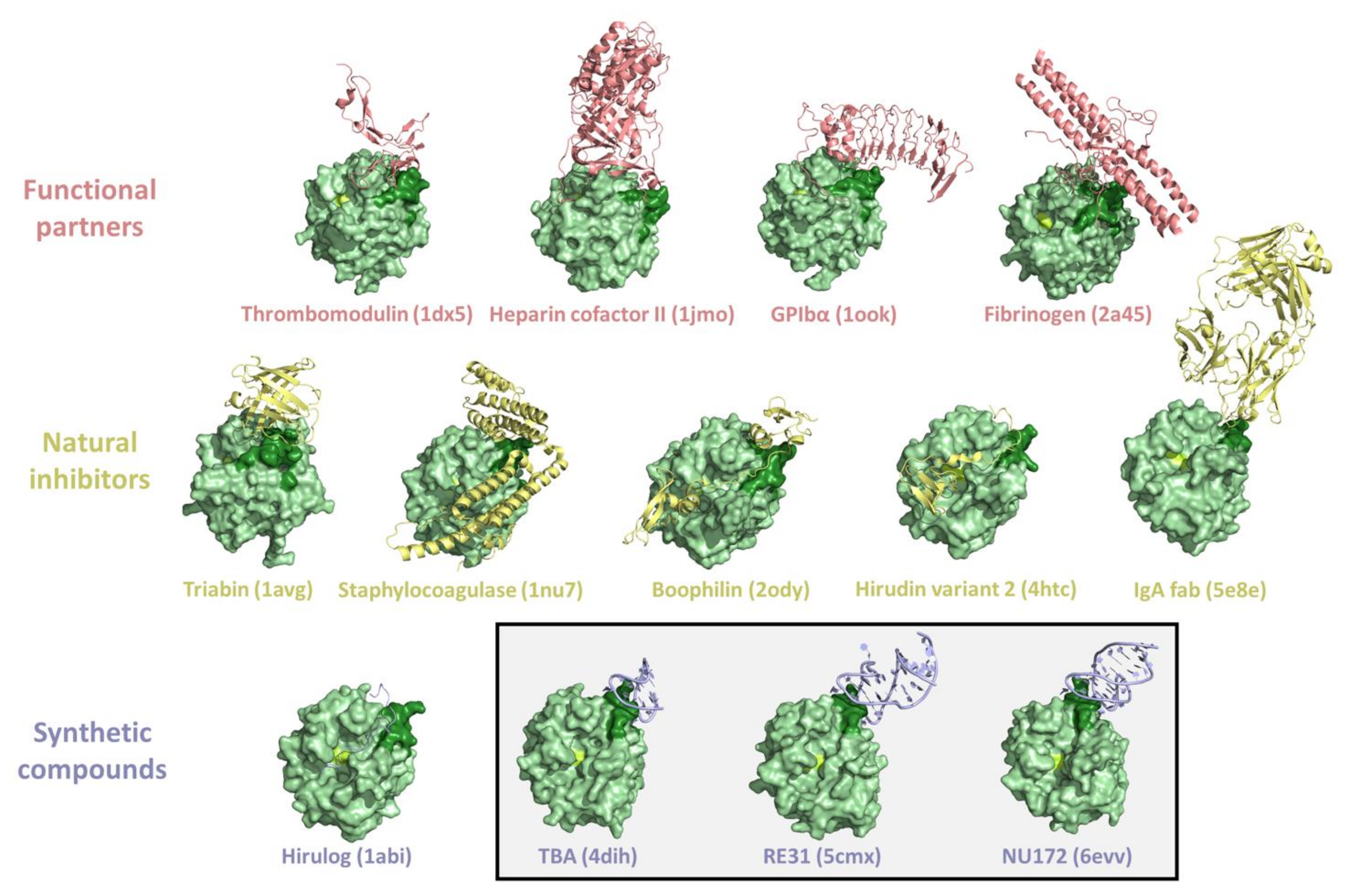
3. Thrombin Recognition of Functional, Natural, and Synthetic Partners: Anatomy of Exosites
4. Beyond a Static View of Thrombin: Functional and Structural Evidence of Exosite Communication
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Di Cera, E. Thrombin. Mol. Aspects Med. 2008, 29, 203–254. [Google Scholar] [CrossRef]
- Palta, S.; Saroa, R.; Palta, A. Overview of the Coagulation System. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef]
- Periayah, M.H.; Halim, A.S.; Mat Saad, A.Z. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int. J. Hematol. Oncol. Stem. Cell Res. 2017, 11, 319–327. [Google Scholar] [PubMed]
- Davie, E.W.; Kulman, J.D. An Overview of the Structure and Function of Thrombin. Semin. Thromb. Hemost. 2006, 32, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Cera, E.D. Thrombin as Procoagulant and Anticoagulant. J. Thromb. Haemost. 2007, 5, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Jesty, J.; Beltrami, E. Positive Feedbacks of Coagulation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2463–2469. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Mechanisms of Fibrin Polymerization and Clinical Implications. Blood 2013, 121, 1712–1719. [Google Scholar] [CrossRef] [Green Version]
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Subcell Biochem. 2017, 82, 405–456. [Google Scholar]
- Simurda, T.; Vilar, R.; Zolkova, J.; Ceznerova, E.; Kolkova, Z.; Loderer, D.; Neerman-Arbez, M.; Casini, A.; Brunclikova, M.; Skornova, I.; et al. A Novel Nonsense Mutation in FGB (c.1421G>A; p.Trp474Ter) in the Beta Chain of Fibrinogen Causing Hypofibrinogenemia with Bleeding Phenotype. Biomedicines 2020, 8, 605. [Google Scholar] [CrossRef] [PubMed]
- Mathews, I.I.; Padmanabhan, K.P.; Ganesh, V.; Tulinsky, A.; Ishii, M.; Chen, J.; Turck, C.W.; Coughlin, S.R.; Fenton, J.W. Crystallographic Structures of Thrombin Complexed with Thrombin Receptor Peptides: Existence of Expected and Novel Binding Modes. Biochemistry 1994, 33, 3266–3279. [Google Scholar] [CrossRef]
- Kawabata, A.; Kuroda, R. Protease-Activated Receptor (PAR), a Novel Family of G Protein-Coupled Seven Trans-Membrane Domain Receptors: Activation Mechanisms and Physiological Roles. Jpn. J. Pharmacol. 2000, 82, 171–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohatgi, T.; Sedehizade, F.; Reymann, K.G.; Reiser, G. Protease-Activated Receptors in Neuronal Development, Neurodegeneration, and Neuroprotection: Thrombin as Signaling Molecule in the Brain. Neuroscientist 2004, 10, 501–512. [Google Scholar] [CrossRef]
- Bah, A.; Chen, Z.; Bush-Pelc, L.A.; Mathews, F.S.; Cera, E.D. Crystal Structures of Murine Thrombin in Complex with the Extracellular Fragments of Murine Protease-Activated Receptors PAR3 and PAR4. PNAS 2007, 104, 11603–11608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, P.S.; Chen, Z.; Di Cera, E. Crystal Structure of Thrombin Bound to the Uncleaved Extracellular Fragment of PAR1. J. Biol. Chem. 2010, 285, 15393–15398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Candia, E.; Hall, S.W.; Rutella, S.; Landolfi, R.; Andrews, R.K.; De Cristofaro, R. Binding of Thrombin to Glycoprotein Ib Accelerates the Hydrolysis of Par-1 on Intact Platelets. J. Biol.Chem. 2001, 276, 4692–4698. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, S. Multifunctional Roles of Thrombin. Ann. Clin. Lab. Sci. 1999, 29, 275–280. [Google Scholar]
- Esmon, C.T. Molecular Events That Control the Protein C Anticoagulant Pathway. Thromb Haemost. 1993, 70, 29–35. [Google Scholar] [CrossRef]
- Rezaie, A.R. Regulation of the Protein C Anticoagulant and Antiinflammatory Pathways. Curr. Med. Chem. 2010, 17, 2059–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapin, J.C.; Hajjar, K.A. Fibrinolysis and the Control of Blood Coagulation. Blood Rev. 2015, 29, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Sillen, M.; Declerck, P.J. Thrombin Activatable Fibrinolysis Inhibitor (TAFI): An Updated Narrative Review. Int. J. Mol. Sci. 2021, 22, 3670. [Google Scholar] [CrossRef]
- Carter, I.S.R.; Vanden Hoek, A.L.; Pryzdial, E.L.G.; MacGillivray, R.T.A. Thrombin A-Chain: Activation Remnant or Allosteric Effector? Thrombosis 2010, 2010, 416167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, N.; Chen, Z.; Gohara, D.W.; Niu, W.; Heyduk, T.; Di Cera, E. Crystal Structure of Prothrombin Reveals Conformational Flexibility and Mechanism of Activation. J. Biol. Chem. 2013, 288, 22734–22744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, N.; Chen, Z.; Pelc, L.A.; Shropshire, D.B.; Di Cera, E. The Linker Connecting the Two Kringles Plays a Key Role in Prothrombin Activation. Proc. Natl. Acad. Sci. USA 2014, 111, 7630–7635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, N.; Chen, Z.; Di Cera, E. How the Linker Connecting the Two Kringles Influences Activation and Conformational Plasticity of Prothrombin. J. Biol. Chem. 2016, 291, 6071–6082. [Google Scholar] [CrossRef] [Green Version]
- Doyle, M.F.; Mann, K.G. Multiple Active Forms of Thrombin. IV. Relative Activities of Meizothrombins. J. Biol. Chem. 1990, 265, 10693–10701. [Google Scholar] [CrossRef]
- Chen, Z.; Pelc, L.A.; Di Cera, E. Crystal Structure of Prethrombin-1. Proc. Natl. Acad. Sci. USA 2010, 107, 19278–19283. [Google Scholar] [CrossRef] [Green Version]
- Krishnaswamy, S. The Transition of Prothrombin to Thrombin. J. Thromb. Haemost. 2013, 11, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, W.; Turk, D.; Karshikov, A. The Refined 1.9-A X-Ray Crystal Structure of D-Phe-Pro-Arg Chloromethylketone-Inhibited Human Alpha-Thrombin: Structure Analysis, Overall Structure, Electrostatic Properties, Detailed Active-Site Geometry, and Structure-Function Relationships. Protein Sci. 1992, 1, 426–471. [Google Scholar] [CrossRef]
- Tanaka-Azevedo, A.M.; Morais-Zani, K.; Torquato, R.J.S.; Tanaka, A.S. Thrombin Inhibitors from Different Animals. J. Biomed. Biotechnol. 2010, 2010, 641025. [Google Scholar] [CrossRef] [Green Version]
- Fuentes-Prior, P.; Noeske-Jungblut, C.; Donner, P.; Schleuning, W.D.; Huber, R.; Bode, W. Structure of the Thrombin Complex with Triabin, a Lipocalin-like Exosite-Binding Inhibitor Derived from a Triatomine Bug. Proc. Natl. Acad. Sci. USA 1997, 94, 11845–11850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenton, J.W. Thrombin Specificity. Ann. NY Acad. Sci. 1981, 370, 468–495. [Google Scholar] [CrossRef]
- Fenton, J.W.; Olson, T.A.; Zabinski, M.P.; Wilner, G.D. Anion-Binding Exosite of Human Alpha-Thrombin and Fibrin(Ogen) Recognition. Biochemistry 1988, 27, 7106–7112. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Di Cera, E. Energetics of Thrombin-Fibrinogen Interaction. Biochemistry 1992, 31, 11567–11571. [Google Scholar] [CrossRef]
- Bouton, M.-C.; Jandrot-Perrus, M.; Bezeaud, A.; Guillin, M.-C. Late-Fibrin(Ogen) Fragment E Modulates Human α-Thrombin Specificity. Eur. J. Biochem. 1993, 215, 143–149. [Google Scholar] [CrossRef]
- Simurda, T.; Brunclikova, M.; Asselta, R.; Caccia, S.; Zolkova, J.; Kolkova, Z.; Loderer, D.; Skornova, I.; Hudecek, J.; Lasabova, Z.; et al. Genetic Variants in the FGB and FGG Genes Mapping in the Beta and Gamma Nodules of the Fibrinogen Molecule in Congenital Quantitative Fibrinogen Disorders Associated with a Thrombotic Phenotype. Int. J. Mol. Sci. 2020, 21, 4616. [Google Scholar] [CrossRef] [PubMed]
- Hofsteenge, J.; Stone, S.R. The Effect of Thrombomodulin on the Cleavage of Fibrinogen and Fibrinogen Fragments by Thrombin. Eur. J. Biochem. 1987, 168, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Nishioka, J.; Hayashi, T. Localization of Thrombomodulin-Binding Site within Human Thrombin. J. Biol. Chem. 1990, 265, 13263–13267. [Google Scholar] [CrossRef]
- Mathews, I.I.; Padmanabhan, K.P.; Tulinksy, A.; Sadler, J.E. Structure of a Nonadecapeptide of the Fifth EGF Domain of Thrombomodulin Complexed with Thrombin. Biochemistry 1994, 33, 13547–13552. [Google Scholar] [CrossRef]
- Srinivasan, J.; Hu, S.; Hrabal, R.; Zhu, Y.; Komives, E.A.; Ni, F. Thrombin-Bound Structure of an EGF Subdomain from Human Thrombomodulin Determined by Transferred Nuclear Overhauser Effects. Biochemistry 1994, 33, 13553–13560. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.K.; Wheaton, V.I.; Hung, D.T.; Charo, I.; Coughlin, S.R. Domains Specifying Thrombin-Receptor Interaction. Nature 1991, 353, 674–677. [Google Scholar] [CrossRef]
- Vu, T.K.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular Cloning of a Functional Thrombin Receptor Reveals a Novel Proteolytic Mechanism of Receptor Activation. Cell 1991, 64, 1057–1068. [Google Scholar] [CrossRef]
- Ishihara, H.; Connolly, A.J.; Zeng, D.; Kahn, M.L.; Zheng, Y.W.; Timmons, C.; Tram, T.; Coughlin, S.R. Protease-Activated Receptor 3 Is a Second Thrombin Receptor in Humans. Nature 1997, 386, 502–506. [Google Scholar] [CrossRef]
- Arni, R.K.; Padmanabhan, K.; Padmanabhan, K.P.; Wu, T.P.; Tulinsky, A. Structures of the Noncovalent Complexes of Human and Bovine Prothrombin Fragment 2 with Human PPACK-Thrombin. Biochemistry 1993, 32, 4727–4737. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.P.; Sadler, J.E. Molecular Mapping of the Heparin-Binding Exosite of Thrombin. Proc. Natl. Acad. Sci. USA 1994, 91, 5518–5522. [Google Scholar] [CrossRef] [Green Version]
- Huntington, J.A. Molecular Recognition Mechanisms of Thrombin. J/ Thromb Haemost. 2005, 3, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Johnson, D.J.D.; Adams, T.E.; Pozzi, N.; De Filippis, V.; Huntington, J.A. Thrombin Inhibition by Serpins Disrupts Exosite II. J. Biol. Chem. 2010, 285, 38621–38629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whinna, H.C.; Church, F.C. Interaction of Thrombin with Antithrombin, Heparin Cofactor II, and Protein C Inhibitor. J Protein Chem. 1993, 12, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Dang, Q.D.; Vindigni, A.; Di Cera, E. An Allosteric Switch Controls the Procoagulant and Anticoagulant Activities of Thrombin. Proc. Natl. Acad. Sci. USA 1995, 92, 5977–5981. [Google Scholar] [CrossRef] [Green Version]
- Di Cera, E.; Page, M.J.; Bah, A.; Bush-Pelc, L.A.; Garvey, L.C. Thrombin Allostery. Phys. Chem. Chem. Phys. 2007, 9, 1291–1306. [Google Scholar] [CrossRef]
- Di Cera, E.; Cantwell, A.M. Determinants of Thrombin Specificity. Ann. NY Acad. Sci. 2001, 936, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.E.; Huntington, J.A. Thrombin-Cofactor Interactions: Structural Insights into Regulatory Mechanisms. Arterioscler. Thromb Vasc. Biol. 2006, 26, 1738–1745. [Google Scholar] [CrossRef]
- Baglin, T.P.; Carrell, R.W.; Church, F.C.; Esmon, C.T.; Huntington, J.A. Crystal Structures of Native and Thrombin-Complexed Heparin Cofactor II Reveal a Multistep Allosteric Mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 11079–11084. [Google Scholar] [CrossRef] [Green Version]
- Huntington, J.A. Thrombin Inhibition by the Serpins. J. Thromb. Haemost. 2013, 11, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kini, R.M. Anticoagulants from Hematophagous Animals. Expert Rev. Hematol. 2008, 1, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Pirone, L.; Ripoll-Rozada, J.; Leone, M.; Ronca, R.; Lombardo, F.; Fiorentino, G.; Andersen, J.F.; Pereira, P.J.B.; Arcà, B.; Pedone, E. Functional Analyses Yield Detailed Insight into the Mechanism of Thrombin Inhibition by the Antihemostatic Salivary Protein CE5 from Anopheles Gambiae. J. Biol. Chem. 2017, 292, 12632–12642. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, A.C.; de Sanctis, D.; Gutiérrez-Gallego, R.; Cereija, T.B.; Macedo-Ribeiro, S.; Fuentes-Prior, P.; Pereira, P.J.B. Unique Thrombin Inhibition Mechanism by Anophelin, an Anticoagulant from the Malaria Vector. PNAS 2012, 109, E3649–E3658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, J.K.; Koh, C.Y.; Kazimirova, M.; Roller, L.; Jobichen, C.; Swaminathan, K.; Mizuguchi, J.; Iwanaga, S.; Nuttall, P.A.; Chan, M.Y.; et al. Avathrin: A Novel Thrombin Inhibitor Derived from a Multicopy Precursor in the Salivary Glands of the Ixodid Tick, Amblyomma Variegatum. FASEB J. 2017, 31, 2981–2995. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Kurihara, K.; Ohnishi, Y.; Tamada, T.; Tomoyori, K.; Masumi, K.; Tanaka, I.; Kuroki, R.; Niimura, N. Neutron and X-Ray Crystallographic Analysis of the Human α-Thrombin–Bivalirudin Complex at PD 5.0: Protonation States and Hydration Structure of the Enzyme–Product Complex. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2013, 1834, 1532–1538. [Google Scholar] [CrossRef]
- Macedo-Ribeiro, S.; Almeida, C.; Calisto, B.M.; Friedrich, T.; Mentele, R.; Stürzebecher, J.; Fuentes-Prior, P.; Pereira, P.J.B. Isolation, Cloning and Structural Characterisation of Boophilin, a Multifunctional Kunitz-Type Proteinase Inhibitor from the Cattle Tick. PLoS ONE 2008, 3, e1624. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, R.; Tulinsky, A.; Vlasuk, G.P.; Pearson, D.; Vallar, P.; Bergum, P.; Brunck, T.K.; Ripka, W.C. Synthesis, Structure, and Structure-Activity Relationships of Divalent Thrombin Inhibitors Containing an α-Keto-Amide Transition-State Mimetic. Protein Sci. 1996, 5, 422–433. [Google Scholar] [CrossRef]
- Richardson, J.L.; Kröger, B.; Hoeffken, W.; Sadler, J.E.; Pereira, P.; Huber, R.; Bode, W.; Fuentes-Prior, P. Crystal Structure of the Human Alpha-Thrombin-Haemadin Complex: An Exosite II-Binding Inhibitor. EMBO J. 2000, 19, 5650–5660. [Google Scholar] [CrossRef] [Green Version]
- Rydel, T.J.; Ravichandran, K.G.; Tulinsky, A.; Bode, W.; Huber, R.; Roitsch, C.; Fenton, J.W. The Structure of a Complex of Recombinant Hirudin and Human Alpha-Thrombin. Science 1990, 249, 277–280. [Google Scholar] [CrossRef]
- Skrzypczak-Jankun, E.; Carperos, V.E.; Ravichandran, K.G.; Tulinsky, A.; Westbrook, M.; Maraganore, J.M. Structure of the Hirugen and Hirulog 1 Complexes of Alpha-Thrombin. J. Mol. Biol. 1991, 221, 1379–1393. [Google Scholar] [CrossRef]
- Qiu, X.; Yin, M.; Padmanabhan, K.P.; Krstenansky, J.L.; Tulinsky, A. Structures of Thrombin Complexes with a Designed and a Natural Exosite Peptide Inhibitor. J. Biol. Chem. 1993, 268, 20318–20326. [Google Scholar] [CrossRef]
- De Simone, G.; Lombardi, A.; Galdiero, S.; Nastri, F.; Della Morte, R.; Staiano, N.; Pedone, C.; Bolognesi, M.; Pavone, V. Hirunorms Are True Hirudin Mimetics. The Crystal Structure of Human Alpha-Thrombin-Hirunorm V Complex. Protein Sci. 1998, 7, 243–253. [Google Scholar] [CrossRef]
- Zdanov, A.; Wu, S.; DiMaio, J.; Konishi, Y.; Li, Y.; Wu, X.; Edwards, B.F.; Martin, P.D.; Cygler, M. Crystal Structure of the Complex of Human Alpha-Thrombin and Nonhydrolyzable Bifunctional Inhibitors, Hirutonin-2 and Hirutonin-6. Proteins 1993, 17, 252–265. [Google Scholar] [CrossRef]
- Baglin, T.P.; Langdown, J.; Frasson, R.; Huntington, J.A. Discovery and Characterization of an Antibody Directed against Exosite I of Thrombin. J. Thromb. Haemost. 2016, 14, 137–142. [Google Scholar] [CrossRef]
- Thompson, R.E.; Liu, X.; Ripoll-Rozada, J.; Alonso-García, N.; Parker, B.L.; Pereira, P.J.B.; Payne, R.J. Tyrosine Sulfation Modulates Activity of Tick-Derived Thrombin Inhibitors. Nat. Chem. 2017, 9, 909–917. [Google Scholar] [CrossRef]
- Bachand, B.; Tarazi, M.; St-Denis, Y.; Edmunds, J.J.; Winocour, P.D.; Leblond, L.; Siddiqui, M.A. Potent and Selective Bicyclic Lactam Inhibitors of Thrombin. Part 4: Transition State Inhibitors. Bioorg. Med. Chem. Lett. 2001, 11, 287–290. [Google Scholar] [CrossRef]
- van de Locht, A.; Stubbs, M.T.; Bode, W.; Friedrich, T.; Bollschweiler, C.; Höffken, W.; Huber, R. The Ornithodorin-Thrombin Crystal Structure, a Key to the TAP Enigma? EMBO J. 1996, 15, 6011–6017. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, R.; Steinmetzer, T.; Huber, R.; Stürzebecher, J.; Bode, W. The Methyl Group of Nα(Me)Arg-Containing Peptides Disturbs the Active-Site Geometry of Thrombin, Impairing Efficient Cleavage. J. Mol. Biol. 2002, 316, 869–874. [Google Scholar] [CrossRef]
- van de Locht, A.; Lamba, D.; Bauer, M.; Huber, R.; Friedrich, T.; Kröger, B.; Höffken, W.; Bode, W. Two Heads Are Better than One: Crystal Structure of the Insect Derived Double Domain Kazal Inhibitor Rhodniin in Complex with Thrombin. EMBO J. 1995, 14, 5149–5157. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, R.; Panizzi, P.; Fuentes-Prior, P.; Richter, K.; Verhamme, I.; Anderson, P.J.; Kawabata, S.-I.; Huber, R.; Bode, W.; Bock, P.E. Staphylocoagulase Is a Prototype for the Mechanism of Cofactor-Induced Zymogen Activation. Nature 2003, 425, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.M.T.R.; Becker, C.F.; Giesel, G.M.; Marques, A.F.; Cargnelutti, M.T.; de Oliveira Neto, M.; Queiroz Monteiro, R.; Verli, H.; Polikarpov, I. Structural and Thermodynamic Analysis of Thrombin:Suramin Interaction in Solution and Crystal Phases. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2009, 1794, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Lange, U.E.W.; Baucke, D.; Hornberger, W.; Mack, H.; Seitz, W.; Höffken, H.W. Orally Active Thrombin Inhibitors. Part 2: Optimization of the P2-Moiety. Bioorg. Med. Chem. Lett. 2006, 16, 2648–2653. [Google Scholar] [CrossRef] [PubMed]
- Slon-Usakiewicz, J.J.; Sivaraman, J.; Li, Y.; Cygler, M.; Konishi, Y. Design of P1′ and P3′ Residues of Trivalent Thrombin Inhibitors and Their Crystal Structures. Biochemistry 2000, 39, 2384–2391. [Google Scholar] [CrossRef] [PubMed]
- Rehse, P.H.; Steinmetzer, T.; Li, Y.; Konishi, Y.; Cygler, M. Crystal Structure of a Peptidyl Pyridinium Methyl Ketone Inhibitor with Thrombin. Biochemistry 1995, 34, 11537–11544. [Google Scholar] [CrossRef] [PubMed]
- Calisto, B.M.; Ripoll-Rozada, J.; Dowman, L.J.; Franck, C.; Agten, S.M.; Parker, B.L.; Veloso, R.C.; Vale, N.; Gomes, P.; de Sanctis, D.; et al. Sulfotyrosine-Mediated Recognition of Human Thrombin by a Tsetse Fly Anticoagulant Mimics Physiological Substrates. Cell Chem. Biol. 2021, 28, 26–33.e8. [Google Scholar] [CrossRef]
- Koh, C.Y.; Kumar, S.; Kazimirova, M.; Nuttall, P.A.; Radhakrishnan, U.P.; Kim, S.; Jagadeeswaran, P.; Imamura, T.; Mizuguchi, J.; Iwanaga, S.; et al. Crystal Structure of Thrombin in Complex with S-Variegin: Insights of a Novel Mechanism of Inhibition and Design of Tunable Thrombin Inhibitors. PLoS ONE 2011, 6, e26367. [Google Scholar] [CrossRef] [Green Version]
- Russo Krauss, I.; Merlino, A.; Randazzo, A.; Novellino, E.; Mazzarella, L.; Sica, F. High-Resolution Structures of Two Complexes between Thrombin and Thrombin-Binding Aptamer Shed Light on the Role of Cations in the Aptamer Inhibitory Activity. Nucleic Acids Res. 2012, 40, 8119–8128. [Google Scholar] [CrossRef] [Green Version]
- Russo Krauss, I.; Merlino, A.; Giancola, C.; Randazzo, A.; Mazzarella, L.; Sica, F. Thrombin-Aptamer Recognition: A Revealed Ambiguity. Nucleic Acids Res. 2011, 39, 7858–7867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pica, A.; Russo Krauss, I.; Merlino, A.; Nagatoishi, S.; Sugimoto, N.; Sica, F. Dissecting the Contribution of Thrombin Exosite I in the Recognition of Thrombin Binding Aptamer. FEBS J. 2013, 280, 6581–6588. [Google Scholar] [CrossRef] [PubMed]
- Dolot, R.; Lam, C.H.; Sierant, M.; Zhao, Q.; Liu, F.-W.; Nawrot, B.; Egli, M.; Yang, X. Crystal Structures of Thrombin in Complex with Chemically Modified Thrombin DNA Aptamers Reveal the Origins of Enhanced Affinity. Nucleic Acids Res. 2018, 46, 4819–4830. [Google Scholar] [CrossRef]
- Smirnov, I.; Kolganova, N.; Troisi, R.; Sica, F.; Timofeev, E. Expanding the Recognition Interface of the Thrombin-Binding Aptamer HD1 through Modification of Residues T3 and T12. Mol. Ther. Nucleic Acids 2021, 23, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Russo Krauss, I.; Spiridonova, V.; Pica, A.; Napolitano, V.; Sica, F. Different Duplex/Quadruplex Junctions Determine the Properties of Anti-Thrombin Aptamers with Mixed Folding. Nucleic Acids Res. 2016, 44, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Troisi, R.; Napolitano, V.; Spiridonova, V.; Russo Krauss, I.; Sica, F. Several Structural Motifs Cooperate in Determining the Highly Effective Anti-Thrombin Activity of NU172 Aptamer. Nucleic Acids Res. 2018, 46, 12177–12185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, S.B.; Long, M.B.; White, R.R.; Sullenger, B.A. Crystal Structure of an RNA Aptamer Bound to Thrombin. RNA 2008, 14, 2504–2512. [Google Scholar] [CrossRef] [Green Version]
- Abeydeera, N.D.; Egli, M.; Cox, N.; Mercier, K.; Conde, J.N.; Pallan, P.S.; Mizurini, D.M.; Sierant, M.; Hibti, F.-E.; Hassell, T.; et al. Evoking Picomolar Binding in RNA by a Single Phosphorodithioate Linkage. Nucleic Acids Res. 2016, 44, 8052–8064. [Google Scholar] [CrossRef]
- Russo Krauss, I.; Pica, A.; Merlino, A.; Mazzarella, L.; Sica, F. Duplex-Quadruplex Motifs in a Peculiar Structural Organization Cooperatively Contribute to Thrombin Binding of a DNA Aptamer. Acta Crystallogr. Biol. Crystallogr. 2013, 69, 2403–2411. [Google Scholar] [CrossRef]
- Tulinsky, A.; Qiu, X. Active Site and Exosite Binding of Alpha-Thrombin. Blood Coagul. Fibrinolysis 1993, 4, 305–312. [Google Scholar] [CrossRef]
- Stubbs, M.T.; Bode, W. A Player of Many Parts: The Spotlight Falls on Thrombin’s Structure. Thromb Res. 1993, 69, 1–58. [Google Scholar] [CrossRef]
- Goldsack, N.R.; Chambers, R.C.; Dabbagh, K.; Laurent, G.J. Thrombin. Int. J. Biochem. Cell Biol. 1998, 30, 641–646. [Google Scholar] [CrossRef]
- Di Cera, E. Thrombin Interactions. Chest 2003, 124, 11S–17S. [Google Scholar] [CrossRef]
- Lane, D.A.; Philippou, H.; Huntington, J.A. Directing Thrombin. Blood 2005, 106, 2605–2612. [Google Scholar] [CrossRef]
- Bode, W. Structure and Interaction Modes of Thrombin. Blood Cells Mol. Dis. 2006, 36, 122–130. [Google Scholar] [CrossRef]
- Bode, W. The Structure of Thrombin: A Janus-Headed Proteinase. Semin Thromb Hemost. 2006, 32, 16–31. [Google Scholar] [CrossRef]
- Tanaka, K.A.; Levy, J.H. Regulation of Thrombin Activity—Pharmacologic and Structural Aspects. Hematol. Oncol. Clin. North Am. 2007, 21, 33–50. [Google Scholar] [CrossRef]
- Di Cera, E. Thrombin as an Anticoagulant. Prog. Mol. Biol. Transl. Sci. 2011, 99, 145–184. [Google Scholar]
- Lechtenberg, B.C.; Freund, S.M.V.; Huntington, J.A. An Ensemble View of Thrombin Allostery. Biol. Chem. 2012, 393, 889–898. [Google Scholar] [CrossRef]
- Huntington, J.A. Thrombin Plasticity. Biochim. Biophys. Acta 2012, 1824, 246–252. [Google Scholar] [CrossRef]
- Guillin, M.C.; Bezeaud, A.; Bouton, M.C.; Jandrot-Perrus, M. Thrombin Specificity. Thromb Haemost. 1995, 74, 129–133. [Google Scholar] [CrossRef]
- Stubbs, M.T.; Bode, W. The Clot Thickens: Clues Provided by Thrombin Structure. Trends Biochem. Sci. 1995, 20, 23–28. [Google Scholar] [CrossRef]
- Tulinsky, A. Molecular Interactions of Thrombin. Semin Thromb Hemost. 1996, 22, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Leung, L.L.; Gibbs, C.S. Modulation of Thrombin’s Procoagulant and Anticoagulant Properties. Thromb Haemost. 1997, 78, 577–580. [Google Scholar] [CrossRef]
- Markwardt, F. Historical Perspective of the Development of Thrombin Inhibitors. PHT 2002, 32, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Stafford, A.R.; Wu, C.; Yeh, C.H.; Kim, P.Y.; Fredenburgh, J.C.; Weitz, J.I. Exosite 2-Directed Ligands Attenuate Protein C Activation by the Thrombin-Thrombomodulin Complex. Biochemistry 2017, 56, 3119–3128. [Google Scholar] [CrossRef] [PubMed]
- Billur, R.; Sabo, T.M.; Maurer, M.C. Thrombin Exosite Maturation and Ligand Binding at ABE II Help Stabilize PAR-Binding Competent Conformation at ABE I. Biochemistry 2019, 58, 1048–1060. [Google Scholar] [CrossRef]
- Olmsted, I.R.; Xiao, Y.; Cho, M.; Csordas, A.T.; Sheehan, J.H.; Meiler, J.; Soh, H.T.; Bornhop, D.J. Measurement of Aptamer-Protein Interactions with Back-Scattering Interferometry. Anal. Chem. 2011, 83, 8867–8870. [Google Scholar] [CrossRef] [PubMed]
- Pica, A.; Russo Krauss, I.; Parente, V.; Tateishi-Karimata, H.; Nagatoishi, S.; Tsumoto, K.; Sugimoto, N.; Sica, F. Through-Bond Effects in the Ternary Complexes of Thrombin Sandwiched by Two DNA Aptamers. Nucleic Acids Res. 2017, 45, 461–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Yu, C.; Feng, F.; Lu, P.; Chai, Y.; Li, Q.; Zhang, D.; Wang, X.; Yao, L. Direct Measurement of Through-Bond Effects in Molecular Multivalent Interactions. Chemistry 2019, 25, 2978–2982. [Google Scholar] [CrossRef]
- Troisi, R.; Balasco, N.; Vitagliano, L.; Sica, F. Molecular Dynamics Simulations of Human α-Thrombin in Different Structural Contexts: Evidence for an Aptamer-Guided Cooperation between the Two Exosites. J. Biomol. Struct. Dyn. 2021, 39, 2199–2209. [Google Scholar] [CrossRef]
- Troisi, R.; Balasco, N.; Santamaria, A.; Vitagliano, L.; Sica, F. Structural and Functional Analysis of the Simultaneous Binding of Two Duplex/Quadruplex Aptamers to Human α-Thrombin. Int. J. Biol. Macromol. 2021, 181, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Bode, W.; Mayr, I.; Baumann, U.; Huber, R.; Stone, S.R.; Hofsteenge, J. The Refined 1.9 A Crystal Structure of Human Alpha-Thrombin: Interaction with D-Phe-Pro-Arg Chloromethylketone and Significance of the Tyr-Pro-Pro-Trp Insertion Segment. EMBO J. 1989, 8, 3467–3475. [Google Scholar] [CrossRef]
- Fuentes-Prior, P.; Iwanaga, Y.; Huber, R.; Pagila, R.; Rumennik, G.; Seto, M.; Morser, J.; Light, D.R.; Bode, W. Structural Basis for the Anticoagulant Activity of the Thrombin-Thrombomodulin Complex. Nature 2000, 404, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Johnson, D.J.D.; Esmon, C.T.; Huntington, J.A. Structure of the Antithrombin-Thrombin-Heparin Ternary Complex Reveals the Antithrombotic Mechanism of Heparin. Nat. Struct. Mol. Biol. 2004, 11, 857–862. [Google Scholar] [CrossRef]
- Carter, W.J.; Cama, E.; Huntington, J.A. Crystal Structure of Thrombin Bound to Heparin. J. Biol. Chem. 2005, 280, 2745–2749. [Google Scholar] [CrossRef] [Green Version]
- Celikel, R.; McClintock, R.A.; Roberts, J.R.; Mendolicchio, G.L.; Ware, J.; Varughese, K.I.; Ruggeri, Z.M. Modulation of Alpha-Thrombin Function by Distinct Interactions with Platelet Glycoprotein Ibalpha. Science 2003, 301, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Dumas, J.J.; Kumar, R.; Seehra, J.; Somers, W.S.; Mosyak, L. Crystal Structure of the GpIbalpha-Thrombin Complex Essential for Platelet Aggregation. Science 2003, 301, 222–226. [Google Scholar] [CrossRef]
- Pechik, I.; Yakovlev, S.; Mosesson, M.W.; Gilliland, G.L.; Medved, L. Structural Basis for Sequential Cleavage of Fibrinopeptides upon Fibrin Assembly. Biochemistry 2006, 45, 3588–3597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pineda, A.O.; Chen, Z.-W.; Marino, F.; Mathews, F.S.; Mosesson, M.W.; Di Cera, E. Crystal Structure of Thrombin in Complex with Fibrinogen Gamma’ Peptide. Biophys. Chem. 2007, 125, 556–559. [Google Scholar] [CrossRef]
- Padmanabhan, K.; Padmanabhan, K.P.; Ferrara, J.D.; Sadler, J.E.; Tulinsky, A. The Structure of Alpha-Thrombin Inhibited by a 15-Mer Single-Stranded DNA Aptamer. J. Biol. Chem. 1993, 268, 17651–17654. [Google Scholar] [CrossRef]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of Single-Stranded DNA Molecules That Bind and Inhibit Human Thrombin. Nature 1992, 355, 564–566. [Google Scholar] [CrossRef]
- Rydel, T.J.; Tulinsky, A.; Bode, W.; Huber, R. Refined Structure of the Hirudin-Thrombin Complex. J. Mol. Biol. 1991, 221, 583–601. [Google Scholar] [CrossRef]
- Liu, C.C.; Brustad, E.; Liu, W.; Schultz, P.G. Crystal Structure of a Biosynthetic Sulfo-Hirudin Complexed to Thrombin. J. Am. Chem. Soc. 2007, 129, 10648–10649. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Padmanabhan, K.P.; Carperos, V.E.; Tulinsky, A.; Kline, T.; Maraganore, J.M.; Fenton, J.W. Structure of the Hirulog 3-Thrombin Complex and Nature of the S’ Subsites of Substrates and Inhibitors. Biochemistry 1992, 31, 11689–11697. [Google Scholar] [CrossRef]
- Steinmetzer, T.; Renatus, M.; Künzel, S.; Eichinger, A.; Bode, W.; Wikström, P.; Hauptmann, J.; Stürzebecher, J. Design and Evaluation of Novel Bivalent Thrombin Inhibitors Based on Amidinophenylalanines. Eur. J. Biochem. 1999, 265, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, A.; De Simone, G.; Nastri, F.; Galdiero, S.; Della Morte, R.; Staiano, N.; Pedone, C.; Bolognesi, M.; Pavone, V. The Crystal Structure of Alpha-Thrombin-Hirunorm IV Complex Reveals a Novel Specificity Site Recognition Mode. Protein Sci. 1999, 8, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Corral-Rodríguez, M.Á.; Bock, P.E.; Hernández-Carvajal, E.; Gutiérrez-Gallego, R.; Fuentes-Prior, P. Structural Basis of Thrombin-Mediated Factor V Activation: The Glu666-Glu672 Sequence Is Critical for Processing at the Heavy Chain-B Domain Junction. Blood 2011, 117, 7164–7173. [Google Scholar] [CrossRef] [Green Version]
- De Filippis, V.; Colombo, G.; Russo, I.; Spadari, B.; Fontana, A. Probing the Hirudin-Thrombin Interaction by Incorporation of Noncoded Amino Acids and Molecular Dynamics Simulation. Biochemistry 2002, 41, 13556–13569. [Google Scholar] [CrossRef] [PubMed]
- De Silva, V.A.; Cargnelutti, M.T.; Giesel, G.M.; Palmieri, L.C.; Monteiro, R.Q.; Verli, H.; Lima, L.M.T.R. Structure and Behavior of Human α-Thrombin upon Ligand Recognition: Thermodynamic and Molecular Dynamics Studies. PLoS ONE 2011, 6, e24735. [Google Scholar] [CrossRef] [Green Version]
- Fuglestad, B.; Gasper, P.M.; McCammon, J.A.; Markwick, P.R.L.; Komives, E.A. Correlated Motions and Residual Frustration in Thrombin. J. Phys. Chem. 2013, 117, 12857–12863. [Google Scholar] [CrossRef]
- de Amorim, H.L.N.; Netz, P.A.; Guimarães, J.A. Thrombin Allosteric Modulation Revisited: A Molecular Dynamics Study. J. Mol. Model 2010, 16, 725–735. [Google Scholar] [CrossRef]
- Kurisaki, I.; Takayanagi, M.; Nagaoka, M. Toward Understanding Allosteric Activation of Thrombin: A Conjecture for Important Roles of Unbound Na(+) Molecules around Thrombin. J. Phys. Chem. 2015, 119, 3635–3642. [Google Scholar] [CrossRef] [PubMed]
- Kurisaki, I.; Nagaoka, M. Na+ Binding Is Ineffective in Forming a Primary Substrate Pocket of Thrombin. J. Phys. Chem. 2016, 120, 11873–11879. [Google Scholar] [CrossRef]
- Bowerman, S.; Wereszczynski, J. Detecting Allosteric Networks Using Molecular Dynamics Simulation. Methods Enzymol. 2016, 578, 429–447. [Google Scholar]
- Xiao, J.; Melvin, R.L.; Salsbury, F.R. Mechanistic Insights into Thrombin’s Switch between “Slow” and “Fast” Forms. Phys. Chem. Chem. Phys. 2017, 19, 24522–24533. [Google Scholar] [CrossRef]
- Xiao, J.; Salsbury, F.R. Na+-Binding Modes Involved in Thrombin’s Allosteric Response as Revealed by Molecular Dynamics Simulations, Correlation Networks and Markov Modeling. Phys. Chem. Chem. Phys. 2019, 21, 4320–4330. [Google Scholar] [CrossRef] [PubMed]
- Kahler, U.; Kamenik, A.S.; Kraml, J.; Liedl, K.R. Sodium-Induced Population Shift Drives Activation of Thrombin. Sci. Rep. 2020, 10, 1086. [Google Scholar] [CrossRef]
- Wu, D.; Xiao, J.; Salsbury, F.R. Light Chain Mutation Effects on the Dynamics of Thrombin. J. Chem. Inf. Model 2021, 61, 950–965. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.L.; Mei, Y.; Han, K.; Zhang, J.Z.H. Quantum and Molecular Dynamics Study for Binding of Macrocyclic Inhibitors to Human Alpha-Thrombin. Biophys. J. 2007, 92, 4244–4253. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, J.E.; Huber, R.G.; Waldner, B.J.; Kahler, U.; von Grafenstein, S.; Kramer, C.; Liedl, K.R. Dynamics Govern Specificity of a Protein-Protein Interface: Substrate Recognition by Thrombin. PLoS ONE 2015, 10, e0140713. [Google Scholar]
- Mena-Ulecia, K.; Tiznado, W.; Caballero, J. Study of the Differential Activity of Thrombin Inhibitors Using Docking, QSAR, Molecular Dynamics, and MM-GBSA. PLoS ONE 2015, 10, e0142774. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Melvin, R.L.; Salsbury, F.R. Probing Light Chain Mutation Effects on Thrombin via Molecular Dynamics Simulations and Machine Learning. J. Biomol. Struct. Dyn. 2019, 37, 982–999. [Google Scholar] [CrossRef]
- Shcherbinin, D.S.; Veselovsky, A.V. Investigation of Interaction of Thrombin-Binding Aptamer with Thrombin and Prethrombin-2 by Simulation of Molecular Dynamics. BIOPHYSICS 2013, 58, 315–323. [Google Scholar] [CrossRef]
- Seelam Prabhakar, P.; Manderville, R.A.; Wetmore, S.D. Impact of the Position of the Chemically Modified 5-Furyl-2′-Deoxyuridine Nucleoside on the Thrombin DNA Aptamer-Protein Complex: Structural Insights into Aptamer Response from MD Simulations. Molecules 2019, 24, E2908. [Google Scholar] [CrossRef] [Green Version]
- Aviñó, A.; Jorge, A.F.; Huertas, C.S.; Cova, T.F.G.G.; Pais, A.; Lechuga, L.M.; Eritja, R.; Fabrega, C. Aptamer-Peptide Conjugates as a New Strategy to Modulate Human α-Thrombin Binding Affinity. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1619–1630. [Google Scholar] [CrossRef]
- Stone, S.R.; Hofsteenge, J. Effect of Heparin on the Interaction between Thrombin and Hirudin. Eur. J. Biochem. 1987, 169, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Fredenburgh, J.C.; Stafford, A.R.; Weitz, J.I. Evidence for Allosteric Linkage between Exosites 1 and 2 of Thrombin. J. Biol. Chem. 1997, 272, 25493–25499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeppe, J.R.; Seitova, A.; Mather, T.; Komives, E.A. Thrombomodulin Tightens the Thrombin Active Site Loops to Promote Protein C Activation. Biochemistry 2005, 44, 14784–14791. [Google Scholar] [CrossRef] [PubMed]
- Sabo, T.M.; Farrell, D.H.; Maurer, M.C. Conformational Analysis of Gamma’ Peptide (410-427) Interactions with Thrombin Anion Binding Exosite II. Biochemistry 2006, 45, 7434–7445. [Google Scholar] [CrossRef]
- Sabo, T.M.; Maurer, M.C. Biophysical Investigation of GpIbalpha Binding to Thrombin Anion Binding Exosite II. Biochemistry 2009, 48, 7110–7122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrera, N.S.; Stafford, A.R.; Leslie, B.A.; Kretz, C.A.; Fredenburgh, J.C.; Weitz, J.I. Long Range Communication between Exosites 1 and 2 Modulates Thrombin Function. J. Biol. Chem. 2009, 284, 25620–25629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malovichko, M.V.; Sabo, T.M.; Maurer, M.C. Ligand Binding to Anion-Binding Exosites Regulates Conformational Properties of Thrombin. J. Biol. Chem. 2013, 288, 8667–8678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, J.; Freitag, D.; Mayer, G.; Pötzsch, B. Anticoagulant Characteristics of HD1-22, a Bivalent Aptamer That Specifically Inhibits Thrombin and Prothrombinase. J. Thromb Haemost. 2008, 6, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Nimjee, S.M.; Oney, S.; Volovyk, Z.; Bompiani, K.M.; Long, S.B.; Hoffman, M.; Sullenger, B.A. Synergistic Effect of Aptamers That Inhibit Exosites 1 and 2 on Thrombin. RNA 2009, 15, 2105–2111. [Google Scholar] [CrossRef] [Green Version]
- Yoshitomi, T.; Wakui, K.; Miyakawa, M.; Yoshimoto, K. Design Strategy of Antidote Sequence for Bivalent Aptamer: Rapid Neutralization of High-Anticoagulant Thrombin-Binding Bivalent DNA Aptamer-Linked M08 with HD22. Res. Pract. Thromb. Haemost. 2021, 5, e12503. [Google Scholar] [CrossRef]
- Xiao, J.; Salsbury, F.R. Molecular Dynamics Simulations of Aptamer-Binding Reveal Generalized Allostery in Thrombin. J. Biomol. Struct. Dyn. 2017, 35, 3354–3369. [Google Scholar] [CrossRef]
- Cooper, A.; Dryden, D.T. Allostery without Conformational Change. A Plausible Model. Eur. Biophys. J. 1984, 11, 103–109. [Google Scholar] [CrossRef]
- Tandon, H.; de Brevern, A.G.; Srinivasan, N. Transient Association between Proteins Elicits Alteration of Dynamics at Sites Far Away from Interfaces. Structure 2021, 29, 371–384.e3. [Google Scholar] [CrossRef] [PubMed]
- Aptamers for Medical Applications: From Diagnosis to Therapeutics; Dong, Y. (Ed.) Springer: Singapore, 2021. [Google Scholar]

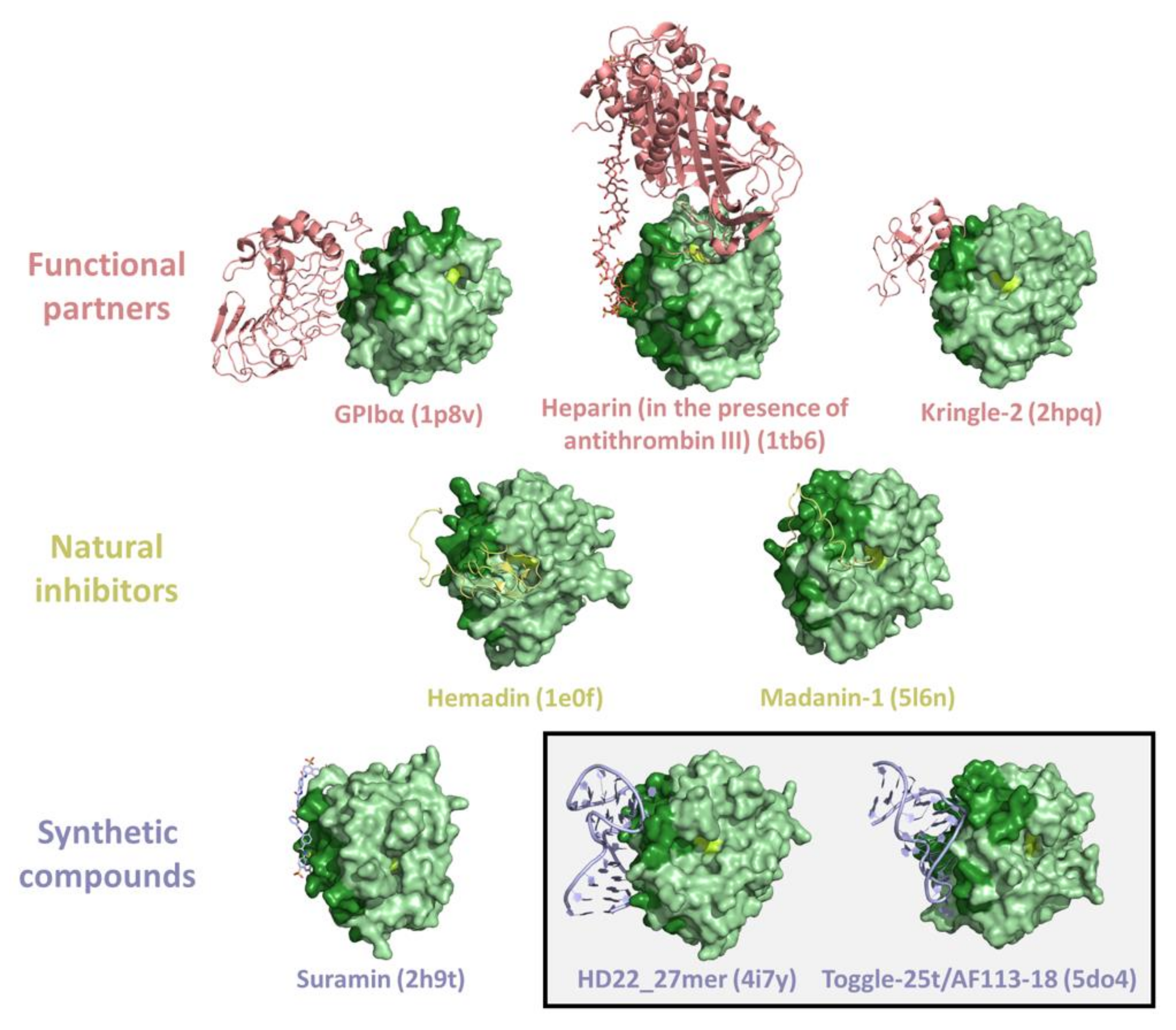

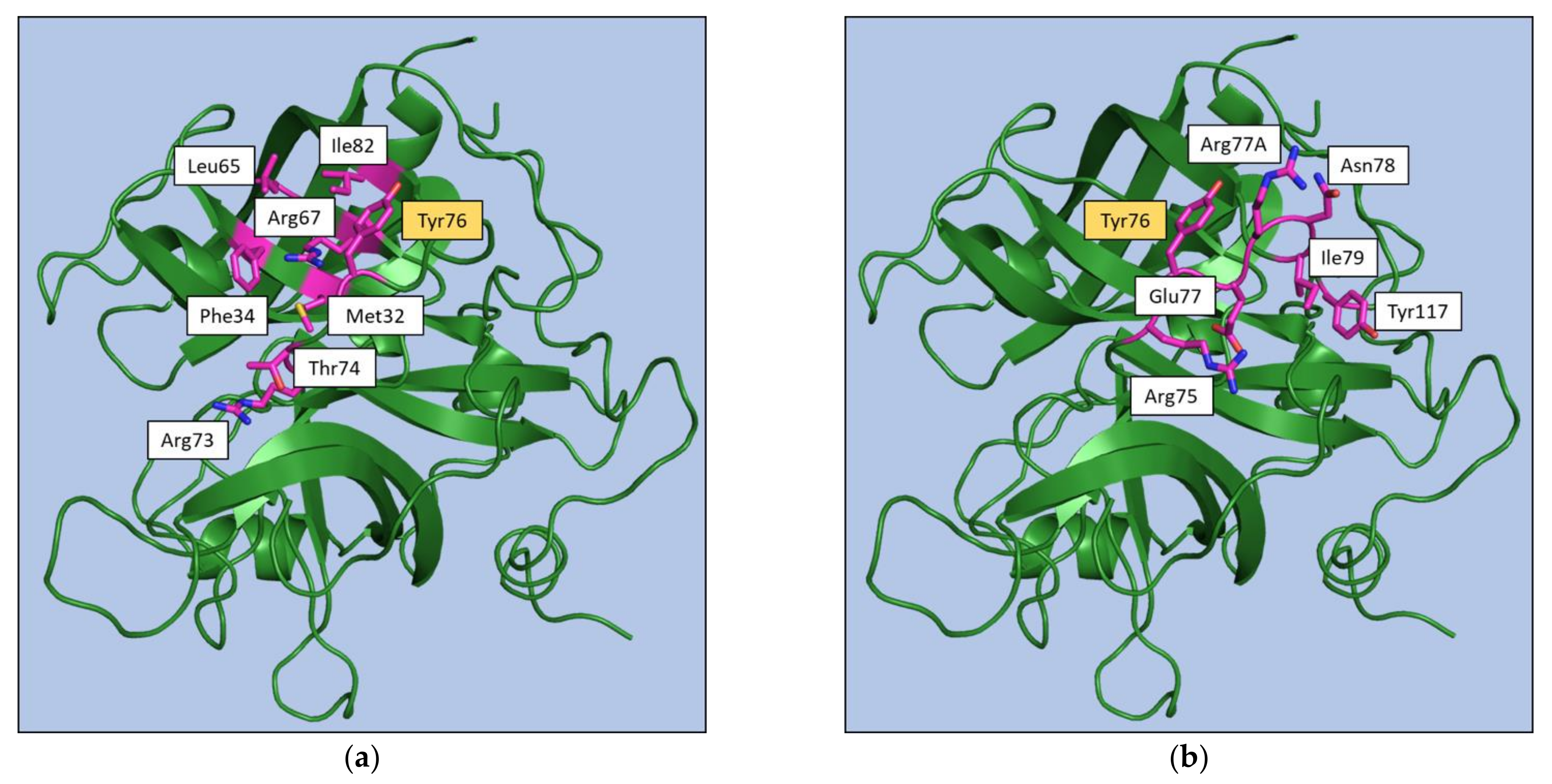
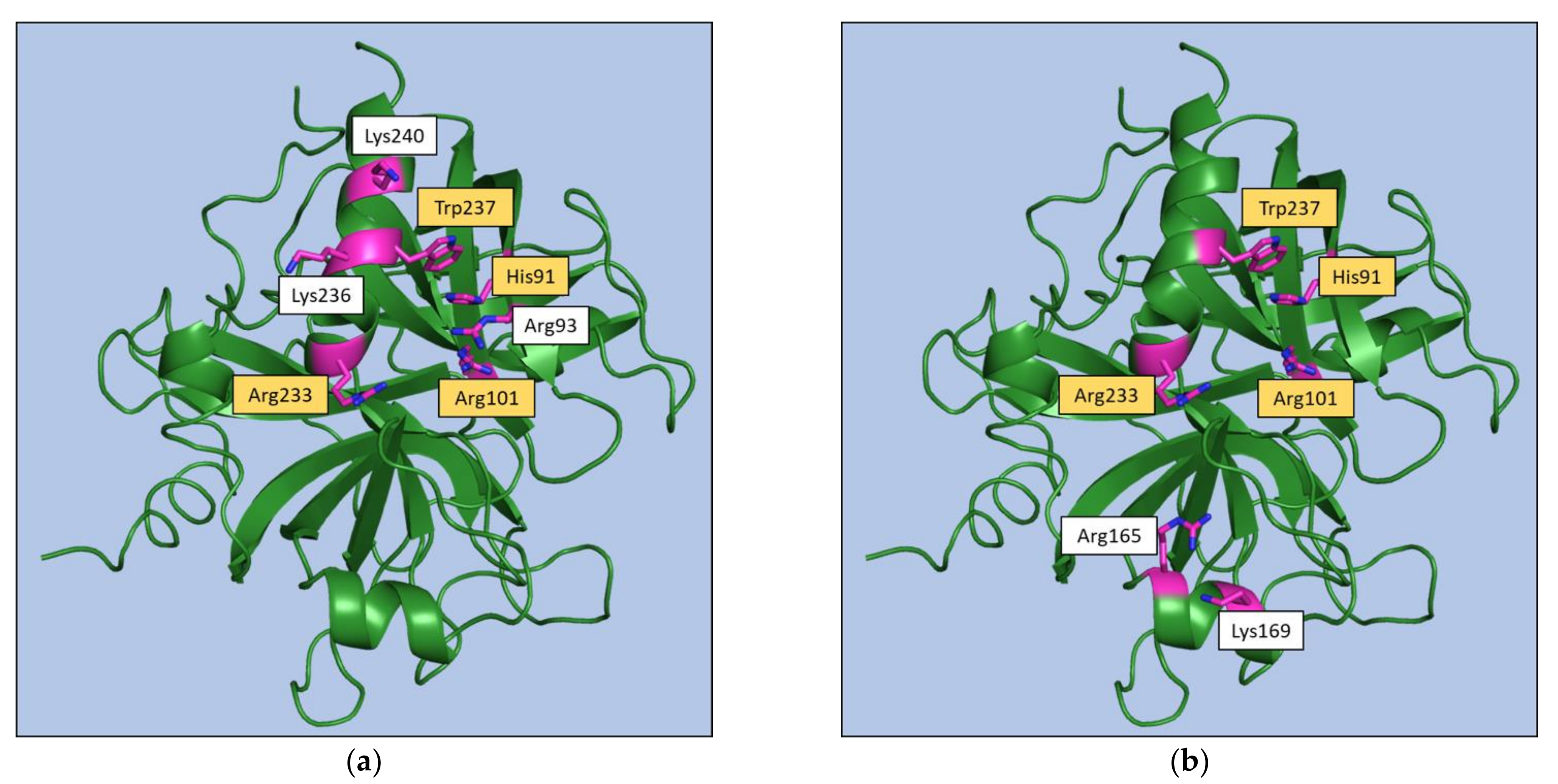


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cofactor | Interactor/Substrate | Function |
|---|---|---|
| Heparin-like glycosaminoglycans (GAGs) | Antithrombin III | Thrombin inhibition |
| Heparin-like glycosaminoglycans (GAGs) | Heparin Cofactor II | Thrombin inhibition |
| - | Fibrinogen | Conversion to fibrin |
| Fibrin | Factor XIII | Activation to factor XIIIa, critical for stabilization of the clot through covalent cross-linking of the fibrin polymers |
| Thrombomodulin | Thrombin activatable fibrinolysis inhibitor (TAFI) | Activation of TAFI |
| Thrombomodulin | Protein C (PC) | Activation to activated protein C (APC) that inactivates cofactors Va and VIIIa |
| Platelet glycoprotein (GP) Ibα (GpIbα) | PAR-1 | Cleavage of the platelet receptor PAR-1, resulting in platelet activation |
| Platelet glycoprotein (GP) Ibα (GpIbα) | Factor XI | Activation to factor XIa, stimulating its own generation |
| - | Factor V | Activation to factor Va, stimulating its own generation |
| - | Factor VIII | Activation to factor VIIIa, stimulating its own generation |
| Ligand | Description | Reference |
|---|---|---|
| AGAP008004-PA | Recombinant P5 fragment of cE5, a salivary protein from the major African malaria vector Anopheles gambiae | [55] |
| Anophelin | Anticoagulant from the malaria vector Anopheles albimanus | [56] |
| Avathrin | Thrombin inhibitor derived from a multicopy precursor in the salivary glands of the ixodid tick Amblyomma variegatum | [57] |
| Bivalirudin | Artificial anticoagulant peptide, mimic of hirudin, composed of 20 residues | [58] |
| Boophilin | Multifunctional kunitz-type proteinase inhibitor from the cattle tick Rhipicephalus microplus | [59] |
| CVS995 synthetic peptide | Divalent thrombin inhibitor that contains an alpha-keto-amide transition-state mimetic linking an active site binding group and a group that binds to the fibrinogen-binding exosite | [60] |
| Hemadin | Thrombin inhibitor from the land-living leech Haemadipsa sylvestris | [61] |
| Hirudin | Protein isolated from the salivary glands of the medicinal leech Hirudo medicinalis | [62] |
| Hirugen | N-acetyl-Hir53′-64′ with sulfato-Tyr63′, where Hir53′-64′ corresponds to the C-terminal residues of hirudin | [63] |
| Hirullin | A natural COOH-terminal peptide isolated from the leech Hirudinaria manillensis | [64] |
| Hirulog | Synthetic hirudin peptide analog | [63] |
| Hirunorm | Synthetic hirudin peptide analog | [65] |
| Hirutonin | Synthetic hirudin peptide analog | [66] |
| IgA fab | Randomly acquired antithrombin antibody found in a patient who presented a traumatic subdural hematoma | [67] |
| Madanin-1 | Small cysteine-free thrombin inhibitor that facilitate blood feeding in the tick Haemaphysalis longicornis | [68] |
| Nonapeptide inhibitor | Synthetic hirudin peptide analog | [69] |
| Ornithodorin | Protein isolated from the blood sucking soft tick Ornithodoros moubata | [70] |
| Peptide inhibitor | C-terminal hirugen-like segment of a bivalent thrombin inhibitor | [71] |
| Rhodniin | Protein isolated from the assassin bug Rhodnius prolixus | [72] |
| Staphylocoagulase | Cofactor secreted by Staphylococcus aureus that activates prothrombin | [73] |
| Suramin | Hexasulfonated naphthylurea first made by O. Dressel, R. Kothe and B. Heymann at Bayer AG laboratories in Elberfeld | [74] |
| Synthetic inhibitor | Hirudin-based fibrinogen recognition exosite peptide inhibitor | [64] |
| Synthetic peptide | Synthetic peptide; sequence: Sin YEPI Hyp EE Smf Alc Q (Sin, succinic acid; Hyp, 4-hydroxyproline; Smf, 4-sulfomethyl-l-phenylalanine; and Alc, 2-amino-3-cyclohexyl-propionic acid) | [75] |
| Synthetic thrombin inhibitor P798 | Synthetic bivalent thrombin inhibitor that comprises an active site blocking segment, a fibrinogen recognition exosite blocking segment, and a linker | [76] |
| Synthetic thrombin inhibitor P596 | Bivalent peptidyl pyridinium methyl ketone inhibitor | [77] |
| Triabin | Protein from the saliva of the blood-sucking triatomine bug Triatoma pallidipennis | [30] |
| Tsetse thrombin inhibitor | Anticoagulant peptide produced in the salivary glands of the tsetse fly Glossina morsitans | [78] |
| Variegin | Thrombin inhibitor isolated from the tropical bont tick Amblyomma variegatum | [79] |
| Name | DNA or RNA | Sequence (5′ → 3′) | Reference |
|---|---|---|---|
| TBA | DNA | GGTTGGTGTGGTTGG | [80] |
| mTBA | DNA | 3′-GGT-5′-5′-TGGTGTGGTTGG-3′ | [81] |
| TBAΔT3 | DNA | GGNΔTGGTGTGGTTGG | [82] |
| TBAΔT12 | DNA | GGTTGGTGTGGNΔTGG | [82] |
| TBA-T4W | DNA | GGTTWGGTGTGGTTGG | [83] |
| TBA-T4K | DNA | GGTTKGGTGTGGTTGG | [83] |
| TBA-3L | DNA | GGTLTGGTGTGGTTGG | [84] |
| TBA-3G | DNA | GGTGTGGTGTGGTTGG | [84] |
| TBA-3Leu | DNA | GGTLeuTGGTGTGGTTGG | [84] |
| RE31 | DNA | GTGACGTAGGTTGGTGTGGTTGGGGCGTCAC | [85] |
| NU172 | DNA | CGCCTAGGTTGGGTAGGGTGGTGGCG | [86] |
| Toggle-25t | RNA | GGGAACFAAAGCFUFGAAGUFACFUFUFACFCFCF | [87] |
| Toggle-25t/AF113-18 | RNA | GGGAACFAAAGCFUSeGAAGUFS2ACUFUSeACFCFCF | [88] |
| HD22_27mer | DNA | GTCCGTGGTAGGGCAGGTTGGGGTGAC | [89] |
| Ligand (PDB Entry) | Interface Area (Å2) | NHB | NSB |
|---|---|---|---|
| Exosite I | |||
| Coagulation Factor V (3p6z) | 415.0 | 5 | 4 |
| Human Factor V, A2-B domain linker (3p70) | 297.4 | 2 | 0 |
| Fibrinogen (2a45) | 642.5 | 9 | 2 |
| Platelet glycoprotein Ibα-GPIbα (1ook) | 1060.0 | 17 | 10 |
| Proteinase-activated receptor 1-PAR1 (1nrr) | 401.1 | 4 | 0 |
| Proteinase-activated receptor 3-PAR3 (2pux) | 659.5 | 7 | 4 |
| Thrombomodulin (1dx5) | 772.7 | 12 | 0 |
| Avathrin, C-terminal peptide (5gim) | 516.6 | 3 | 1 |
| IgA fab heavy chain (5e8e) | 526.8 | 5 | 2 |
| Hirugen (1aht) | 627.1 | 7 | 3 |
| Hirullin (1thr) | 727.9 | 10 | 5 |
| Staphylocoagulase (1nu7) | 1661.9 | 22 | 15 |
| Triabin (1avg) | 697.4 | 7 | 5 |
| mTBA (3qlp) | 656.6 | 11 | 0 |
| TBA (4dih) | 565.0 | 12 | 0 |
| TBAΔT12 (4lz1) * | 533.8 | 13 | 0 |
| TBA-T4W (6eo6) * | 707.6 | 14 | 0 |
| TBA-T4K (6eo7) * | 640.6 | 13 | 0 |
| TBA-3L (6z8v) * | 605.8 | 14 | 0 |
| TBA-3Leu (6z8x) * | 592.2 | 9 | 0 |
| RE31 (5cmx) | 551.7 | 10 | 0 |
| NU172 (6evv) | 589.8 | 10 | 0 |
| Bivalirudin, C-terminal fragment (3vxe) | 566.5 | 6 | 1 |
| Nonapeptide inhibitor (1g37) | 512.0 | 2 | 0 |
| Peptide inhibitor (1eb1) | 544.8 | 4 | 3 |
| Synthetic inhibitor (1ths) * | 534.8 | 6 | 0 |
| Synthetic peptide (2a2x) * | 698.1 | 9 | 0 |
| Exosite I + active site | |||
| Heparin cofactor II (1jmo) | 1743.8 | 30 | 13 |
| Proteinase-activated receptor 1-PAR1 (3lu9) | 1573.5 | 25 | 6 |
| AGAP008004-PA from A. gambiae (5nhu) | 1468.6 | 24 | 8 |
| Anophelin (4e05) | 1425.4 | 29 | 11 |
| Boophilin (2ody) | 1979.4 | 24 | 15 |
| Hirudin variant 1 (2pw8) | 1531.1 | 21 | 6 |
| Hirudin variant 2 (4htc) | 1652.1 | 20 | 15 |
| Ornithodorin (1toc) | 1763.3 | 15 | 7 |
| Rhodniin (1tbr) | 1703.7 | 16 | 10 |
| Variegin (3b23) | 900.0 | 12 | 5 |
| CVS995 synthetic peptide (1dit) | 1159.2 | 18 | 4 |
| Hirulog (1abi) | 1241.6 | 19 | 4 |
| bza-2 hirulog (1qur) * | 1348.3 | 13 | 2 |
| Hirunorm IV (4thn) | 1038.4 | 11 | 11 |
| Hirunorm V (5gds) | 1030.7 | 9 | 5 |
| Hirutonin 2 (1ihs) * | 1334.8 | 16 | 4 |
| Hirutonin 6 (1iht) * | 1027.2 | 14 | 4 |
| Synthetic thrombin inhibitor P798 (1eoj) * | 1188.8 | 8 | 4 |
| Synthetic thrombin inhibitor P596 (1hbt) | 1143.5 | 16 | 0 |
| Ligand (PDB Entry) | Interface Area (Å2) | NHB | NSB |
|---|---|---|---|
| Exosite II | |||
| Fibrinogen γ’ peptide (2hwl) | 782.3 | 15 | 6 |
| Bovine kringle-2 (2hpp) | 795.2 | 15 | 9 |
| Human kringle-2 (2hpq) | 778.1 | 11 | 10 |
| Heparin (1xmn) * | 396.5 | 2 | 5 |
| Heparin (in the presence of antithrombin III) (1tb6) * | 307.9 | 0 | 6 |
| Platelet glycoprotein Ibα-GPIbα (1p8v) | 934.0 | 11 | 5 |
| Toggle-25t/AF113-18 (5do4) * | 924.9 | 17 | 0 |
| HD22_27mer (4i7y) | 1079.5 | 18 | 0 |
| Suramin (2h9t) | 476.0 | 3 | 0 |
| Exosite II + active site | |||
| Hemadin (1e0f) | 1216.0 | 19 | 7 |
| Madanin-1 (5l6n) | 1365.3 | 24 | 13 |
| Tsetse thrombin inhibitor (6tkl) | 1652.4 | 27 | 11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Troisi, R.; Balasco, N.; Autiero, I.; Vitagliano, L.; Sica, F. Exosite Binding in Thrombin: A Global Structural/Dynamic Overview of Complexes with Aptamers and Other Ligands. Int. J. Mol. Sci. 2021, 22, 10803. https://doi.org/10.3390/ijms221910803
Troisi R, Balasco N, Autiero I, Vitagliano L, Sica F. Exosite Binding in Thrombin: A Global Structural/Dynamic Overview of Complexes with Aptamers and Other Ligands. International Journal of Molecular Sciences. 2021; 22(19):10803. https://doi.org/10.3390/ijms221910803
Chicago/Turabian StyleTroisi, Romualdo, Nicole Balasco, Ida Autiero, Luigi Vitagliano, and Filomena Sica. 2021. "Exosite Binding in Thrombin: A Global Structural/Dynamic Overview of Complexes with Aptamers and Other Ligands" International Journal of Molecular Sciences 22, no. 19: 10803. https://doi.org/10.3390/ijms221910803