1. Introduction
Cadmium (Cd) is a well-known carcinogen, classified by the International Agency for Research on Cancer as a group I human carcinogen; however, the molecular mechanisms leading cells into a malignant phenotype are largely unknown. Still, cadmium is a widespread environmental contaminant, currently released by anthropogenic activities at a rate of 30,000 tons per year. Human uptake is therefore very easy: besides occupational exposure, it can occur through food, drinking water, inhalation of air particles, and cigarette smoking. The absence of any specific excretory way for cadmium leads to its persistence inside the body, with a half-life between 10 and 30 years [
1].
With the exception of a catalytic role in some algal enzymes [
2], cadmium is devoid of all biological roles; however, Cd
2+ ions can easily displace Zn
2+ ions, due to their similar charge and mass [
3,
4,
5,
6]. This similarity also accounts for cadmium uptake by zinc channels and transporters, in what has been named a “Trojan horse mechanism” [
7]. Moreover, cadmium can interfere with zinc binding proteins, whose estimated number within the cell is higher than 3,000. These features lead to a major displacement of Zn in the Zn-proteome, and the substitution with Cd in the Zn-finger regions. Consequences of these chemical properties are the disruption of the correct folding and the inactivation of proteins involved in genomic stability, such as the p53, and of proteins involved in DNA repair (e.g., NER and BER) [
6,
8].
Cell Transformation Assay (CTA), the most advanced in vitro test for the prediction of human chemical carcinogenicity, refers to the induction, in cultured cells, of phenotypic alterations that have long been considered associated with cells exhibiting neoplastic potential in vivo. After exposure to carcinogenic stimuli, such cells show morphological alterations and form discrete anchorage-independent altered colonies, referred to as transformed
foci, atop the confluent monolayer. The transformed
foci are able to produce tumors in vivo. Moreover, the CTA has been shown to closely model some key stages of the in vivo carcinogenic process [
9]. In addition to be widely used for the screening of potential carcinogens [
10,
11], the CTA is also a powerful tool for mechanistic studies of carcinogenesis [
12,
13]. In our experiments, the CTA based on the use of C3H10T1/2Cl8 mouse embryo fibroblasts was adopted, the latter being among the suitable cells suggested by standard protocols [
14].
Foci, obtained at the end of the CTA, are recognized under a microscope and classified by morphological features, such as deep basophilic staining, multilayered growth, random cell orientation at the edge of the focus, and invasiveness of the surrounding normal cells monolayer [
9,
14]. These morphological features are related to molecular changes leading the cells to acquire fully malignant characteristics, which are demonstrated by their ability to develop tumors when injected into susceptible host animals [
15].
In a previous work [
16], we found that morphological evaluations and proliferative assays confirmed the loss of contact inhibition and the higher proliferative rate of transformed clones. Moreover, biochemical analysis of EGFR pathway revealed that, despite the same initial carcinogenic stimulus (1 μM CdCl
2 for 24 h), the different
foci were characterized by the activation of different molecular pathways. In particular, F1
focus showed ERK activation and a high proliferation rate, while F3
focus showed Akt activation and a survival molecular profile. More recently [
12], a toxicogenomic study performed by our group, showed that, upon cadmium administration, the two
foci also developed distinct patterns of up-regulated and down-regulated genes.
This work reports an in-depth characterization of both F1 and F3
foci, with the aim of identifying metabolic alterations caused by gene dysregulation. We investigated oxygen consumption rate and ATP production, as well as mitochondrial morphology and defense mechanisms against oxidative stress; mitochondria are in fact known to play a key role in malignant transformation and metastasis development [
17]. Our results show a different metabolic pattern in each
focus, which could explain their different proliferation rates.
3. Discussion
In this work we compared two different
foci, F1 and F3, obtained at the end of the CTA performed on C3H cells treated with CdCl
2 for 24 h. The results showed that previously observed differences in their proliferative behavior [
16] and gene dysregulation [
12] are accompanied by metabolic differences. This in turn confirms that there are many targets for cadmium at the molecular level, eventually leading to malignant transformation.
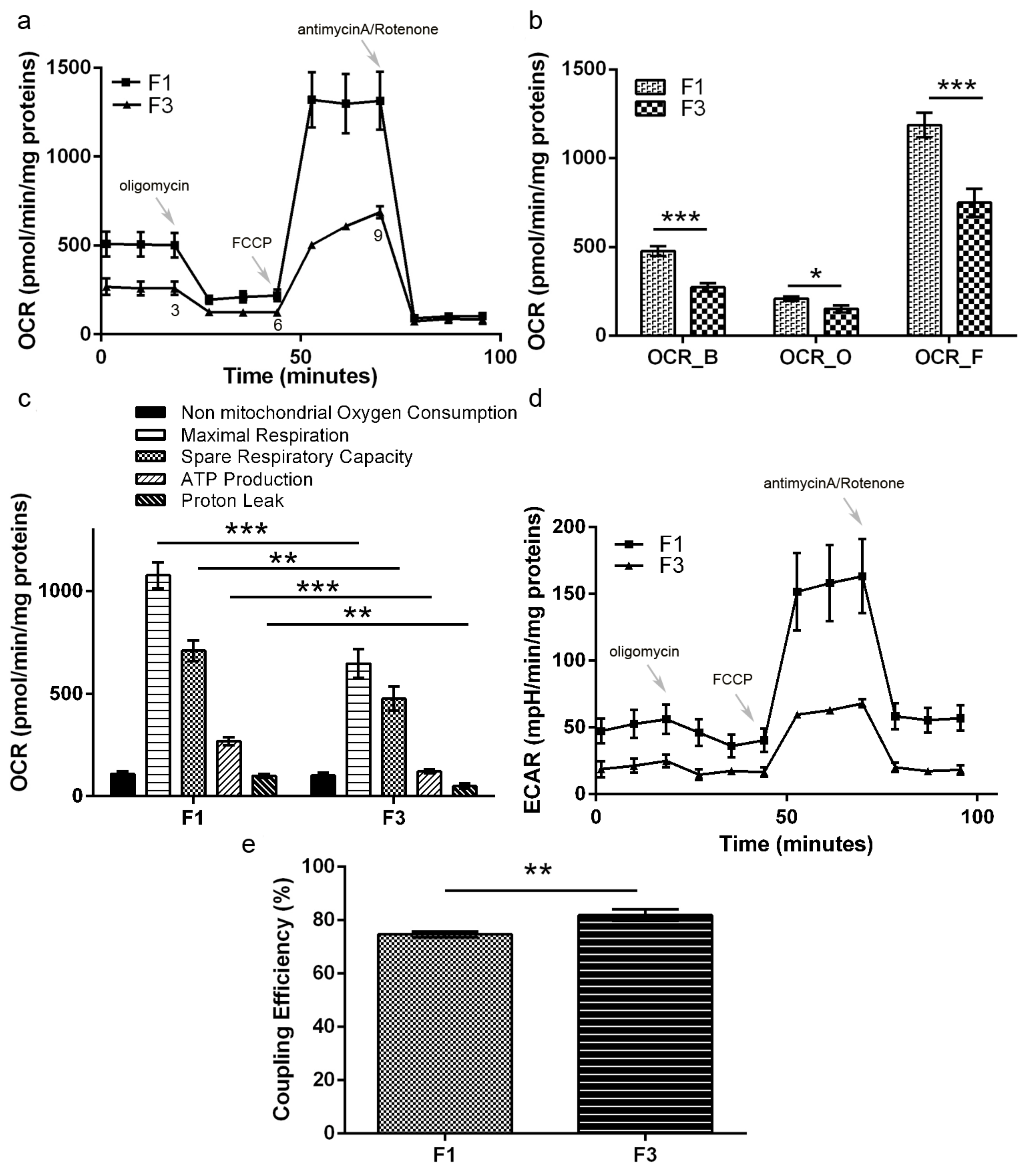
In F1
focus, we observed higher glycolytic, TCA and oxidative phosphorylation rates compared to F3
focus, which are well in accordance with its higher proliferative rate supported by increased mitochondrial functionality. This is likely accomplished through a mitochondrial reorganization in a tightly crowded distribution around the nucleus, as shown by confocal microscopy analysis. A similar picture was previously observed in C3H cells treated with CdCl
2 for 24 h [
18]. However, all the NADH produced by Cd-treated C3H10T1/2Cl8 cells was efficiently reoxidized on the electron transport chain. In F1
focus, NADH production exceeds mitochondrial reoxidative capacity, so that NADH is partly reoxidized by LDH, producing lactate. Another interesting feature of F1
focus is the loss of the Pasteur effect: although most ATP is produced through oxidative phosphorylation, glycolysis is not inhibited. This is likely due to the overexpression of PFKFB3, which, unlike PFK1FBP, is not inhibited by ATP and citrate [
20] and is endowed with a much higher ratio in kinase/phosphatase activity, allowing cells to maintain high glycolytic rates. F1 high glycolytic rate could also sustain other metabolic synthesis, through overexpression of PKM2 isoform, promoting PEP conversion into pyruvate without ATP synthesis. This could lead to the synthesis of many metabolites, essential to sustain high proliferation rates, and particularly to glutathione synthesis, which is in fact more abundant in F1
focus compared to F3. Glutamine is also required for glutathione synthesis and further experiments will assess whether this amino acid contributes to F1 high proliferation.
F3 focus relies mainly on glycolysis for its ATP request, although this pathway is on the whole less active than in F1 focus; NADH produced in the glycolytic pathway is reoxidized by LDH, but lactate production is lower than in F1 focus. The metabolic flux through glycolysis is reduced (as shown by lower PFKFB3, PKM2 and GAPDH expression), compared to F1 focus, justifying F3 lower proliferative rate.
F3
focus showed a higher accumulation of total ROS, with an increased production of mitochondrial H2O2, compared to F1
focus: this is well in accordance with higher specific activities of both glutathione peroxidase and catalase in F3
focus. ROS accumulation is also supported by the GSH/GSSG ratio close to 1, observed in both
foci, while in healthy cells it normally ranges between 200:1 and 30:1 [
21]. In particular, the increase in H
2O
2 production can be due to superoxide dismutation, reflecting in an increase in superoxide production in F3
focus as well. However, it could also be due to electron transfer impairment in F3 damaged mitochondria, as supported by confocal microscopy data and confirmed by Seahorse results. Mitochondrial damage is also supported by previous toxicogenomic data [
12] obtained in our laboratory showing that 13 out of the 15 top up-regulated genes in F3
focus are involved in an interferon mediated inflammatory response, similar to that triggered by viral DNA and RNA [
22]. Furthermore, the upregulation, in F3
focus, of TLR8 gene and IL-6 coding gene [
12] suggests that mtRNA release from damaged mitochondria could be responsible for the observed inflammatory response. A similar result was previously observed in human monocytes, where viral RNA was detected by TLR8 resulting in IL-6 and TNF secretion [
23]. The importance of inflammation in the process of carcinogenesis is widely recognized [
24], indicating that damaged mitochondria-mediated inflammation could be one possible route to transformation. Overall, our data show that, although total ATP level is the same in both
foci, F3 consumes less ATP, due to its low proliferation. Therefore, although glycolysis is less active in F3
focus than in F1, it is still sufficient to support its growth.
F1
focus showed a lower level of total ROS, but a higher production of O
2− compared to F3
focus, although no significant differences were found in mitochondrial O
2− levels. This is in accordance with the higher glutathione S-transferase and glutathione reductase specific activities detected in F1
focus, representing the defense against O
2− accumulation, and is further confirmed by the higher level of reduced glutathione in this
focus; a GSH/GSSG ratio close to 1 suggests that oxidative stress is high in this
focus. The glutathione level is often increased in cancer cells, as a result of increased oxidative stress and glycolysis up-regulation, where it leads to faster growth rates and resistance to a number of chemotherapeutic agents [
25]. Additionally, GSH has been shown to directly reduce O
2− [
26] and may therefore represent a defense mechanism against this ROS, in addition to SOD activity. This could also explain why neither SOD1 nor SOD2 is increased in F1
focus in order to detoxify O
2−.
Previous data obtained in our laboratory [
18] showed that SOD1 activity is impaired by 24 h treatment with CdCl
2, leading to an increased O
2− level; however, toxicogenomic data showed that the SOD1 coding gene is not dysregulated by CdCl
2 treatment neither in C3H cells [
3] nor in human SH-SY5Y neuroblastoma cells [
27]. In
foci, we could not detect any impairment in SOD1 activity, suggesting that this enzyme inhibition is an early event caused by CdCl
2 administration, far before the 4 to 6 weeks needed for the in vitro cell transformation. Finally, no autophagy activation was detected in the two
foci, suggesting that the autophagy observed after cadmium administration for 24 h was part of the early mechanism of defense against this metal. By comparing data reported in this paper with those previously obtained on healthy C3H10T1/2Cl8 cells treated with CdCl
2 for 24 h [
18], we observed that each
focus maintains some but not all the alterations observed in C3H10T1/2Cl8. This finding suggests that the different alterations did not originally occur in all treated cells, but that different cells reacted to CdCl
2 exposure in different ways. In other words, although C3H cells are all genetically identical, much of their fate upon CdCl
2 exposure depends on a plethora of microenvironmental, epigenetic and subcellular factors, such as, for example, local metallothioneins and GST expression and ROS defense enzyme activities.
Although in different ways, both F1 and F3 foci showed uncoordinated glycolysis, TCA and oxidative phosphorylation, each pathway working independently. This is likely a consequence of the loss of the Pasteur effect in F1 focus and of mitochondrial damage in F3 focus, each leading to cell transformation and foci formation.
CdCl2 toxicity is efficiently inactivated in most cells; however, a very small number of cells are damaged, in different ways. These cells proliferate in the absence of CdCl2, giving rise to foci after 4 to 6 weeks. Further research will address the question of how all these effects are irreversibly triggered by CdCl2 uptake, 4–6 weeks earlier than foci detection and collection.
4. Materials and Methods
4.1. Cell and Culture Conditions
The experiments were performed using the cells collected from cadmium-transformed
foci obtained at the end of Cell Transformation Assays (CTAs) on C3H10T1/2 clone 8 mouse embryonic fibroblasts (cell line ATCC, CCL 226 lot. n. 58078542), as previously described [
16]. This cell line was chosen for its high sensitivity to carcinogenic compounds, its low spontaneous transformation rates, and because it represents one of the three cell lines suggested in the Detailed Review Paper on Cell Transformation Assay to be used for detection of chemical carcinogens [
14]. Cells with passages from 9 to 12 were used for cell transformation studies [
14]. The CTA was carried out following cells exposure to 1 μM CdCl
2 for 24 h; after that, the cadmium containing medium was substituted with a fresh medium without cadmium and the cells were grown for 6 weeks, until
foci appeared [
28]. Different
foci were collected at the end of the CTAs, cultured and processed for further analyses, as described in the following sections. Among the different fully transformed
foci, F1 and F3
foci were subjected to further analyses, since they were found to be very different, both morphologically and from the biochemical point of view. Cells were cultured in Basal Medium Eagle (BME, Sigma Chemical Co., St. Louis, MO, USA) enriched with 10% heat-inactivated fetal bovine serum (FBS, Euroclone, Pero, Italy), 1 % glutamine, 0.5% HEPES 2 M and 25 μg/mL gentamicin (all purchased from Sigma) at 37 °C in a humidified incubator supplied with a constant flow of 5% CO
2 in air throughout each experiment. Cells were routinely seeded in 100 mm Ø cell culture Petri dishes, the medium was changed every 3 days and cells grown until 80% confluence maximum was reached. The cells were stored in ampoules, frozen at –80 °C with 10% sterile dimethyl sulfoxide (DMSO) as a preservative.
4.2. Cell Lysis and Preparation of Protein Samples
Cells from both F1 and F3 foci were harvested by trypsinization at 80% confluence, rinsed with ice-cold PBS and lysed in 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 10% glycerol, 1% NP40 buffer, containing 1 μM leupeptin, 2 μg/mL aprotinin, 1 μg/mL pepstatin and 1 mM phenylmethylsulfonyl fluoride (PMSF). After lysis on ice, homogenates were obtained by passing the cells 5 times through a blunt 20-gauge needle fitted to a syringe and then centrifuging at 15,000× g for 30 min at 4 °C. Enzyme activities were assayed on supernatants.
For cytosolic fraction, cells were rinsed with ice-cold PBS and lysed in PBS, containing 1 μM leupeptin, 2 μg/mL aprotinin, 1 μg/mL pepstatin and 1 mM PMSF; homogenates were obtained by passing the cells 5 times through a blunt 20-gauge needle fitted to a syringe, incubating on ice for 15 min and sonicating 2 times (10 s cycle). The supernatant was obtained by centrifugation at 15,000× g for 10 min at 4 °C and used to measure enzyme activities.
For mitochondrial fraction, cell lysate was centrifuged at 800× g for 5 min at 4 °C to remove nuclei and unbroken cells. The post-nuclear supernatant was centrifuged at 10,000× g for 15 min to collect mitochondria. After centrifugation, the supernatant was removed and pellets were resuspended in 50 mM Tris-HCl 50, pH 7.4, 150 mM NaCl, 5 mM EDTA, 10% glycerol, 1% NP40 buffer, containing 1 μM leupeptin, 2 μg/mL aprotinin, 1 μg/mL pepstatin and 1 mM PMSF and used to measure enzymatic activities. The cytosolic and mitochondrial fractions were analyzed through Western blot to confirm mitochondria isolation.
4.3. Cell Lysis and Preparation of Metabolite Samples
Both cell clones were harvested by trypsinization at 80% confluence; the pellets were resuspended in 3 mL PBS, harvested by a centrifugation in the above conditions, weighed, and resuspended with 5 volumes of 5% perchloric acid. The suspension was passed 5 times through a blunt 20-gauge needle fitted to a syringe, incubated on ice for 15 min and centrifuged at 3000× g at 4 °C for 10 min. The resulting supernatant was neutralized with 2.5 M K2CO3 to pH 6.5, centrifuged at 3000× g at 4 °C for 10 min to eliminate potassium perchlorate and kept at −80 °C for metabolite analyses.
4.4. Enzyme and Metabolite Assays
The enzymes and metabolites were assayed using the following procedures. All assays were performed in triplicate at 30 °C in a Cary3 Spectrophotometer and analyzed by the Cary Win UV application software for Windows. Enzymatic activities were expressed in international units and referred to protein concentration as determined by the Bradford method [
29]. Metabolite concentrations were expressed in nmol/mg cells.
Hexokinase (HK) activity was determined by following NADPH formation at 340 nm by coupled assay with glucose-6-phosphate dehydrogenase (G6PDH) according to Bergmeyer [
30]. The protein samples were incubated with 100 mM Tris-HCl pH 7.6, 1 mM ATP, 0.6 mM NADP
+, 2 mM glucose, 10 mM MgCl
2, 1 U/mL G6PDH.
In glyceraldeyde-3-phosphate dehydrogenase (GAPDH) assay was measured the disappearance of NADH at 340 nm according to Bergmeyer [
30]. The protein samples were incubated with 80 mM HEPES-Tris pH 7.6, 6 mM glycerate 3-phosphate, 0.9 mM EDTA, 1.1 mM ATP, 0.2 mM NADH, 1.7 mM MgSO
4, 15 U/mL phosphoglycerate kinase (PGK). In lactate dehydrogenase (LDH) assay was measured the disappearance of NADH at 340 nm according to Bergmeyer [
30]. The protein samples were incubated with 85 mM potassium phosphate buffer, 0.2 mM NADH, 0.6 mM pyruvate. Pyruvate kinase (PK) activity was determined by following NADH disappearance at 340 nm by coupled assay with lactate dehydrogenase (LDH) according to Bergmeyer [
30]. The protein samples were incubated with 50 mM HEPES-Tris pH 7.6, 75 mM KCl, 8 mM MgCl2, 0.2 mM NADH, 8 mM 2- phosphoenolpyruvate, 1.5 mM ADP, 9 U/mL LDH. In malate dehydrogenase (MDH) assay was measured the disappearance of NADH at 340 nm according to Bergmeyer [
30]. The protein samples were incubated with 50 mM HEPES-Tris pH 7.6, 0.5 mM oxaloacetate, 0.2 mM NADH. In glutamate dehydrogenase (GLDH) assay was measured the disappearance of NADH at 340 nm according to Bergmeyer [
30]. The protein samples were incubated with 50 mM HEPES-Tris pH 7.6, 100 mM NH
4Cl, 0.2 mM NADH, 1 mM ADP, 1 mM EDTA, 7 mM 2-oxoglutarate. Glucose-6-phosphate dehydrogenase (G6PDH) activity was determined by following NADPH formation at 340 nm according to Bergmeyer [
30]. The protein samples were incubated with Tris-HCl 90 mM, 7 mM MgCl
2, 1 mM glucose-6-phosphate, 0.4 mM NADP
+.
NADP
+ dependent isocitrate dehydrogenase (ICDH) activity was determined by following NADPH formation at 340 nm according to Bergmeyer [
30]. The protein samples were incubated with 50 mM HEPES-Tris pH 7.6, 4 mM MnCl
2, 3.7 mM isocitrate, 0.32 mM NADP
+. Malic enzyme (ME) activity was determined by following NADPH formation at 340 nm according to Bergmeyer [
30]. The protein samples were incubated with 50 mM HEPES-Tris pH 7.6, 10 mM MgSO
4, 4 mM malate, 0.5 mM NADP
+. Citrate synthase (CS) was assayed according to Shepherd [
31]. In detail, the protein samples were incubated with 100 mM Tris-HCl pH 8, 0.1 mM 5,5′- dithiobis-(2-nitrobenzoic acid), 0.05 mM acetyl-CoA and 0.25 mM oxaloacetate. The reaction was monitored at 412 nm. Catalase (CAT) activity was assayed according to Bergmeyer [
32], using 12 mM H
2O
2 as substrate in the presence of 50 mM sodium phosphate buffer, pH 7.5. The reaction was monitored at 240 nm. Glutathione S-transferase (GST) was measured as reported in Habig [
33], using 1 mM reduced glutathione (GSH) and 1 mM 1-chloro-2,4-dinitrobenzene (CDNB) as substrates in the presence of 90 mM potassium phosphate buffer pH 6.5. The reaction was monitored at 340 nm. The glutathione peroxidase (GPx) activity is based on the oxidation of GSH using H
2O
2 as substrate, coupled to the disappearance of NADPH by glutathione reductase (GR), according to Nakamura [
34]. The protein samples were incubated with 50 mM sodium phosphate buffer pH 7.5, 0.16 mM NADPH, 1 mM NaN
3, 0.4 mM EDTA, 1 mM GSH, 0.2 mM H
2O
2, 2 U/mL GR.
Glutathione reductase (GR) was measured following the disappearance of NADPH at 340 nm according to Wang [
35]. The protein samples were incubated with 100 mM potassium phosphate buffer pH 7.6, 0.16 mM NADPH, 1 mM EDTA, 1mg/mL BSA, 4.6 mM oxidized glutathione (GSSG). Superoxide dismutase 1 (SOD1) and superoxide dismutase 2 (SOD2) were measured using an indirect method according to Vance [
36]. This technique is based on the ability of superoxide dismutase to compete with ferricytochrome c for superoxide anions generated by the xanthine oxidase system and thus to inhibit the reduction of ferricytochrome c. Briefly, the protein samples were incubated with 0.01 mM ferricytochrome c in 10 mM HEPES-Tris pH 7.5, 0.1 mM EDTA, 0.01 mM xanthine in 1 mM NaOH and xanthine oxidase at final concentration of 0.006 U/mL. Under these conditions, one unit of SOD is the amount of enzyme able to yield a 50% decrease in the rate of ferricytochrome c reduction followed at 550 nm.
Lactate and total intracellular ATP were measured using standard enzymatic tests [
30]. In detail, lactate was measured following NADH formation in the presence of 380 mM Tris-Glycine pH 9, 2.6 mM NAD
+, 15 U/mL lactate dehydrogenase; ATP was measured following NADPH formation in the presence of 25 mM HEPES-Tris pH 7.6, 3.3 mM MgCl
2, 0.23 mM NADP
+, 17 mM glucose, 0.23 U/mL G6PDH, 0.46 U/mL HK.
4.5. GSH Assay
Both
foci were harvested by trypsinization at 80% confluence; the pellets were washed in 3 mL PBS, harvested by a centrifugation and weighed to normalize the results to mg of cells. Pellets were resuspended in 500 μL cold 5% 5-sulfosalicylic acid (SSA), lysed by vortexing and by passing through a blunt 20-gauge needle fitted to a syringe 5 times. All the samples were incubated for 10 min at 4 °C and then centrifuged at 14,000×
g for 10 min at 4 °C. The supernatant was prepared and used for the analysis following the instructions of Glutathione Colorimetric Detection Kit (catalog number EIAGSHC, Invitrogen, Carlsbad, CA, USA). The Kit is designed to measure oxidized glutathione (GSSG), total glutathione (GSH tot) and reduced glutathione (GSH tot—GSSG) concentrations through enzymatic recycling assay based on glutathione reductase and reduction of Ellman reagent (5,5-dithiobis(2-nitrobenzoic acid)) and using 2-vinylpyridine as reagent for the derivatization of glutathione [
37]. Therefore, it was possible to obtain GSH/GSSG ratio, a critical indicator of cell health. The absorbance was measured at 405 nm using a micro plate reader. The values of absorbance were compared to standard curves (GSH tot and GSSG, respectively) and normalized to mg of cells. Final concentrations were expressed in nmol/mg cells.
4.6. SDS-PAGE and Western Blotting
For sample preparation for Western-blot analysis, both cell clones were harvested by trypsinization at 80% confluence. The cells were then rinsed with ice-cold PBS and lysed in RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) containing 1 μM leupeptin, 2 μg/mL aprotinin, 1 μg/mL pepstatin, 1 mM PMSF and phosphatase inhibitors. After lysis on ice, homogenates were obtained by passing them through a blunt 20-gauge needle fitted to a syringe 5 times; they were then centrifuged at 15,000×
g for 30 min. Supernatants were analyzed for protein content by the BCA protein assay [
38]. SDS-PAGE and Western blot were carried out by standard procedures [
39]. Sixty micrograms of proteins were separated on a 10% or 15% acrylamide/bis-acrylamide SDS-PAGE, transferred onto a nitrocellulose membrane (Millipore, Billerica, MA, USA), probed with the appropriated antibodies and visualized using ECL detection system (Millipore). Protein levels were quantified by densitometry of immunoblots using Scion Image software (Scion Corp., Frederick, MD, USA). The following primary antibodies were used (all purchased by Cell Signaling Technology, Danvers, MA, USA): anti-PKM2 (catalog number #4053, dilution 1:1000), anti-PFKFB3 (catalog number #13123, dilution 1:1000), anti P-AMPK (Thr172) (catalog number #2535, dilution 1:1000), anti-AMPK (catalog number #2532, dilution 1:1000), anti- LC3B (catalog number #2775, dilution 1:500), anti-GAPDH (catalog number #2118, dilution 1:10,000), anti-tubulin (catalog number #2125S, dilution 1:1000) (all purchased by Cell Signaling Technology, Danvers, MA, USA) and anti-vinculin (catalog number #V9131, dilution 1:10,000) (purchased by Sigma Chemical Co., St. Louis, MO, USA). IgG HRP anti-rabbit (catalog number #7074) and anti-mouse (catalog number #7076) conjugated secondary antibodies (purchased by Cell Signaling Technology, Danvers, MA, USA) were diluted 1:10,000.
4.7. Detection of Intracellular Reactive Oxygen Species
The generation of intracellular reactive oxygen species (ROS) was detected by the oxidation of 2′,7′-Dichlorofluorescin diacetate (H
2DCFDA) or Dihydroethidium (DHE). H
2DCFDA is an indicator for both reactive oxygen species and nitric oxide (•NO); the second probe is more selective towards superoxide anion (O
2−). The cells were plated at a density of 2.5 × 10
5 cells per well into six-well plates in 2 mL of complete culture medium and incubated 24 h after seeding with H
2DCFDA (5 μΜ final concentration in PBS) or DHE (10 μΜ final concentration in complete medium) for 20 min in the dark at 37 °C. At the end of incubation, cells were washed by warm PBS, trypsinized (500 μL of trypsin/well) and harvested by centrifugation (5 min at 2000×
g) at room temperature. The pellet was resuspended in 500 μL/tube of PBS and ROS generation of 10,000 cells was measured by the fluorescence intensity. FL-1 channel (530 nm) was utilized to detect the fluorescence intensity of DCF; HE fluorescence can be measured at 585 nm, or FL-2 channel, band-pass filter. Logarithmic amplification, which produces an output signal proportional to the logarithm of the input signal, was used to detect probe fluorescence. Data quality is enhanced when the brightness levels of all probes excited off a single laser are balanced within one log scale of fluorescence intensity [
40]. Flow cytometry data were analyzed using CytExpert 2.3 Software (Beckman Coulter, Inc., Brea, CA, USA).
4.8. Detection of Mitochondrial ROS (mtROS)
MitoSOX ™ Red (ThermoFisher Scientific, Massachusetts, USA) and MitoPY1 (Tocris Bioscience, Bristol, UK) indicators were used to detect the mitochondrial superoxide anion and mitochondrial hydrogen peroxide production, respectively, in intact adherent cells. The oxidation of these probes forms intermediate probe-derived radicals that are successively oxidized to generate the corresponding fluorescent products [
41]. The cells were plated at a density of 2.5 × 10
5 cells per well into six-well plates in 2 mL of complete culture medium and stained 24 h after seeding with MitoSOX™ Red or MitoPY1 (5 μΜ final concentration in 1 mL of PBS) for 20 min in the dark at 37 °C. After staining, the cells were washed by warm PBS, trypsinized (500 μL of trypsin /well) and harvested by centrifugation (5 min at 2000×
g) at room temperature. The pellet was resuspended in 500 μL/tube of PBS and mtROS generation of 10,000 cells was measured by the fluorescence intensity. FL-1 channel (530 nm) was utilized to detect the fluorescence intensity of MitoPY1; FL-2 channel (585 nm) band-pass filter was utilized to measure the fluorescence intensity of MitoSOX ™ Red. Logarithmic amplification, which produces an output signal proportional to the logarithm of the input signal, was used to detect probe fluorescence. Data quality is enhanced when the brightness levels of all probes excited off a single laser are balanced within one log scale of fluorescence intensity [
40]. Flow cytometry data were analyzed using CytExpert 2.3 Software (Beckman Coulter, Inc., Brea, CA, USA).
4.9. Mitochondrial Transmembrane Potential (MTP) Assay
MTP alterations were assessed by flow cytometry, using the mitochondrial potential sensitive dye 3,3′-dihexyloxacarbocyanine Iodide (DiOC6), which accumulates in mitochondria due to their negative membrane potential and can be applied to monitor the mitochondrial membrane potential using flow cytometry detection. The cells were plated at a density of 2.5 × 10
5 cells per well into six-well plates in 2 mL of complete culture medium, harvested 24 h after seeding by centrifugation (5 min at 2000×
g) at room temperature and stained with DiOC6 (40 nM in PBS, 20 min at 37 °C and 5% CO2 in the dark). Loss in DiOC6 fluorescence indicates disruption of the mitochondrial inner transmembrane potential. The probe was excited at 488 nm and emission was measured through a 530 nm (FL-1 channel) band-pass filter. Logarithmic amplification, which produces an output signal proportional to the logarithm of the input signal, was used to detect probe fluorescence. Data quality is enhanced when the brightness levels of all probes excited off a single laser are balanced within one log scale of fluorescence intensity [
40]. Flow cytometry data were analyzed using CytExpert 2.3 Software (Beckman Coulter, Inc., Brea, CA, USA).
4.10. Oxygen Consumption Rate and Extra-Cellular Acidification Rate Measurements
Oxygen consumption rate (OCR) and extra-cellular acidification rate (ECAR) were measured in F1 and F3 foci with Seahorse XFe24 Analyzer (Seahorse Bioscience, Billerica, MA, USA) using Seahorse XF Cell Mito Stress Test Kit (catalog number #103015-100, purchased by Agilent Technologies, Santa Clara, CA, USA) and Agilent Seahorse XF Glycolytic Rate Assay Kit (catalog number #103344-100, purchased by Agilent Technologies, Santa Clara, CA, USA). The cells were seeded in Agilent Seahorse XF24 cell culture microplates at density of 3 × 104 cells/well in 250 μL of Basal Medium Eagle and 24 h after seeding the growth medium was replaced with 525 μL/well of Seahorse XF Base Medium containing 1 mM pyruvate, 2 mM glutamine and 10 mM glucose for the Cell Mito Stress Test Kit or 1 mM pyruvate, 2 mM glutamine, 10 mM glucose and 5 mM HEPES for the Glycolytic Rate Assay Kit. Then, the plate was incubated into 37 °C non-CO2 incubator for 1 h, before starting the experiment procedure.
The sensor cartridge was calibrated by Seahorse XFe24 Analyzer. Pre-warmed oligomycin, FCCP, rotenone and antimycin A were loaded into injector ports A, B and C of sensor cartridge, to reach working concentration of 1 μM, 2 μM and 0.5 μM, respectively, for the Cell Mito Stress Test Kit.
Pre-warmed rotenone and antimycin A and 2-deoxy-D-glucose (2-DG) were loaded into injector ports A and B of sensor cartridge, to reach working concentration of 0.5 μM and 50 mM for the Glycolytic Rate Assay Kit.
OCR and ECAR were detected under basal conditions followed by the sequential addition of the drugs, to measure non-mitochondrial respiration, maximal respiration, proton leak, ATP respiration, respiratory capacity, coupling efficiency for the Cell Mito Stress Test Kit and basal glycolysis, basal proton efflux rate, compensatory glycolysis and post 2-DG acidification for the Glycolytic Rate Assay Kit [
42,
43,
44]. After assay performance, cells were lysed and the total cellular proteins were quantified using Bradford method [
29] in order to normalize the data.
4.11. Confocal Microscopy
Mitochondria fluorescence was studied by laser scanning confocal microscopy, using a Bio-Rad MRC-600 confocal microscope (Bio-Rad, Hemel Hempstead, UK) equipped with a 25 mW argon laser. The scanning head was coupled with an upright epifluorescence microscope Nikon Optiphot-2 (Nikon, Tokyo, Japan) with a 60× oil immersion objective Nikon Planapochromat (N.A. = 1.4). The fluorescence was excited at 488 nm and the emission was collected through a long pass filter above 515 nm. High sensitivity photon counting detection was used to minimize the excitation power (0.1 mW at the entry of the optical head) and preserve cell viability.
The cells were plated in 35 mm Ø Petri dishes at density of 6 × 104 cells/well and 24 h after seeding the medium was removed, the cells were washed twice with phosphate buffer saline (PBS) and incubated for 10 min in 1 μM rhodamine 123 (R123) solution at 37 °C and 5% CO2. After incubation, the cells were rinsed twice with PBS and few microliters of PBS were left in the Petri dish to avoid cell drying. A coverslip was placed over the cells that were immediately imaged by confocal microscope.
To measure mitochondrial interconnectivity and elongation from confocal microscope images we used the macro designed by Dagda and colleagues [
45].
4.12. Statistical Analysis
The data were tested by Student’s
t-test. All calculations were conducted using the R statistics programming environment [
46].


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}