
Loops around the Heme Pocket Have a Critical Role in the Function and Stability of BsDyP from Bacillus subtilis

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Deletion of Tat-Signal Peptide from BsDyP
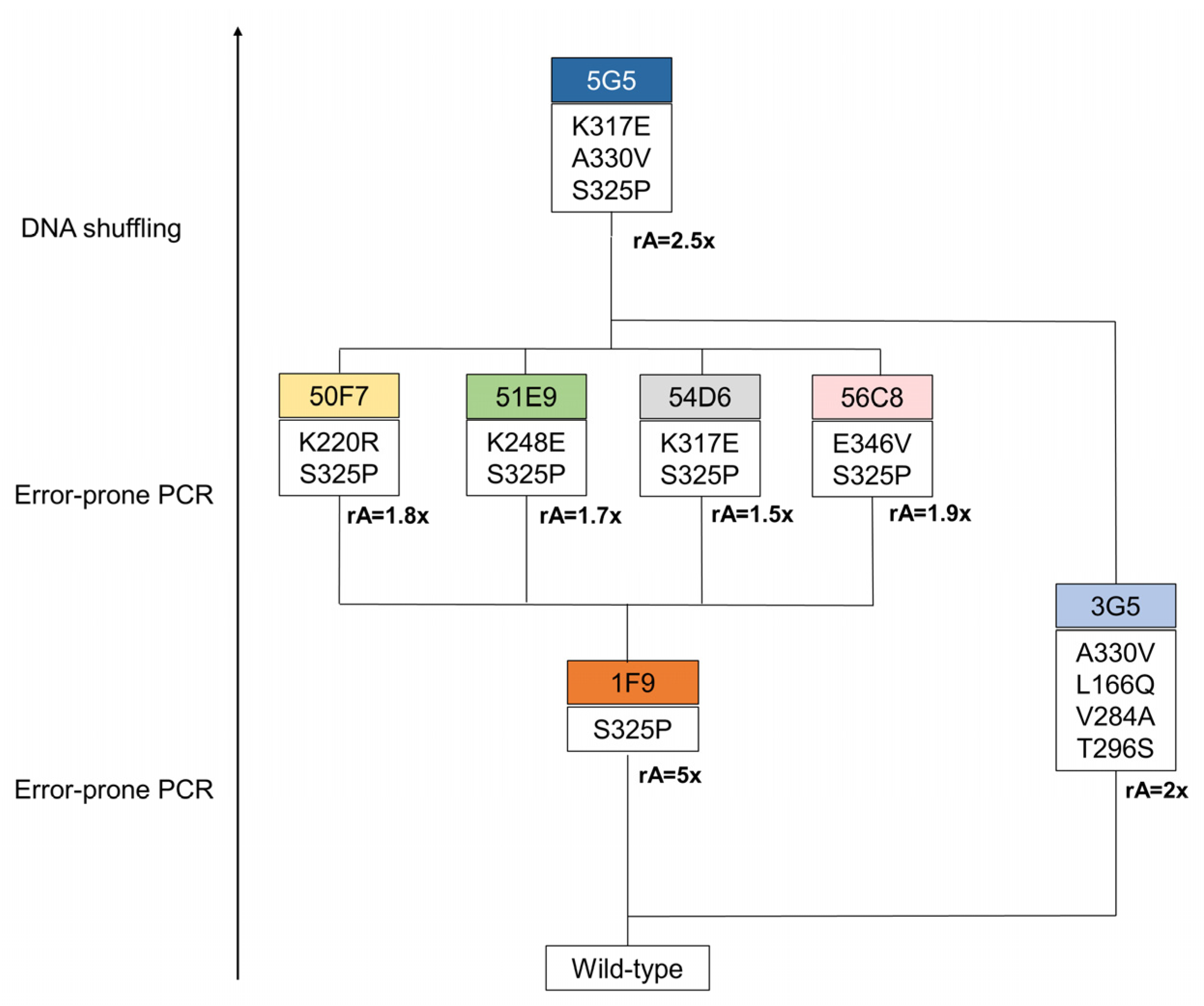
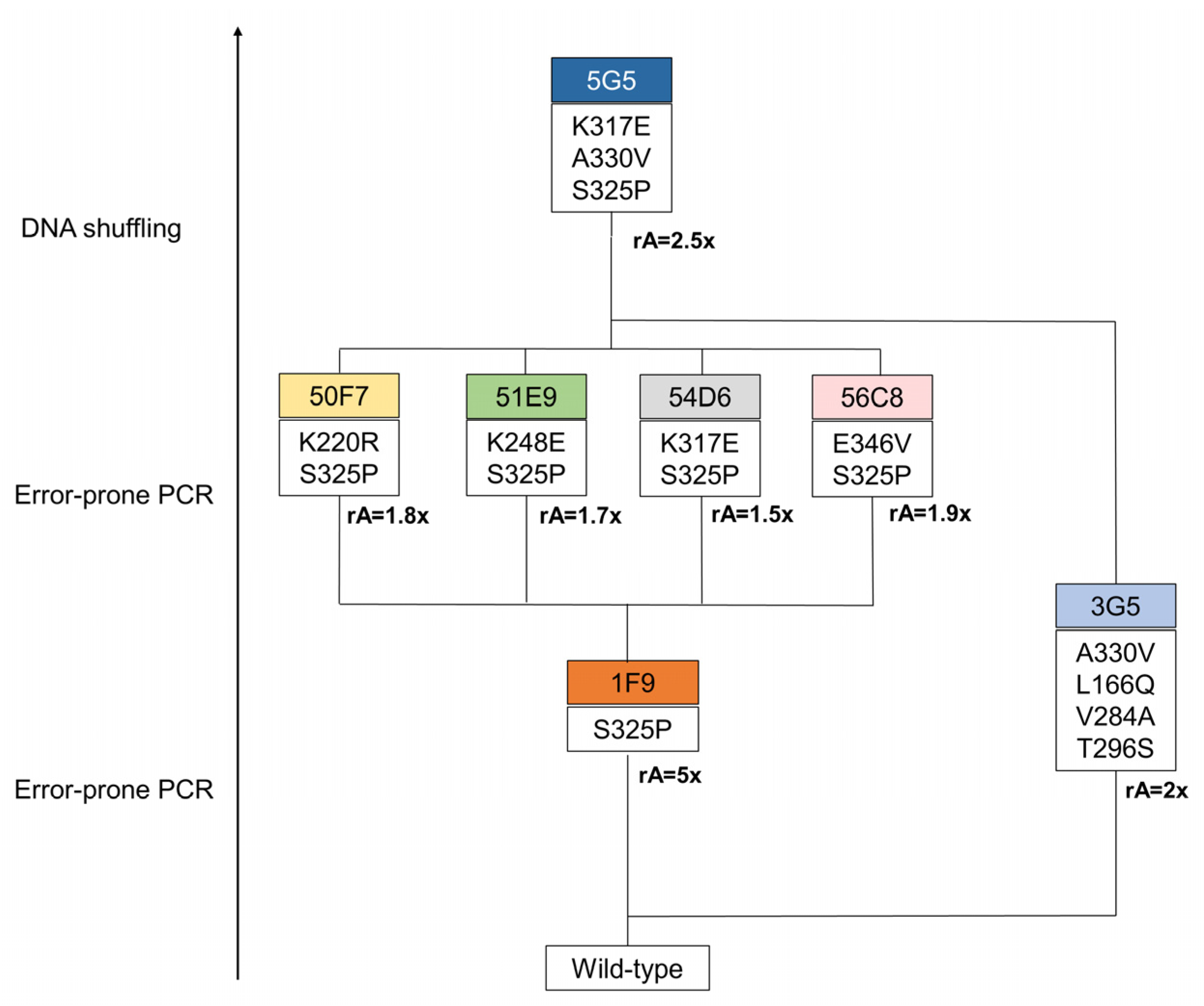
2.2. Directed Evolution of BsDyP for Improved DMP Oxidation
2.3. Biochemical and Kinetic Characterization
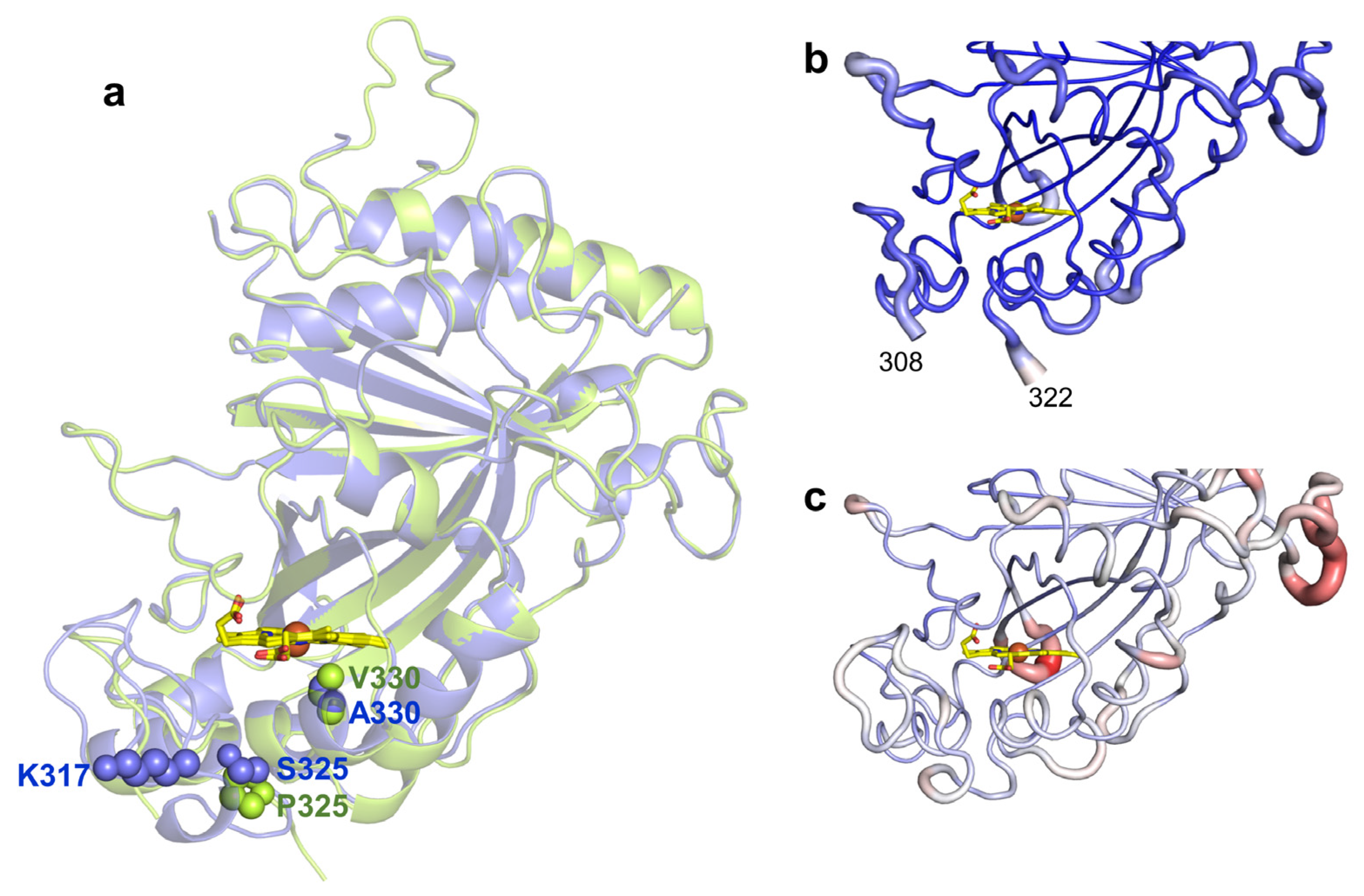
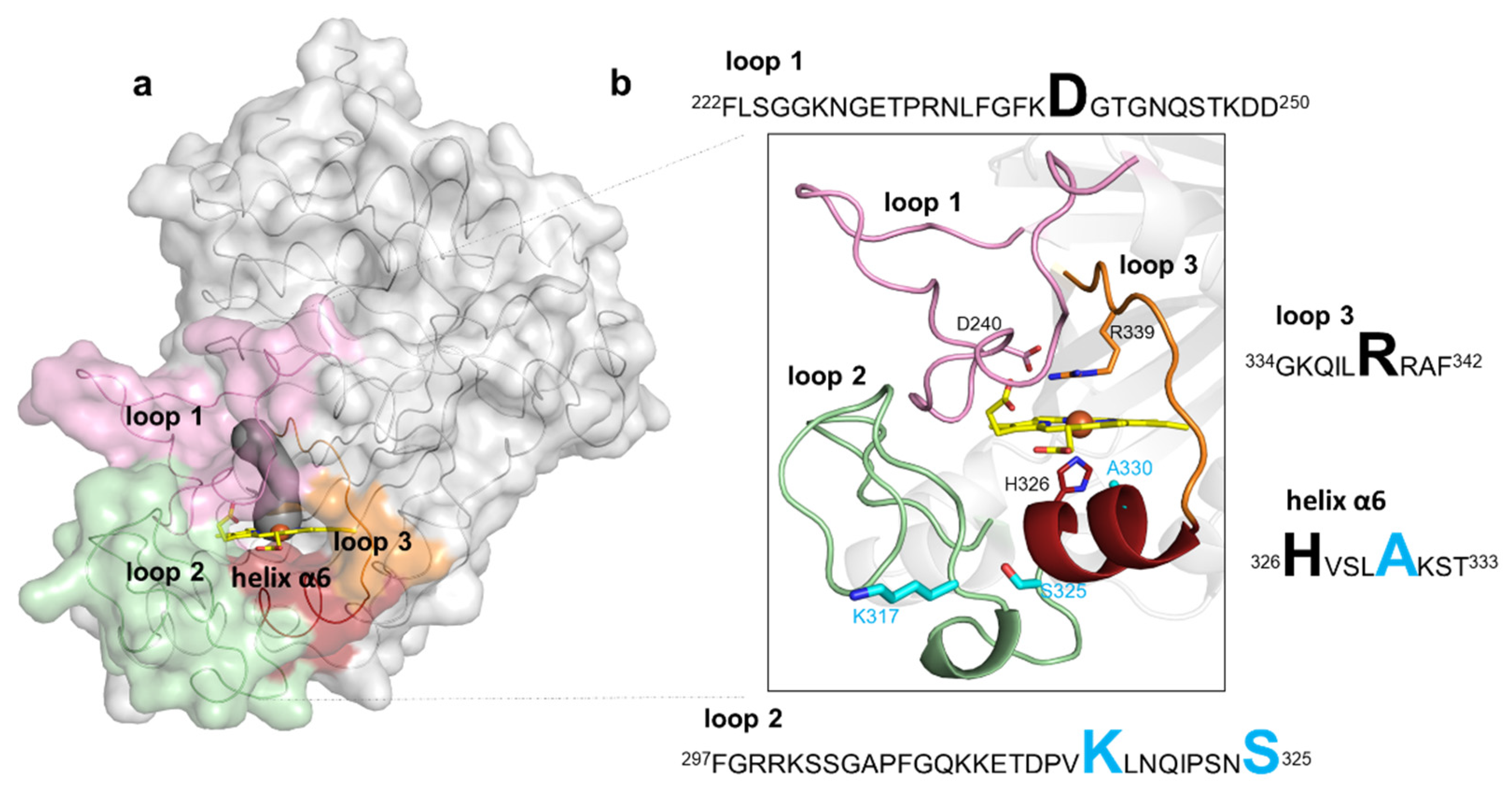
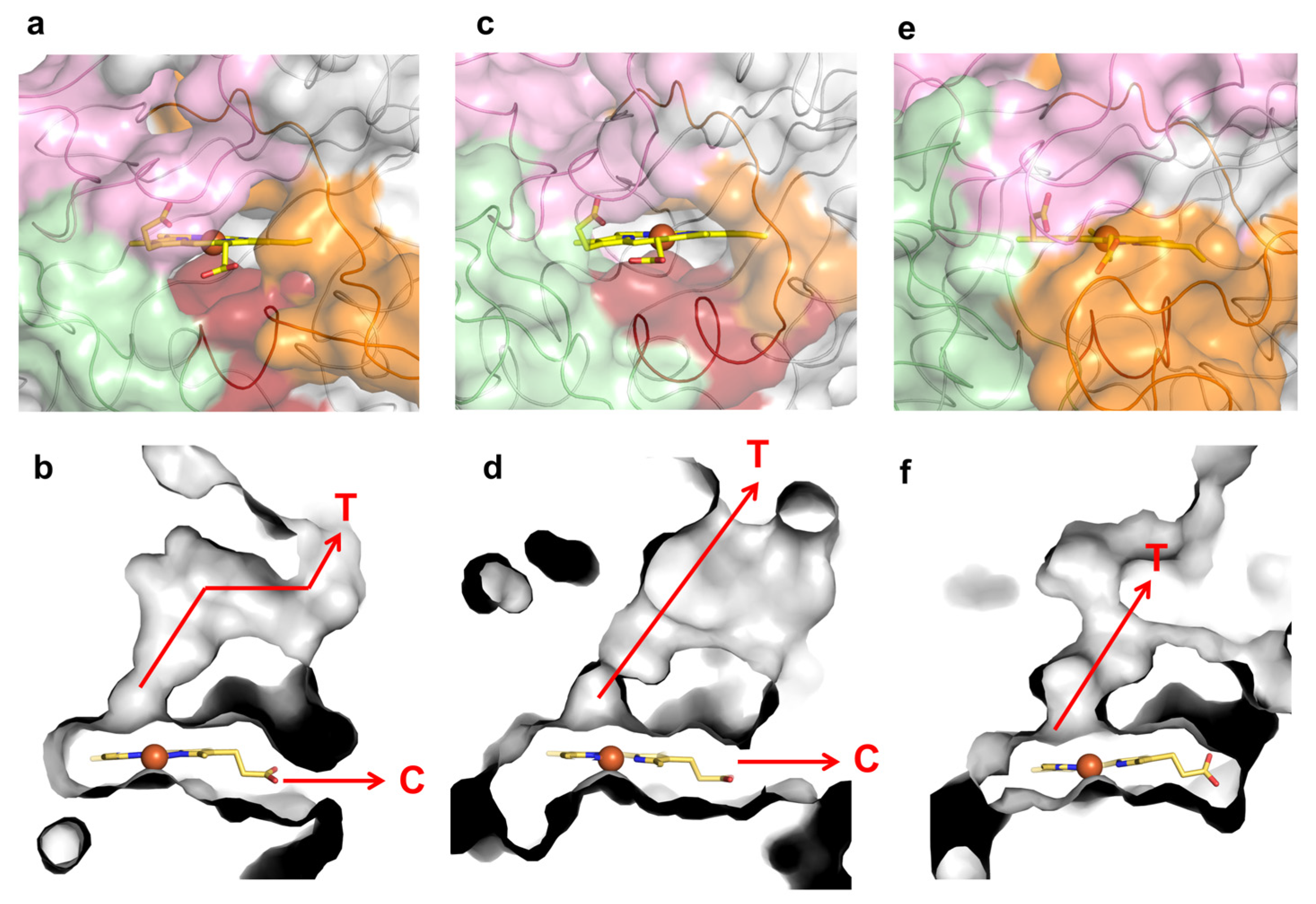
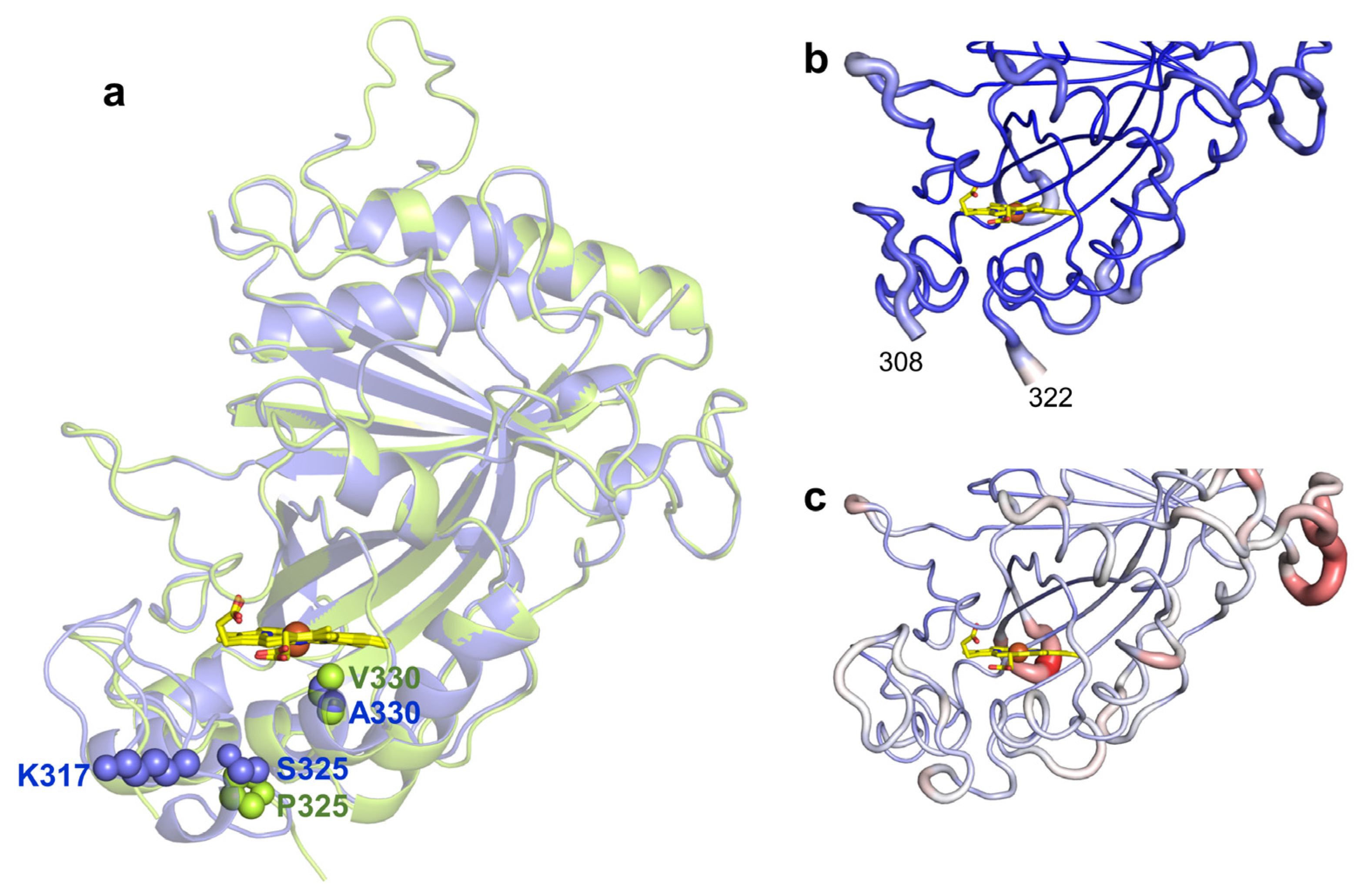
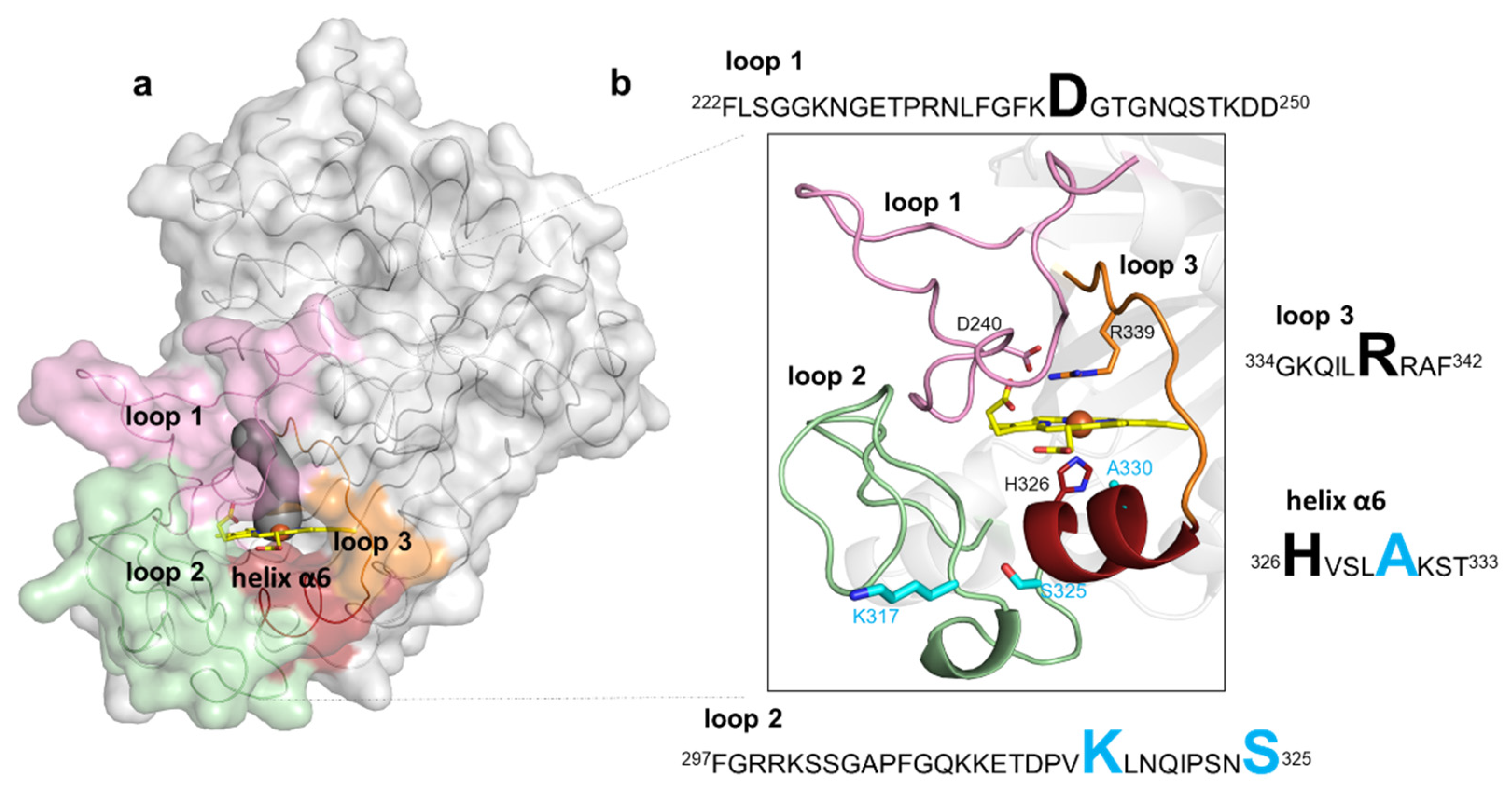
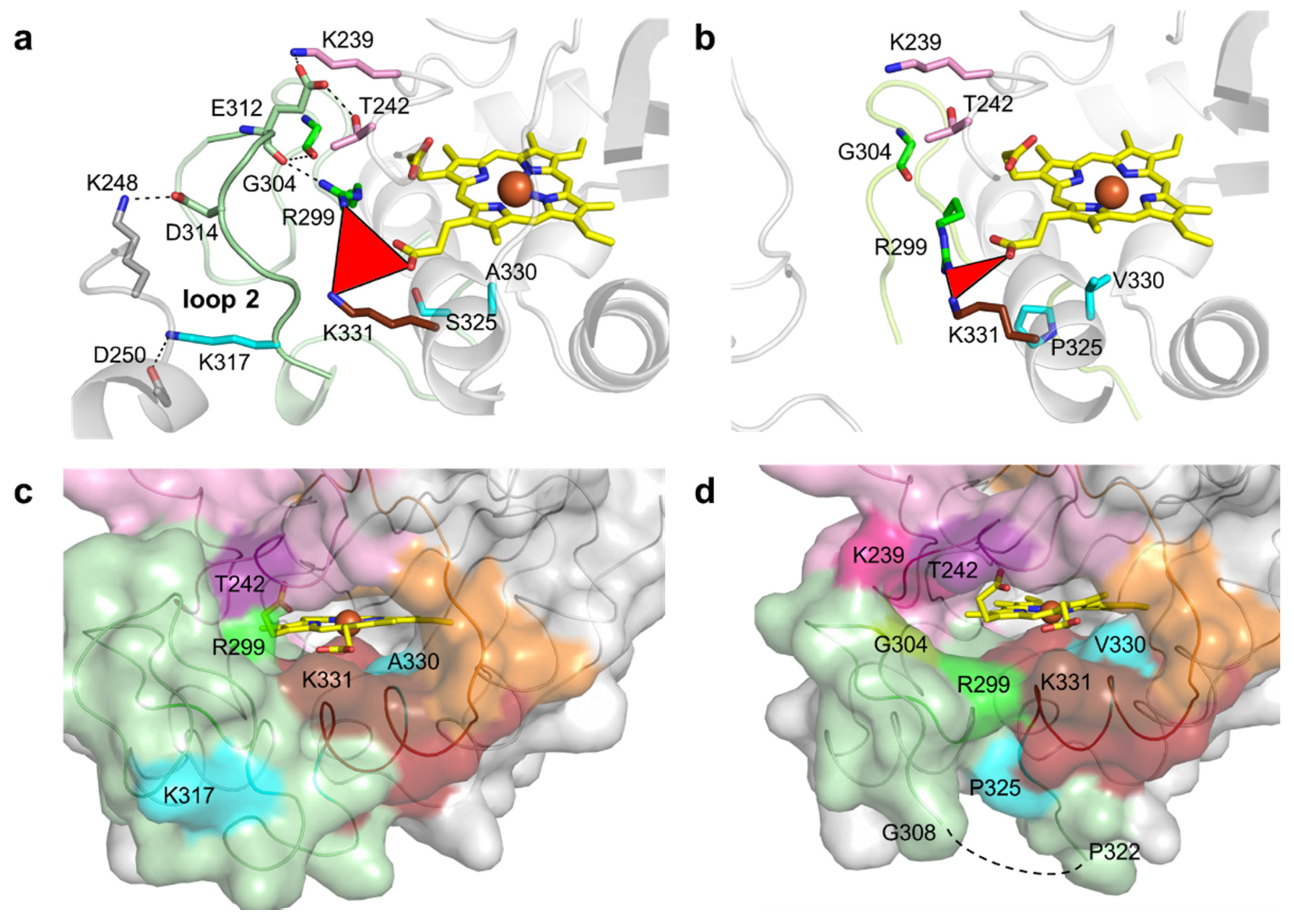
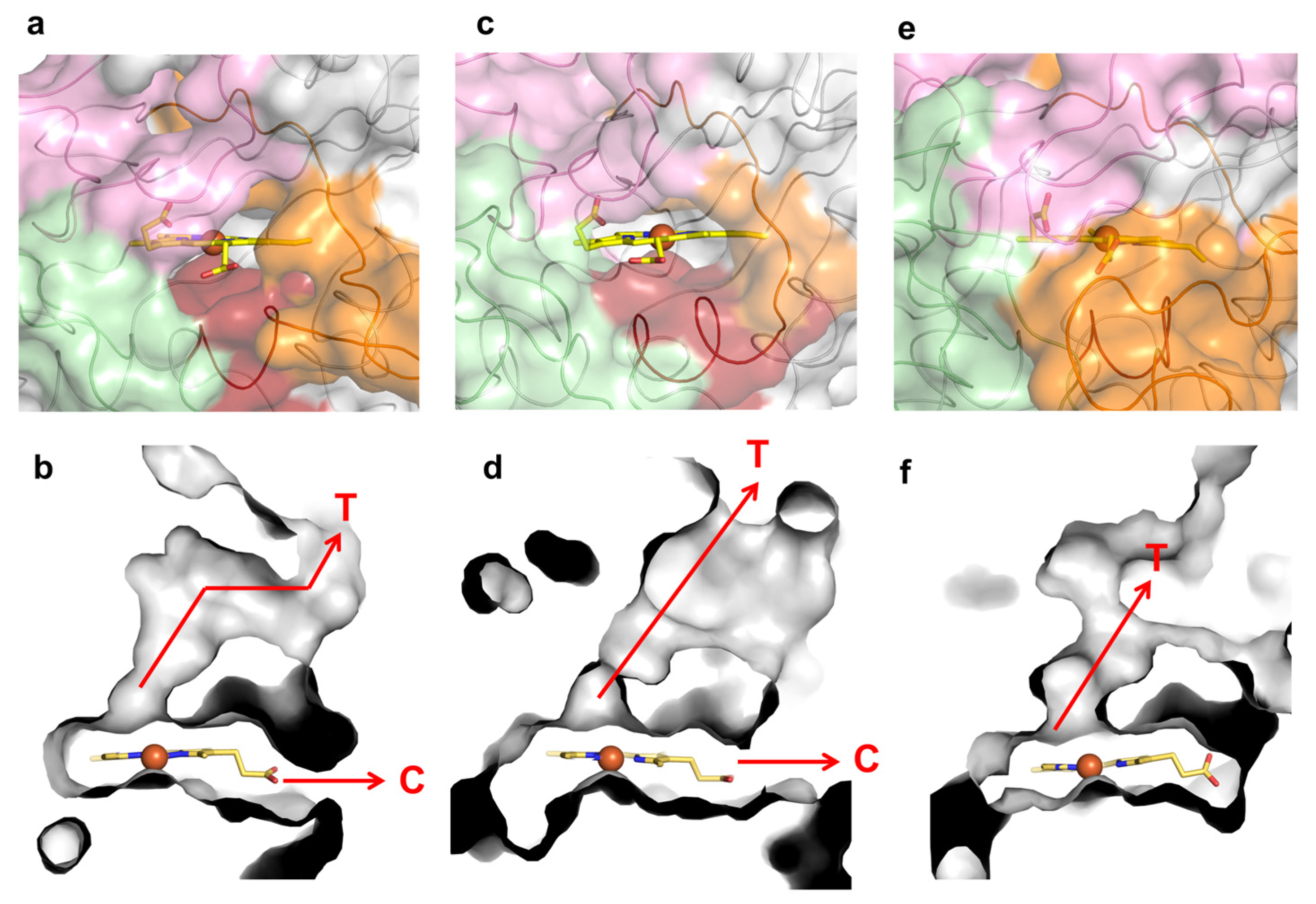
2.4. Role of Mutations in the Optimization of 5G5 for DMP Oxidation
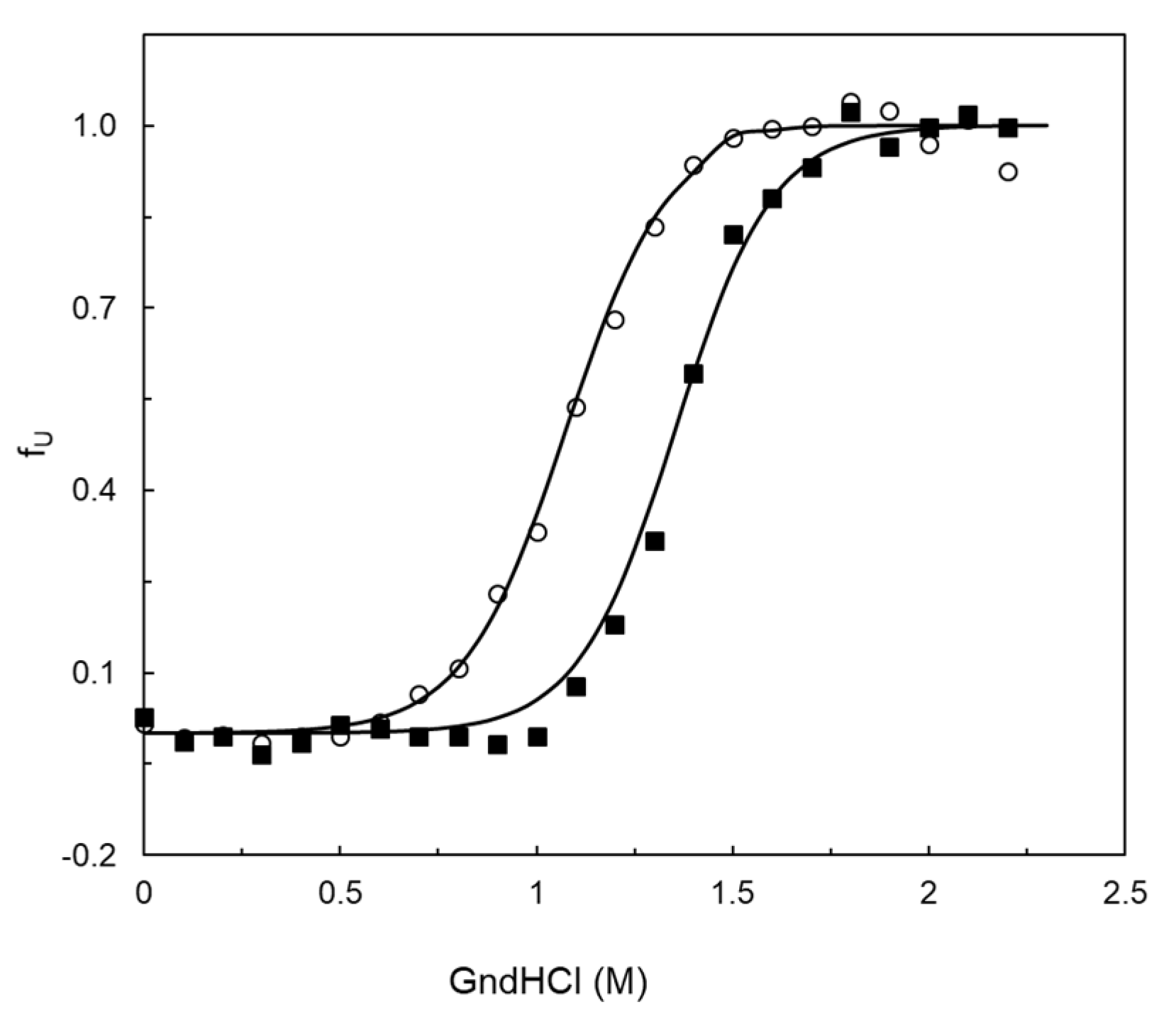
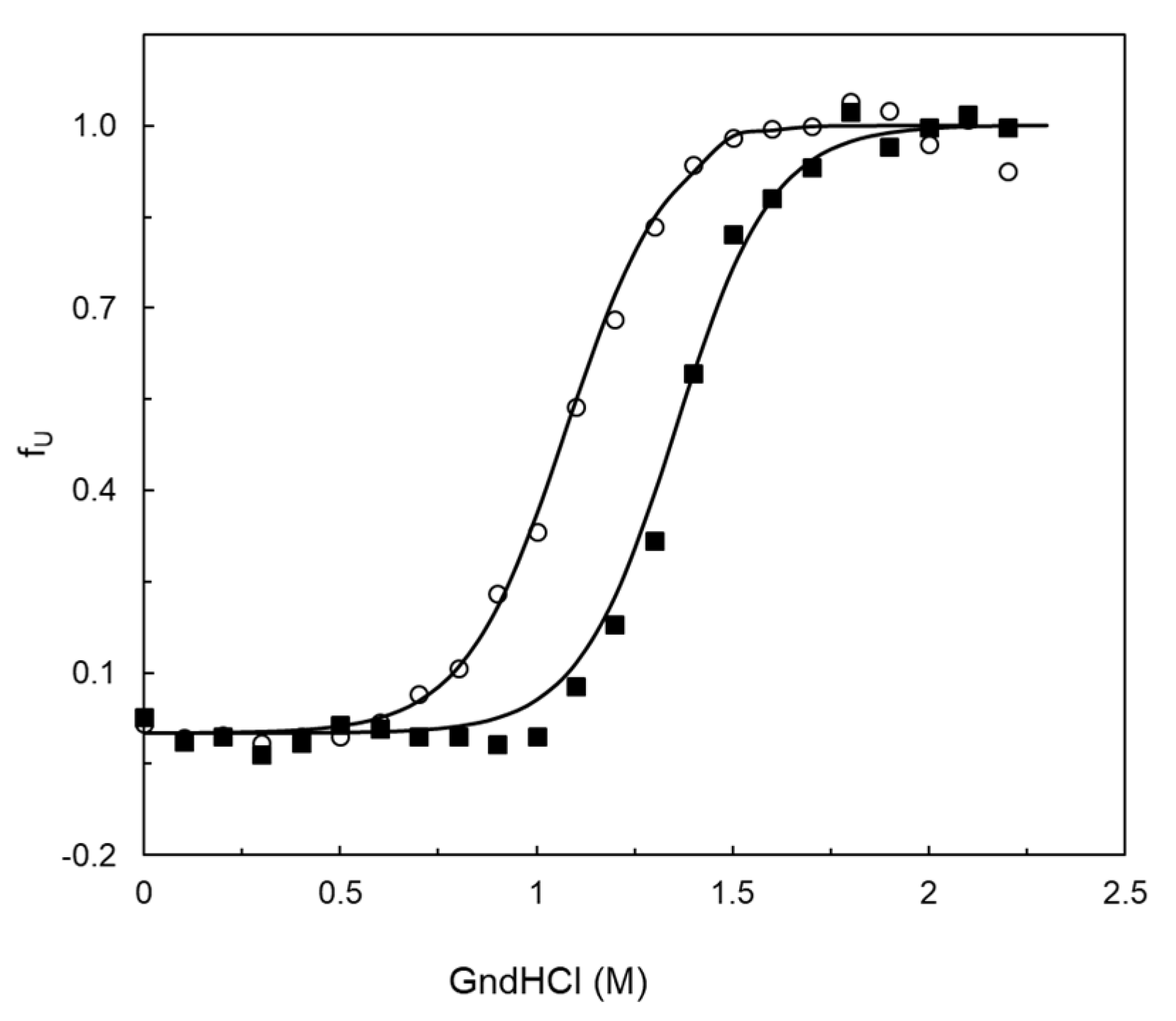
2.5. Stability Properties of 5G5 as Compared to Wild Type
3. Materials and Methods
3.1. Bacterial Strains, Plasmids and Cultivation Media
3.2. Construction of BsDyP Wild Type without Signal Peptide Sequence
3.3. Random Mutagenesis by Error-Prone PCR and Mutant Library Construction
3.4. Recombination by DNA Shuffling and Mutant Library Construction
3.5. “Activity-On-Plate” Initial High-Throughput Screening
3.6. Activity Screening in 96-Well Plates
3.7. Activity Re-Screenings at Large Scale
3.8. Production and Purification of Wild-Type BsDyP and Variants
3.9. Apparent Steady-State Kinetic Analysis
3.10. Crystallization
3.11. Data Collection
3.12. Structure Determination and Refinement
3.13. Enzyme Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, Z.; Fridrich, B.; de Santi, A.; Elangovan, S.; Barta, K. Bright side of lignin depolymerization: Toward new platform chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef] [Green Version]
- Hamalainen, V.; Gronroos, T.; Suonpaa, A.; Hekkila, M.W.; Romein, B.; Ihalainen, P.; Malandra, S.; Birikh, K.R. Enzymatic processes to unlock the lignin value. Front. Bioeng. Biotechnol. 2018, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Van den Bosch, S.; Koelewijn, S.F.; Renders, T.; Van den Bossche, G.; Vangeel, T.; Schutyser, W.; Sels, B.F. Catalytic strategies towards lignin-derived chemicals. Top. Curr. Chem. 2018, 376, 36. [Google Scholar] [CrossRef] [PubMed]
- Runeberg, P.A.; Brusentsev, Y.; Rendon, S.M.K.; Eklund, P.C. Oxidative transformations of lignans. Molecules 2019, 24, 300. [Google Scholar] [CrossRef] [Green Version]
- Llevot, A.; Grau, E.; Carlotti, S.; Grelier, S.; Cramail, H. From Lignin-derived Aromatic compounds to novel biobased polymers. Macromol. Rapid Commun. 2016, 37, 9–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natte, K.; Narani, A.; Goyal, V.; Sarki, N.; Jagadeesh, R.V. Synthesis of functional chemicals from lignin-derived monomers by selective organic transformations. Adv. Synth. Catal. 2020, 362, 5143–5169. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Bugg, T.D.H.; Williamson, J.J.; Rashid, G.M.M. Bacterial enzymes for lignin depolymerisation: New biocatalysts for generation of renewable chemicals from biomass. Curr. Opin. Chem. Biol. 2020, 55, 26–33. [Google Scholar] [CrossRef] [PubMed]
- de Gonzalo, G.; Colpa, D.I.; Habib, M.H.; Fraaije, M.W. Bacterial enzymes involved in lignin degradation. J. Biotechnol. 2016, 236, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Pollegioni, L.; Tonin, F.; Rosini, E. Lignin-degrading enzymes. FEBS J. 2015, 282, 1190–1213. [Google Scholar] [CrossRef]
- Martinez, A.T.; Ruiz-Duenas, F.J.; Martinez, M.J.; del Rio, J.C.; Gutierrez, A. Enzymatic delignification of plant cell wall: From nature to mill. Curr. Opin. Biotechnol. 2009, 20, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Hofbauer, S.; Pfanzagl, V.; Michlits, H.; Schmidt, D.; Obinger, C.; Furtmuller, P.G. Understanding molecular enzymology of porphyrin-binding α + β barrel proteins—One fold, multiple functions. BBA Proteins Proteom. 2020, 1869, 1400536. [Google Scholar] [CrossRef]
- Hofrichter, M.; Ullrich, R.; Pecyna, M.J.; Liers, C.; Lundell, T. New and classic families of secreted fungal heme peroxidases. Appl. Microbiol. Biotechnol. 2010, 87, 871–897. [Google Scholar] [CrossRef]
- Sugano, Y. DyP-type peroxidases comprise a novel heme peroxidase family. Cell Mol. Life Sci. 2009, 66, 1387–1403. [Google Scholar] [CrossRef]
- Colpa, D.I.; Fraaije, M.W.; van Bloois, E. DyP-type peroxidases: A promising and versatile class of enzymes. J. Ind. Microbiol. Biotechnol. 2014, 41, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Sugano, Y. A structural and functional perspective of DyP-type peroxidase family. Arch. Biochem. Biophys. 2015, 574, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, T. Bacterial dye-decolorizing peroxidases: Biochemical properties and biotechnological opportunities. Phys. Sci. Rev. 2016, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Eltis, L.D. The multihued palette of dye-decolorizing peroxidases. Arch. Biochem. Biophys. 2015, 574, 56–65. [Google Scholar] [CrossRef]
- Brissos, V.; Tavares, D.; Sousa, A.C.; Robalo, M.P.; Martins, L.O. Engineering a bacterial dyp-type peroxidase for enhanced oxidation of lignin-related phenolics at alkaline pH. ACS Catal. 2017, 7, 3454–3465. [Google Scholar] [CrossRef]
- Barbosa, C.; Silveira, C.M.; Silva, D.; Brissos, V.; Hildebrandt, P.; Martins, L.O.; Todorovic, S. Immobilized dye-decolorizing peroxidase (DyP) and directed evolution variants for hydrogen peroxide biosensing. Biosens. Bioelectron. 2020, 153, 112055. [Google Scholar] [CrossRef] [PubMed]
- Pour, R.R.; Ehibhatiomhan, A.; Huang, Y.; Ashley, B.; Rashid, G.M.; Mendel-Williams, S.; Bugg, T.D.H. Protein engineering of Pseudomonas fluorescens peroxidase Dyp1B for oxidation of phenolic and polymeric lignin substrates. Enz. Microb. Technol. 2019, 123, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.; Mendes, S.; Brissos, V.; Martins, L.O. New dye-decolorizing peroxidases from Bacillus subtilis and Pseudomonas putida MET94: Towards biotechnological applications. Appl. Microbiol. Biotechnol. 2014, 98, 2053–2065. [Google Scholar] [CrossRef]
- Sezer, M.; Santos, A.; Kielb, P.; Pinto, T.; Martins, L.O.; Todorovic, S. Distinct structural and redox properties of heme active in bacterial DyP-type peroxidases from two subfamilies: Resonance Raman and electrochemistry study. Biochemistry 2013, 52, 3074–3084. [Google Scholar] [CrossRef]
- Min, K.; Gong, G.; Woo, H.M.; Kim, Y.; Um, Y. A dye-decolorizing peroxidase from Bacillus subtilis exhibiting substrate-dependent optimum temperature for dyes and beta-ether lignin dimer. Sci. Rep. 2015, 5, 8245. [Google Scholar] [CrossRef] [PubMed]
- Mendes, S.; Catarino, T.; Silveira, C.; Todorovic, S.; Martins, L.O. Catalytic mechanism of BsDyP an A-type dye-decolourising peroxidase: Neither aspartate nor arginine is individually essential for peroxidase activity. Catal. Sci. Technol. 2015, 5, 5196–5207. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Hauer, B.; Jaeger, K.E.; Schwaneberg, U. Directed Evolution Empowered Redesign of Natural Proteins for the Sustainable Production of Chemicals and Pharmaceuticals. Angew. Chem. Int. Ed. 2019, 58, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, E.; Serrer, K.; Liers, C.; Ullrich, R.; Hofrichter, M.; Piontek, K.; Schleicher, E.; Plattner, D.A. The toolbox of Auricularia auricula-judae dye-decolorizing peroxidase—Identification of three new potential substrate-interaction sites. Arch. Biochem. Biophys. 2015, 574, 75–85. [Google Scholar] [CrossRef]
- Martinez, A.T.; Ruiz-Duenas, F.J.; Camarero, S.; Serrano, A.; Linde, D.; Lund, H.; Vind, J.; Tovborg, M.; Herold-Majumdar, O.M.; Hofrichter, M.; et al. Oxidoreductases on their way to industrial biotransformations. Biotechnol. Adv. 2017, 35, 815–831. [Google Scholar] [CrossRef] [Green Version]
- Jongbloed, J.D.; Grieger, U.; Antelmann, H.; Hecker, M.; Nijland, R.; Bron, S.; van Dijl, J.M. Two minimal Tat translocases in Bacillus. Mol. Microbiol. 2004, 54, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- van der Ploeg, R.; Mader, U.; Homuth, G.; Schaffer, M.; Denham, E.L.; Monteferrante, C.G.; Miethke, M.; Marahiel, M.A.; Harwood, C.R.; Winter, T.; et al. Environmental salinity determines the specificity and need for Tat-dependent secretion of the YwbN protein in Bacillus subtilis. PLoS ONE 2011, 6, e18140. [Google Scholar] [CrossRef] [Green Version]
- van Bloois, E.; Torres Pazmino, D.E.; Winter, R.T.; Fraaije, M.W. A robust and extracellular heme-containing peroxidase from Thermobifida fusca as prototype of a bacterial peroxidase superfamily. Appl. Microbiol. Biotechnol. 2010, 86, 1419–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, A.; Schierhorn, A.; Lindenstrauss, U.; Lilie, H.; Bruser, T. YcdB from Escherichia coli reveals a novel class of Tat-dependently translocated hemoproteins. J. Biol. Chem. 2006, 281, 13972–13978. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, K.; Nishihashi, Y.; Narioka, T.; Yoshida, T.; Morita, M.; Sugano, Y. Characterization of a novel DyP-type peroxidase from Streptomyces avermitilis. J. Biosci. Bioeng. 2017, 123, 425–430. [Google Scholar] [CrossRef]
- Liers, C.; Bobeth, C.; Pecyna, M.; Ullrich, R.; Hofrichter, M. DyP-like peroxidases of the jelly fungus Auricularia auricula-judae oxidize nonphenolic lignin model compounds and high-redox potential dyes. Appl. Microbiol. Biotechnol. 2010, 85, 1869–1879. [Google Scholar] [CrossRef]
- Dhankhar, P.; Dalal, V.; Mahto, J.K.; Gurjar, B.R.; Tomar, S.; Sharma, A.K.; Kumar, P. Characterization of dye-decolorizing peroxidase from Bacillus subtilis. Arch. Biochem. Biophys. 2020, 693, 108590. [Google Scholar] [CrossRef]
- Roberts, J.N.; Singh, R.; Grigg, J.C.; Murphy, M.E.; Bugg, T.D.; Eltis, L.D. Characterization of dye-decolorizing peroxidases from Rhodococcus jostii RHA1. Biochemistry 2011, 50, 5108–5119. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, R.; Huang, G.C.; Meekins, D.A.; Geisbrecht, B.V.; Li, P. Mechanistic insights into dye-decolorizing peroxidase revealed by solvent isotope and viscosity effects. Acs. Catal. 2017, 7, 6352–6364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, T.; Sasaki, M.; Tanaka, Y.; Ishimorit, K. A dye-decolorizing peroxidase from Vibrio cholerae. Biochemistry 2015, 54, 6610–6621. [Google Scholar] [CrossRef]
- Yoshida, T.; Tsuge, H.; Konno, H.; Hisabori, T.; Sugano, Y. The catalytic mechanism of dye-decolorizing peroxidase DyP may require the swinging movement of an aspartic acid residue. FEBS J. 2011, 278, 2387–2394. [Google Scholar] [CrossRef]
- Strittmatter, E.; Liers, C.; Ullrich, R.; Wachter, S.; Hofrichter, M.; Plattner, D.A.; Piontek, K. First crystal structure of a fungal high-redox potential dye-decolorizing peroxidase: Substrate interaction sites and long-range electron transfer. J. Biol. Chem. 2013, 288, 4095–4102. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Tsuge, H.; Hisabori, T.; Sugano, Y. Crystal structures of dye-decolorizing peroxidase with ascorbic acid and 2,6-dimethoxyphenol. FEBS Lett. 2012, 586, 4351–4356. [Google Scholar] [CrossRef] [Green Version]
- Linde, D.; Ruiz-Duenas, F.J.; Fernandez-Fueyo, E.; Guallar, V.; Hammel, K.E.; Pogni, R.; Martinez, A.T. Basidiomycete DyPs: Genomic diversity, structural-functional aspects, reaction mechanism and environmental significance. Arch. Biochem. Biophys. 2015, 574, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Liers, C.; Aranda, E.; Strittmatter, E.; Piontek, K.; Plattner, D.A.; Zorn, H.; Ullrich, R.; Hofrichter, M. Phenol oxidation by DyP-type peroxidases in comparison to fungal and plant peroxidases. J. Mol. Cat B Enz. 2014, 103, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Strittmatter, E.; Wachter, S.; Liers, C.; Ullrich, R.; Hofrichter, M.; Plattner, D.A.; Piontek, K. Radical formation on a conserved tyrosine residue is crucial for DyP activity. Arch. Biochem. Biophys. 2013, 537, 161–167. [Google Scholar] [CrossRef]
- Fernandez-Fueyo, E.; Linde, D.; Almendral, D.; Lopez-Lucendo, M.F.; Ruiz-Duenas, F.J.; Martinez, A.T. Description of the first fungal dye-decolorizing peroxidase oxidizing manganese(II). Appl. Microbiol. Biotechnol. 2015, 99, 8927–8942. [Google Scholar] [CrossRef] [Green Version]
- Baratto, M.C.; Sinicropi, A.; Linde, D.; Saez-Jimenez, V.; Sorace, L.; Ruiz-Duenas, F.J.; Martinez, A.T.; Basosi, R.; Pogni, R. Redox-active sites in Auricularia auricula-judae dye-decolorizing peroxidase and several directed variants: A multifrequency EPR study. J. Phys. Chem. 2015, 119, 13583–13592. [Google Scholar] [CrossRef] [PubMed]
- Linde, D.; Pogni, R.; Canellas, M.; Lucas, F.; Guallar, V.; Baratto, M.C.; Sinicropi, A.; Saez-Jimenez, V.; Coscolin, C.; Romero, A.; et al. Catalytic surface radical in dye-decolorizing peroxidase: A computational, spectroscopic and site-directed mutagenesis study. Biochem. J. 2015, 466, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Nys, K.; Furtmuller, P.G.; Obinger, C.; Van Doorslaer, S.; Pfanzagl, V. On the track of long-range electron transfer in b-type dye-decolorizing peroxidases: Identification of a tyrosyl radical by computational prediction and electron paramagnetic resonance spectroscopy. Biochemistry 2021, 60, 1226–1241. [Google Scholar] [CrossRef] [PubMed]
- Karplus, P.A.; Diederichs, K. Linking Crystallographic Model and Data Quality. Science 2012, 336, 1030–1033. [Google Scholar] [CrossRef] [Green Version]
- Arndt, U.W.; Crowther, R.A.; Mallett, J.F.W. A computer-linked cathode-ray tube microdensitometer for X-ray crystallography. J. Phys. E Sci. Instrum. 1968, 1, 510–516. [Google Scholar] [CrossRef]
- Diederichs, K.; Karplus, P.A. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 1997, 4, 269–275. [Google Scholar] [CrossRef]
- Weiss, M.S. Global indicators of X-ray data quality. J. Appl. Crystallogr. 2001, 34, 130–135. [Google Scholar] [CrossRef]
- Nestl, B.M.; Hauer, B. Engineering of flexible loops in enzymes. ACS Catal. 2014, 4, 3201–3211. [Google Scholar] [CrossRef]
- Heinemann, P.M.; Armbruster, D.; Hauer, B. Active-site loop variations adjust activity and selectivity of the cumene dioxygenase. Nat. Commun. 2021, 12, 1095. [Google Scholar] [CrossRef] [PubMed]
- Pabis, A.; Risso, V.A.; Sanchez-Ruiz, J.M.; Kamerlin, S.C.L. Cooperativity and flexibility in enzyme evolution. Curr. Opin. Strut. Biol. 2018, 48, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Berry, E.A.; Trumpower, B.L. Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal. Biochem. 1987, 161, 1–15. [Google Scholar] [CrossRef]
- Juanhuix, J.; Gil-Ortiz, F.; Cuni, G.; Colldelram, C.; Nicolas, J.; Lidon, J.; Boter, E.; Ruget, C.; Ferrer, S.; Benach, J. Developments in optics and performance at BL13-XALOC, the macromolecular crystallography beamline at the Alba Synchrotron. J. Synchrotron Radiat. 2014, 21, 679–689. [Google Scholar] [CrossRef]
- von Stetten, D.; Carpentier, P.; Flot, D.; Beteva, A.; Caserotto, H.; Dobias, F.; Guijarro, M.; Giraud, T.; Lentini, M.; McSweeney, S.; et al. ID30A-3 (MASSIF-3)—A beamline for macromolecular crystallography at the ESRF with a small intense beam. J. Synchrotron Radiat. 2020, 27, 844–851. [Google Scholar] [CrossRef]
- Kabsch, W. Xds. Acta Crystallogr. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Kantardjieff, K.A.; Rupp, B. Matthews coefficient probabilities: Improved estimates for unit cell contents of proteins, DNA, and protein-nucleic acid complex crystals. Protein Sci. 2003, 12, 1865–1871. [Google Scholar] [CrossRef]
- Matthews, B.W. Solvent Content of Protein Crystals. J. Mol. Biol. 1968, 33, 491–497. [Google Scholar] [CrossRef]
- Vagin, A.; Lebedev, A. MoRDa, an automatic molecular replacement pipeline. Acta Crystallogr. A 2015, 71, s19. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.; Afonine, P.; Echols, N.; Headd, J.; Grosse-Kunstleve, R.; Moriarty, N. New tools for structure refinement in Phenix. Acta Crystallogr. A 2010, 66, s15. [Google Scholar] [CrossRef] [Green Version]
- Oeffner, R.D.; Bunkoczi, G.; McCoy, A.J.; Read, R.J. Improved estimates of coordinate error for molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 2209–2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D 2012, 68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.W.; Read, R.J.; Adams, P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D 2008, 64, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Painter, J.; Merritt, E.A. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D 2006, 62, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Engh, R.A.; Huber, R. Accurate Bond and Angle Parameters for X-Ray Protein-Structure Refinement. Acta Crystallog. A 1991, 47, 392–400. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Meth. Mol. Biol. 2014, 1137, 1–15. [Google Scholar]
- Chovancova, E.; Pavelka, A.; Benes, P.; Strnad, O.; Brezovsky, J.; Kozlikova, B.; Gora, A.; Sustr, V.; Klvana, M.; Medek, P.; et al. CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 2012, 8, e1002708. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: Palo Alto, CA, USA, 2002. [Google Scholar]
- Schrodinger, L.L.C. The PyMOL Molecular Graphics System; Version 1.7.2.2; Schrödinger, LLC.: New York, NY, USA, 2010. [Google Scholar]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Richards, F.M. Interpretation of Protein Structures—Estimation of Static Accessibility. J. Mol. Biol. 1971, 55, 379. [Google Scholar] [CrossRef]
- Saff, E.B.; Kuijlaars, A.B.J. Distributing many points on a sphere. Math. Intell. 1997, 19, 5–11. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Tsai, C.J.; Nussinov, R. Temperature Range of Thermodynamic Stability for the Native State of Reversible Two-State Proteins. Biochemistry 2003, 42, 4864–4873. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Production (mg L−1) | Rz | λmax (nm) | ε (mM−1 cm−1) | Heme Content |
|---|---|---|---|---|---|
| Wild Type | 8.3 ± 0.4 | 2.3 | 406 | 82 | 0.4 ± 0.1 |
| 1F9 | 11.1 ± 0.8 | 2.5 | 407 | 92 | 0.7 ± 0.2 |
| 3G5 | 6.5 ± 0.5 | 1.6 | 406 | 60 | 0.6 ± 0.2 |
| 54D6 | 16.0 ± 0.9 | 1.8 | 407 | 66 | 0.6 ± 0.1 |
| 5G5 | 15.0 ± 1.0 | 2.1 | 407 | 77 | 0.7 ± 0.1 |
| Enzyme | H2O2 | ABTS | ||||
|---|---|---|---|---|---|---|
| kcatapp (s−1) | Kmapp (mM) | kcatapp/Kmapp (M−1s−1) | Ki (mM) | Kmapp (mM) | kcatapp/Kmapp (M−1s−1) | |
| Wild Type | 14.2 ± 1.0 | 0.06 ± 0.01 | (2.2 ± 0.4) × 105 | 6.5 ± 1.9 | 0.42 ± 0.03 | (3.6 ± 0.3) × 104 |
| 1F9 | 50.4 ± 1.4 | 0.19 ± 0.05 | (2.7 ± 0.7) × 105 | 2.6 ± 0.2 | 0.89 ± 0.10 | (5.8 ± 0.7) × 104 |
| 3G5 | 28.1 ± 2.1 | 0.06 ± 0.01 | (4.9 ± 0.7) × 105 | 8.5 ± 0.4 | 1.37 ± 0.25 | (2.5 ± 0.5) × 104 |
| 54D6 | 47.7 ± 2.9 | 0.15 ± 0.02 | (3.3 ± 0.6) × 105 | 8.1 ± 0.7 | 0.80 ± 0.14 | (6.0 ± 1.1) × 104 |
| 5G5 | 56.1 ± 1.9 | 0.18 ± 0.02 | (3.3 ± 0.7) × 105 | 3.9 ± 0.6 | 1.29 ± 0.19 | (4.2 ± 0.6) × 104 |
| Enzyme | kcatapp (s−1) | Kmapp (mM) | kcatapp/Kmapp (M−1s−1) |
|---|---|---|---|
| Wild Type | 0.42 ± 0.03 | 0.06 ± 0.01 | (7 ± 1) × 103 |
| 1F9 | 0.83 ± 0.04 | 0.15 ± 0.01 | (5.5 ± 0.4) × 103 |
| 3G5 | 0.8 ± 0.1 | 0.09 ± 0.01 | (9 ± 1) × 103 |
| 54D6 | 1.0 ± 0.1 | 0.60 ± 0.01 | (1.6 ± 0.5) × 103 |
| 5G5 | 2.8 ± 0.1 | 0.7 ± 0.1 | (3.8 ± 0.2) × 103 |
| BsDyP-Wild-Type | BsDyP-5G5 | |
|---|---|---|
| Data Collection | ||
| Beamline | BL13-XALOC | ID30A-3 |
| Wavelength (Å) | 0.97926 | 0.9680 |
| Space group | P 31 2 1 | P 1 21 1 |
| Unit cell parameters (Å) | a = 95.3, b = 95.3, c = 181.2 | a = 63.9, b = 114.8, c = 116.8 |
| Resolution (Å) | 75.09–2.49 (2.59–2.49) | 61.96–2.10 (2.20–2.10) |
| Number of observations | 215,157 (26,986) | 285,233 (46,534) |
| Unique reflections | 33,999 (5374) | 92,499 (15,038) |
| Completeness (%) | 99.8 (99.2) | 99.1 (98.0) |
| Multiplicity | 6.3 (5.0) | 3.1 (3.1) |
| Mosaicity (ᵒ) | 0.12 | 0.09 |
| CC1/2 (%) a | 99.7 (30.6) | 99.6 (47.8) |
| Rsym (%) b | 13.0 (78.8) | 8.8 (53.2) |
| Rmeas (%) c | 16.8 (243.4) | 12.2 (111.2) |
| Rpim (%) d | 5.6 (39.5) | 5.8 (35.2) |
| <I/σ(I)> | 10.37 (0.67) | 9.02 (1.39) |
| Wilson B-factor (Å2) | 61.4 | 42.4 |
| VM (Å3 Da−1) | 2.99 | 2.27 |
| Estimated solvent content (%) | 58.9 | 45.8 |
| Refinement | ||
| Rwork (%) e | 20.7 | 19.1 |
| Rfree (%) e | 23.2 | 21.5 |
| rmsd for bond lengths (Å) | 0.008 | 0.002 |
| rmsd for bond angles (°) | 0.922 | 0.627 |
| Average B-factor (Å2) | 68.3 (chain A), 68.2 (chain B) | 37.8 (chain A), 39.5 (chain B), 43.2 (chain C), 47.6 (chain D) |
| Ramachandran plot | ||
| Residues in favored regions (%) | 97.6 | 97.4 |
| Residues in allowed regions (%) | 2.4 | 2.6 |
| Residues in disallowed regions (%) | 0 | 0 |
| PDB code | 7PKX | 7PL0 |
| Enzyme | Tm (°C) | Half-Life 40 °C (h) |
|---|---|---|
| WT | 62.4 ± 0.7 | 109 ± 1 |
| 1F9 | 59.2 ± 0.4 | 21 ± 1 |
| 3G5 | 58.1 ± 0.2 | 27 ± 3 |
| 54D6 | 58.2 ± 0.4 | 10 ± 2 |
| 5G5 | 58.6 ± 0.4 | 20 ± 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, C.F.; Borges, P.T.; Scocozza, M.F.; Silva, D.; Taborda, A.; Brissos, V.; Frazão, C.; Martins, L.O. Loops around the Heme Pocket Have a Critical Role in the Function and Stability of BsDyP from Bacillus subtilis. Int. J. Mol. Sci. 2021, 22, 10862. https://doi.org/10.3390/ijms221910862
Rodrigues CF, Borges PT, Scocozza MF, Silva D, Taborda A, Brissos V, Frazão C, Martins LO. Loops around the Heme Pocket Have a Critical Role in the Function and Stability of BsDyP from Bacillus subtilis. International Journal of Molecular Sciences. 2021; 22(19):10862. https://doi.org/10.3390/ijms221910862
Chicago/Turabian StyleRodrigues, Carolina F., Patrícia T. Borges, Magali F. Scocozza, Diogo Silva, André Taborda, Vânia Brissos, Carlos Frazão, and Lígia O. Martins. 2021. "Loops around the Heme Pocket Have a Critical Role in the Function and Stability of BsDyP from Bacillus subtilis" International Journal of Molecular Sciences 22, no. 19: 10862. https://doi.org/10.3390/ijms221910862
APA StyleRodrigues, C. F., Borges, P. T., Scocozza, M. F., Silva, D., Taborda, A., Brissos, V., Frazão, C., & Martins, L. O. (2021). Loops around the Heme Pocket Have a Critical Role in the Function and Stability of BsDyP from Bacillus subtilis. International Journal of Molecular Sciences, 22(19), 10862. https://doi.org/10.3390/ijms221910862